

Significance
Maintenance of cell volume and ionic homeostasis are fundamental to cell function. Cells shrink when extracellular osmolality is increased, and although mechanisms controlling cell volume recovery are well documented, the mechanisms underlying changes in membrane trafficking that occur during adaptation to high osmolality are not well understood. Molecular crowding arising from cell shrinkage can alter protein structure and has been proposed to provoke changes in trafficking events. However, cell volume recovery involves a rapid influx of ions, and the effects of ionic imbalance on trafficking dynamics in living cells remain largely unexplored. We found that high levels of chloride and loss of ATP, in addition to molecular crowding, contribute to altered cytoskeletal and vesicular dynamics during hypertonic stress.
Keywords: electrolyte, cell volume, hypertonicity, membrane trafficking, mitochondria
Abstract
Cell volume homeostasis is vital for the maintenance of optimal protein density and cellular function. Numerous mammalian cell types are routinely exposed to acute hypertonic challenge and shrink. Molecular crowding modifies biochemical reaction rates and decreases macromolecule diffusion. Cell volume is restored rapidly by ion influx but at the expense of elevated intracellular sodium and chloride levels that persist long after challenge. Although recent studies have highlighted the role of molecular crowding on the effects of hypertonicity, the effects of ionic imbalance on cellular trafficking dynamics in living cells are largely unexplored. By tracking distinct fluorescently labeled endosome/vesicle populations by live-cell imaging, we show that vesicle motility is reduced dramatically in a variety of cell types at the onset of hypertonic challenge. Live-cell imaging of actin and tubulin revealed similar arrested microfilament motility upon challenge. Vesicle motility recovered long after cell volume, a process that required functional regulatory volume increase and was accelerated by a return of extracellular osmolality to isosmotic levels. This delay suggests that, although volume-induced molecular crowding contributes to trafficking defects, it alone cannot explain the observed effects. Using fluorescent indicators and FRET-based probes, we found that intracellular ATP abundance and mitochondrial potential were reduced by hypertonicity and recovered after longer periods of time. Similar to the effects of osmotic challenge, isovolumetric elevation of intracellular chloride concentration by ionophores transiently decreased ATP production by mitochondria and abated microfilament and vesicle motility. These data illustrate how perturbed ionic balance, in addition to molecular crowding, affects membrane trafficking.
A basic challenge for all animal cells is to maintain constant volume and intracellular electrolyte composition (1, 2). K+ is the principal intracellular cation, and Na+ is extruded from the cell interior. Cellular concentration of these ions is controlled by energy-consuming transport mechanisms, primarily the Na+/K+-ATPase. Cl− is a major anion in the extracellular milieu that is maintained at relatively low concentrations (5–40 mM) within the cytosol by a variety of plasma membrane transporters, including the CFTR and members of the chloride channel, voltage-senstitive (ClC) family of Cl− channels (3). Together, these ions not only maintain cell volume and a biologically compatible intracellular milieu but also allow passive diffusional movement of ions down their electrochemical gradients that is central to cell function, from general cell division to the more specialized processes of secretion and contractibility. Various types of environmental challenge lead to electrolyte imbalance, as illustrated by variations of environmental tonicity that immediately alter cell volume and intracellular ion composition (4–6). Cells shrink rapidly in hypertonic environments and swell in hypotonic environments. Cell volume is reestablished quickly—within minutes—in mammalian cells by salt (Na+ and Cl−) influx (in response to hypertonic stress) or K+ efflux (in response to hypotonic stress), but such changes occur at the expense of electrolyte imbalance. Prechallenge intracellular ion composition is reestablished gradually over time (hours to days) by readjusting the intracellular levels of nonionic solutes, principally amino acids and other organic compounds (2, 6) that have far fewer effects on enzyme activity.
Numerous organisms across the animal kingdom are routinely exposed to fast variations of environmental osmolality. In mammals, many cell types must deal with hypertonic challenge as part of their daily function. These include blood cells passing through the renal medulla and intestinal epithelial cells after food ingestion (7). Moreover, hypertonic fluids are routinely administered to treat disorders such as hyponatremia, cystic fibrosis, and hemorrhagic shock (8–10). Hypertonicity affects a wide range of cellular processes. For example, it causes DNA damage (11) and protein damage and aggregation (5, 12), promotes autophagy (13), and leads to apoptosis when challenge is overwhelming (4). Membrane trafficking is a major process affected by hypertonicity that probably contributes significantly to the change incurred upon challenge. Indeed, we and others have observed that endocytosis, exocytosis, and vesicular trafficking are reduced from the onset of challenge, and that the reduction can persist hours after challenge (14–19). However, how immediate changes in the cell’s biophysical parameters influence these events is unclear. By inducing cell shrinkage, hypertonicity increases intracellular component density and molecular crowding (20). Increased crowding of macromolecules and organelles by water efflux results in cytoplasm stiffening (21), protein misfolding and aggregation (12), and reduces protein diffusion (22). Therefore it is reasonable to suppose that molecular crowding may interfere with at least some trafficking events. On the other hand, because osmotically induced cell shrinkage is invariably accompanied by increased intracellular salt concentration, the relative contribution of each occurrence on membrane trafficking is unclear.
High levels of intracellular salt after hypertonic challenge may perturb a number of parameters that collectively affect membrane trafficking. Microtubules (MT) and actin filaments play paramount roles in membrane trafficking. Their dynamic nature creates forces that help move cargo, and they supply “tracks” for long-range delivery of cargo (23). A key feature of MT biology is dynamic instability, so that tubules continuously switch between episodes of steady growth and rapid shrinkage (24), allowing the cell to probe the cytoplasm constantly and adapt quickly to environmental change. A hallmark feature of hypertonicity is that it induces actin cytoskeleton reorganization (4), and we recently have shown that it also induces fast MT remodeling (13). Because the dynamics of microfilament assembly/disassembly are sensitive to ionic conditions (25, 26), a sudden increase in intracellular salt could alter their dynamics and impinge on membrane trafficking. High levels of intracellular salt also could affect membrane trafficking by modulating available energy. Increased ion concentration decreases mitochondrial matrix volume and disrupts key parameters of the respiratory chain, as shown in isolated mitochondria (27, 28) and as predicted by mathematical models of mitochondrial bioenergetics (29). Thus, hypertonicity conceivably could affect membrane trafficking by decreasing ATP production by mitochondria. However, the influence of hypertonicity on mitochondrial function in intact cells has been investigated mostly in the context of apoptosis induced by severe hypertonic stress, which disrupts mitochondrial potential (30–32).
In the present study, we examined the effects of molecular crowding, high intracellular Cl− concentration [Cl−]i, microfilament remodeling, and altered mitochondrial function by hypertonicity on vesicle motility. We show that strong but sustainable hypertonic challenge to mammalian cells immediately halts both microfilament motility and the motility of a wide variety of endosome/vesicle subpopulations. We demonstrate that vesicle motility recovers significantly more slowly than cell volume and that an isovolumetric increase in [Cl−]i by ionophores triggers mitochondrial depolarization and decreases ATP levels, microfilament dynamics, and vesicle motility. Thus, these data illustrate how intracellular ionic imbalance, in addition to molecular crowding, affects membrane trafficking and cell function.
Results
Hypertonicity Induces a Transient and Ubiquitous Decrease in Vesicle Motility.
We have shown previously that hypertonicity reduces endocytosis and exocytosis and affects vesicle distribution concomitant with autophagy in porcine LLC-PK1 renal proximal tubule-like cells (13, 18). We also showed that the great majority of cells survive when medium osmolality is increased from 300 to 500 mOsmol/kg but not to 700 mOsmol/kg (13). To explore further the mechanisms underlying these changes in intracellular trafficking, we analyzed the motility of FITC-dextran–loaded endosomes in live LLC-PK1 cells by spinning-disk confocal imaging (Fig. 1). About 90% of FITC-dextran–loaded endosomes were mobile under isosmotic conditions, indicating that the dextran was incorporated into functional endosomes efficiently. Consistent with expected patterns (33), movement consisted of either quick, short trajectories that frequently changed direction or longer, more linear trajectories that sometimes spanned the entire length of cells. Endosome velocity was broadly distributed, ranging from the fast motility characteristic of MT-based movement [30–60 µm/min (34, 35)] to the slower motility distinctive of movement mediated by myosin motor proteins and actin treadmilling [1.2–12 µm/min (36–40)]. Because of their large size, movement of dextran-loaded endosomes can be tracked easily by live-cell imaging (Fig. 1A). We quantified their motility by measuring the mean endosome displacement over 2 min at 12-s intervals. We considered only mobile endosomes that remained in the focal plane during the entire 2-min time interval, thus limiting our analysis to slower-moving endosomes. The distribution of the average velocity (displacement/time) of individual endosomes during a 2-min time interval under isotonic conditions is depicted in Fig. 1B. Despite the high variability (SD) in the velocity of individual endosomes (10.6 ± 3.2 µm/min), the average velocity of tracked endosomes (30 for each 2 min time interval) was highly reproducible among experiments (10.6 ± 1.1 µm/min) (Fig. S1A).
Fig. 1.

Endosome motility is transiently abolished by hypertonic challenge. (A) The mean velocity of FITC-dextran–loaded endosomes in LLC-PK1 cells was quantified by measuring mean endosome displacement over 2 min at 12-s intervals. Shown are examples of movement occurring under isotonic conditions. (B) Histogram of endosome motility occurring under isotonic conditions. Data were gathered from 120 endosomes. (C) Kymograph illustrating endosome motility occurring in a single focal plane (x axis) over time (y axis) 360 s before and 1,080 s after exposure to hypertonic medium (500 mOsmol/kg). Endosomes that were mobile under isotonic conditions or after long periods of exposure to hypertonic medium produced dim or jagged lines, whereas endosomes immobilized upon challenge produced straight lines. (D) Displacement of endosomes at 360 and 1 s before (Ctl) and 12, 360, and 720 s after NaCl challenge. Arrows show examples of immobilized endosomes. (Scale bar: 10 µm.) (E and F) Endosome motility is shown normalized to the average displacement under isotonic conditions (first 6 min). Motility at 12 s after challenge is shown also to illustrate nearly instantaneous endosome immobilization. Endosome motility was quantified before and after (arrow) NaCl challenge (350, 400, 500 mOsmol/kg) (E) and before and after (arrow) NaCl, KCl, glucose, mannitol, or urea challenge (500 mOsmol/kg) (F). (Also see Movie S1). Error bars show mean ± SEM from four independent experiments.
Fig. S1.
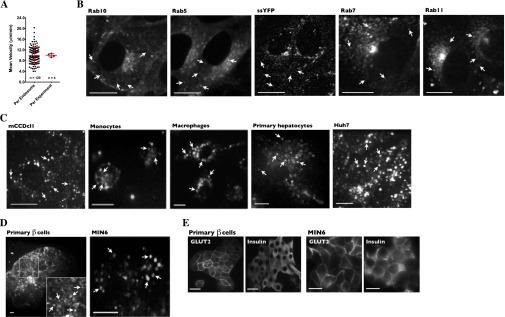
Fluorescent labeling of endosome/vesicle subpopulations. (A) Although the motility of individual FITC-dextran–loaded endosomes (n = 120) varies greatly, the variance of the average endosome motility between experiments (n = 4) is much smaller. Error bars show means ± SD. (B) LLC-PK1 cells were transiently transfected with cDNA encoding various fluorescent proteins used as markers to trace motilities of distinct endosome/vesicle subpopulations, as described in the legend of Fig. 1. (C and D) Various primary cells and stable cell lines were loaded with FITC-dextran to investigate early/late endosome motility. Arrows indicate examples of fluorescently labeled endosomes. (E) GLUT2 and insulin expression in primary β cells (Left) and MIN6 cells (Right). Cell clusters of primary pancreatic cell preparations consisted principally of β cells, as reflected by high levels of GLUT2 at the plasma membrane and intracellular insulin. (Scale bars: 10 µm.)
The motility of at least 95% of all labeled endosomes was abolished almost completely immediately after NaCl challenge (500 mOsmol/kg) and recovered partly after longer periods of time (Fig. 1 C–F and Movie S1). We examined in detail the effects of hypertonicity on the motility of tracked endosomes (Fig. 1 E and F) and drew several conclusions from these studies. First, half-maximal recovery (Ret1/2) increased with the increasing intensity of NaCl challenge, e.g., 2.56 ± 0.12 min for NaCl-350 challenge compared with 9.44 ± 1.15 min for NaCl-500 challenge (Fig. 1E). Increased Ret1/2 was accompanied by decreased recovery of maximal motility at steady state (Remax), e.g., 0.87 ± 0.01 for NaCl-350 challenge compared with 0.53 ± 0.05 for NaCl-500 challenge (Fig. 1E). Under these conditions, the plateau levels reached at steady state after longer periods of time depended on the size of the challenge. Second, an increase in osmolality of 200 mOsmol/kg by either KCl (another ionic compound) or a nonelectrolyte mannitol produced effects similar to those induced by NaCl (Fig. 1F). On the other hand, endosome motility recovered significantly faster in cells challenged with cell-permeable urea, although the level of motility at the steady-state plateau still was slightly smaller (0.86 ± 0.01) than that observed in cells exposed to isotonic conditions (300 mOsm/kg; 0.91 ± 0.05) for the same amount of time (Fig. 1 E and F), possibly as a consequence of increased intracellular osmolality. Third, NaCl challenge reduced the motility of numerous endosome/vesicle subpopulations. Because dextran is distributed quickly to early, late, and recycling endosomes, our analysis cannot distinguish possible differences in motility among these intracellular compartments. Therefore we investigated the effects of NaCl (500 mOsmol/kg) on other vesicular compartments by following the motilities of various GFP-tagged Rab isoforms and the motility of soluble secreted YFP (ssYFP) (41), each expressed in specific endosome/vesicle subpopulations (Fig. 2 A and B and Movie S2). Images of these markers are shown in Fig. S1B. Although NaCl challenge initially abolished motility of all endosome/vesicle subpopulations, their recovery rates varied. This variation is illustrated by the differences in Ret1/2 and Remax values: Some, but not all, vesicular compartments reached a steady-state plateau at lower levels of motility than those exposed to isotonic conditions for the same amount of time (Fig. 2A). This result indicates that each vesicular compartment reacts differently to NaCl challenge, possibly as part of the cell adaptation process to osmotic stress. Interestingly, the average recovered motility of Rab isoforms used as markers for early (Rab5), late (Rab7), and recycling (Rab11) endosomes was similar to that found for FITC-dextran–loaded endosomes (Fig. S2E), supporting the use of dextran as a tool for studying generic vesicle populations.
Fig. 2.

Reduced vesicle motility by hypertonicity is a general phenomenon affecting numerous endosome/vesicle subpopulations in various cell types. (A) Endosome/vesicle motility before and after NaCl challenge (500 mOsmol/kg) in LLC-PK1 cells transfected with various GFP-tagged Rab isoforms (Rab5, -7, -10, and -11) or ssYFP, each used as a marker for specific endosome/vesicle subpopulations as shown in the table at right. The motility of all subpopulations was abolished immediately after challenge, whereas recovery rates varied, as shown in the table at right. (B) Kymographs illustrate different rates of recovery in cells expressing GFP-Rab10, ssYFP, or GFP-Rab11. (C–E) The effects of NaCl challenge on the motility of dextran-loaded endosomes in primary hepatocytes (C), primary pancreatic β cells (D), and cultured pancreatic MIN6 cells (E). (Also see Figs. S1 and S2 and Movies S2 and S3.) Error bars show mean ± SEM from four independent experiments.
Fig. S2.

Hypertonicity reduces vesicle motility in various cell types. (A–D) The effects of various osmolytes on FITC-dextran–loaded endosome motility were analyzed in mCCDcl1 cells (A), HeLa cells (B), freshly isolated human monocytes and differentiated macrophages (C), and cultured Huh7 hepatocytes (D). (E) Comparison of the motility of FITC-dextran–loaded endosomes and the averaged motility of Rab5, -7, and -11 endosomes (data are shown in Fig. 2A) illustrating their similar recovery kinetics. Ret1/2 and Remax values are shown at right. Arrows indicate the time when cells were challenged. Error bars show mean ± SEM from four independent experiments.
Decreased endosome motility by NaCl challenge also was found to be a generalized phenomenon, occurring in all cultured and primary cell types tested (Fig. S1 C and D). Compared with LLC-PK1 cells, dextran-loaded endosomes recovered motility more slowly in both primary hepatocytes and insulin-producing pancreatic β cells and also in cultured Huh7 and MIN6 cells, well-established hepatocyte and pancreatic β cell models, respectively (Fig. 2 C–E, Fig. S2D, and Movie S3) (42–44). Endosome motility also was strongly reduced by NaCl in cultured renal cortical collecting duct principal cells (mCCDcl1) (Fig. S2A) and HeLa cells (Fig. S2B). Similarly, in primary human monocytes and macrophages, NaCl-350 mOsmol/kg challenge dramatically reduced endosome motility, which recovered fully after longer periods of time (Fig. S2C). Stronger challenge in these cells led to excessive cell shrinkage that precluded further analysis. Interestingly, the effect of glucose, a nonelectrolyte, on endosome motility was different from that produced by NaCl and varied among cell types. In LLC-PK1 cells, glucose challenge initially immobilized endosomes (Fig. 1F). However, although motility recovered more slowly (Ret1/2 of 14.11 ± 0.95 min), it recovered to a greater extent. Similar to the effect of cell-permeable urea in LLC-PK1 cells (Fig. 1F), motility in both primary and cultured pancreatic β cells recovered significantly faster after glucose challenge than after NaCl challenge and reached near-prechallenge levels (Fig. 2 D and E). This faster recovery may result from fast glucose uptake into pancreatic β cells compared with LLC-PK1 cells (Fig. S3A), possibly reflecting high cell-surface expression of glucose transporter GLUT2 (Fig. S1E), whose high transport capacity allows faster equilibration of extracellular and intracellular glucose (45). In primary and cultured hepatocytes, another glucose-transporting cell type, the effects of glucose on endosome motility were significantly stronger than those observed in pancreatic β cells (Fig. 2C and Fig. S2D). However, as in LLC-PK1 cells, endosome motility in Huh7 cells did not plateau at lower levels after long periods of glucose challenge, in contrast to the effects of NaCl (Fig. S2D).
Fig. S3.

Effects of glucose and NaCl on MIN6 and LLC-PK1 cell volume. (A) Intracellular glucose was measured by the FRET probe FLII12Pglu-700uDelta6 (76) in MIN6 and LLC-PK1 cells incubated in glucose-free Hank’s medium, before and after exchange to iso-osmolar Hank’s medium containing 5 mM glucose (arrow). Glucose uptake was significantly faster and stronger in MIN6 cells than in LLC-PK1 cells. FRET emission was quenched at 500 mOsmol/kg challenge, possibly as a result of cell shrinkage-induced misfolding of the probe, precluding determination of glucose influx kinetics during hyperosmotic glucose challenge. (B and C) Measurement of MIN6 (B) and LLC-PK1 (C) cell volume using calcein-AM under isosmotic conditions and after NaCl (500 mOsmol/kg) or glucose (500 mOsmol/kg) challenge (arrow). MIN6 cell volume recovers faster after glucose challenge than after NaCl challenge, possibly as a consequence of fast glucose uptake. Error bars show mean ± SEM from four independent experiments.
Taken together, these data show that hypertonicity transiently immobilizes vesicle motility and that this immobilization is a generalized event affecting different vesicle subpopulations in a variety of cell types. Additionally, endosome motility recovered quickly in response to urea challenge in LLC-PK1 cells and to glucose challenge in pancreatic β cells but not after NaCl challenge in all cell types tested. This finding indicates that, in addition to molecular crowding, ionic imbalance may contribute to perturbed vesicle trafficking.
Hypertonicity Affects MT and Actin Dynamics.
Vesicle trafficking relies on microfilament networks. We previously have observed that NaCl challenge not only decreases vesicle motility but perturbs microfilament dynamics as well (13). In that study, we showed that NaCl (500 mOsmol/kg) transiently decreases MT polymerization and induces fast MT remodeling. Confocal microscopy performed on live LLC-PK1 cells expressing either GFP-tagged β-tubulin or GFP-tagged β-actin revealed that NaCl (500 mOsmol/kg) also decreases microfilament motility immediately (Fig. 3A and Movie S4). The effect on actin is readily illustrated by visualizing the movement of membrane ruffles in which undulations are driven principally by protrusive forces that arise from polymerization of actin filaments near the cell surface (46). Although mobile microfilaments appeared as rainbow colors using a time-lapse, pseudocoloring methodology, nonmobile microfilaments appeared white because of the superimposition of differently colored time-lapse frames. We quantified these qualitative observations using a method based on spatiotemporal image correlation spectroscopy (STICS) flow mapping, which estimates motility based on the calculation of relative local velocities of intensity maxima (Materials and Methods and ref. 47). Actin motility remained low, whereas MT motility recovered after long periods of NaCl challenge (Fig. 3A). The movement of each microfilament network was affected differently by cell-permeable urea (500 mOsmol/kg) (Fig. S4 B and C). Although MT motility generally was decreased by urea, actin motility increased above baseline levels shortly after challenge and then returned to baseline levels.
Fig. 3.

Reduced vesicle motility by hypertonicity relies on reduced microfilament dynamics rather than sudden shifts of their polymerization states. (A) Time-lapse pseudocolored images of GFP-β-actin (Lower Row) and GFP-α-tubulin (Upper Row) live-cell movies illustrate the extent of microfilament motility in LLC-PK1 cells. (Left) Six minutes prechallenge (−6 min to −1 s). (Center) Zero seconds to six minutes (+1 s to 6 min) after NaCl challenge (500 mOsmol/kg). (Right) Fourteen to twenty minutes (+14 min to +20 min) postchallenge. (Scale bars: 10 µm.) Representative images are shown on the left, and motility quantification is shown on the right. Data were gathered from three independent experiments. (B) FITC-dextran–loaded endosome motility in LLC-PK1 cells before and after (arrow) the addition of latrunculin B (2.5 µM), jasplakinolide (0.4 µM), or NaCl (500 mOsmol/kg) or before and after NaCl challenge followed by the addition of jasplakinolide (orange arrow) once steady-state levels were reached. *P < 0.05 vs. control. (C) FITC-dextran–loaded endosome motility in LLC-PK1 cells before and after the addition of taxol (10 µM), colchicine (10 µM), nocodazole (10 µM), or NaCl (500 mOsmol/kg) or before and after (arrow) NaCl challenge followed by the addition of nocodazole (NaCl/Noco; orange arrow) once steady-state levels were reached. Error bars show mean ± SEM from four independent experiments. (D) Recapitulative table of the effects of NaCl and drugs on vesicle motility and microfilament polymerization and motility. Agents that decrease microfilament motility also decrease vesicle motility. ND, not determined. (Also see Figs. S4–S6 and Movies S4–S6.)
Fig. S4.

Effects of urea on MT and actin polymerization and motility. Live-cell spinning-disk images of GFP-tagged β-actin and GFP-tagged α-tubulin were collected before and after urea challenge (500 mOsmol/kg). (A) Single-frame images of LLC-PK1 cells 1 s prechallenge and 2 and 20 min postchallenge. Urea induced strong MT depolymerization and actin polymerization upon challenge. (B) Images were color-coded as described in Materials and Methods. MT and actin motility were not as severely decreased or increased, respectively, after urea challenge as after NaCl challenge (see Fig. 3A). Representative images from three independent experiments are shown. (C) Quantification of microtubule (Left) and actin (Right) motility. Of note, because motility values shown immediately (≤2 min) after urea challenge also incorporated whole-cell movement that could not be avoided, local cytoskeletal movement values at ≤2 min probably are overestimated. Data were gathered from three independent experiments. *P < 0.05 and **P < 0.01 vs. control. (Scale bars: 10 μm.)
To provide a framework within which shifts of microfilament motility caused by hypertonic challenge and their effects on vesicle motility can be compared, we next investigated how sudden shifts of microfilament polymerization by NaCl challenge affect vesicle motility. We previously observed that decreased MT motility by NaCl is associated with immediate MT depolymerization, with repolymerization occurring within minutes of challenge (13). Cell-permeable urea also induced strong MT depolymerization upon challenge (Fig. S4A). Unlike MTs, actin polymerizes quickly after challenge by either NaCl or urea and remains polymerized for long periods of time, as revealed by Western blot analysis of fractionated G- vs. F-actin and by TRITC-phalloidin fluorescence analysis (Fig. S5). Then, we compared the effects of NaCl with those of chemicals that alter microfilament polymerization or polymer stability (Fig. 3 B and C). We previously showed that taxol, an agent that stabilizes MT polymers, and nocodazole, which interferes with tubulin polymerization, immediately increase and decrease tubulin polymerization, respectively (13). Taxol also increased actin polymerization (Fig. S5 B and C) but had no effect on either actin (Fig. S6 B and C) or vesicle motility (Fig. 3C). Similar to taxol, nocodazole increased actin polymerization (Fig. S5 B and C) but strongly decreased both actin (Fig. S6 B and C) and vesicle motility (Fig. 3C). It also reduced Remax after NaCl challenge (Fig. 3C). Colchicine, another chemical agent that interferes with tubulin polymerization, also decreased vesicle motility (Fig. 3C). Jasplakinolide, an agent that stabilizes actin filaments by binding to F-actin, immediately increased actin polymerization (Fig. S5) and decreased actin filament motility (Fig. S6 B and C) but had no effect on MT motility (Fig. S6 A and C). It immediately reduced, but did not abolish, vesicle motility and, interestingly, had no effect on Remax when applied after NaCl challenge (Fig. 3B). Latrunculin B, an agent that complexes with G-actin, thereby inhibiting actin polymerization, immediately induced actin depolymerization (Fig. S5) and had no effect on either MT (Fig. S6 A and C) or vesicle motility (Fig. 3B).
Fig. S5.
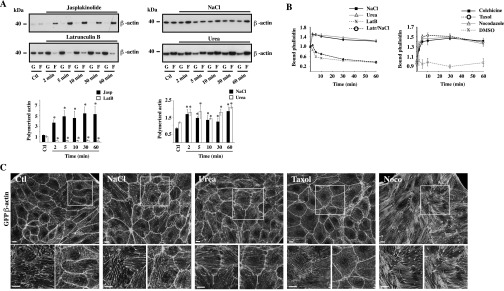
Actin remodeling by NaCl, urea, and chemicals that interfere with microfilament stability. (A) Time-dependent effects of jasplakinolide (0.4 µM), latrunculin B (2.5 µM), and NaCl and urea challenge (500 mOsmol/kg) on actin polymerization dynamics in LLC-PK1 cells were analyzed by Western blot. F-actin/G-actin ratios are shown below. Data depict fast effects by chemicals and hyperosmotic challenge. (B) TRITC-phalloidin fluorescence depicting increased actin polymerization in LLC-PK1 cells by NaCl challenge, urea challenge, and chemicals that interfere with MT polymerization. Latrunculin B, used as a control of actin depolymerization, strongly reduced actin polymerization by NaCl. Error bars show mean ± SEM from four independent experiments. *P < 0.05 vs. control. (C) Maximum projection of TRITC-phalloidin confocal z-stacks show actin organization under isotonic conditions (Ctl), after 30 min of NaCl or urea challenge (500 mOsmol/kg), and after the addition of taxol (10 µM, 30 min) or nocodazole (Noco) (10 µM, 30 min). Boxes below each image show enlarged maximum projections (Right) and an image of a single, basal confocal plane that depicts actin stress fibers (Left). (Scale bars: 5 µm.) Representative images from three independent experiments are shown.
Fig. S6.

Microfilament motility is differentially affected by chemicals that interfere with microfilament stability. (A and B) Live-cell spinning-disk images of GFP-tagged α-tubulin (A) and GFP-tagged β-actin (B) were collected before and after the addition of taxol (10 µM), nocodazole (10 µM) (Noco), jasplakinolide (0.4 µM) (Jasp), or latrunculin B (2.5 µM) (Lat) to LLC-PK1 cells and are color-coded as described in Materials and Methods. Images depicting motility during a 6-min timeframe prechallenge and 14 min postchallenge indicate that taxol interferes moderately with MT motility, and nocodazole and jasplakinolide appear to reduce actin motility. (Scale bars: 10 µm.) Representative images from three independent experiments are shown. (C) Quantification of microtubule and actin motility. Data were gathered from three independent experiments. *P < 0.05, **P < 0.01 vs. control. ND, not determined.
These data show that chemical agents that decrease microfilament motility reduce vesicle motility (Fig. 3D) and indicate that a decrease in microfilament dynamics, rather than sudden shifts in their polymerization states per se that conceivably could interfere with membrane trafficking events, may be implicated in the reduction of vesicle motility by NaCl challenge. Our data also suggest that neither jasplakinolide nor latrunculin B alters MT remodeling, at least not to the levels achieved by hyperosmotic stress. This finding is supported by our observation that neither chemical agent affects end-binding protein 1 (EB1) comet-like structure or motility, which can be used as a means to follow MT growth (Movies S5 and S6; the effects of NaCl on EB1 motility are described in ref. 13). On the other hand, the stabilization or destabilization of MT networks by taxol and nocodazole, respectively, induces actin polymerization, as previously observed (48–53).
Full Recovery of Vesicle Motility Following NaCl Hypertonic Challenge Occurs Long After Cell Volume Recovery.
We next examined how changes in cell volume caused by either NaCl or cell-permeable urea affect vesicle motility by measuring the fluorescence intensity of calcein-AM. NaCl spontaneously reduced LLC-PK1 cell volume in a dose-dependent manner (Fig. 4A). Equiosmolar urea produced a similar loss of cell volume (Fig. 4A), indicating that water efflux upon urea challenge precedes influx of urea in LLC-PK1 cells. As expected, cell volume recovered significantly faster after urea challenge than after NaCl challenge, with Ret1/2 occurring at 1.0 ± 0.03 min for urea challenge and at 2.5 ± 0.03 min for NaCl challenge. Cell volume recovered almost completely (96 ± 0.1%) after NaCl challenge and recovered completely following urea challenge (100 ± 0.1%). FITC-dextran–loaded endosome motility recovered simultaneously with cell volume after urea challenge. On the other hand, it recovered significantly more slowly than cell volume after NaCl challenge (Fig. 4A) and remained low well after cell volume was restored to near-baseline levels. Plotting the curve fit of vesicle displacement as a function of cell volume variation (Fig. 4B) shows that reducing cell volume by ≥12% with either NaCl or urea is associated with endosome immobilization. However, when the loss of cell volume caused by either osmolyte is <12%, endosome motility is decreased to a larger extent by NaCl than by urea challenge. LLC-PK1 cell volume also recovered significantly faster than endosome motility after glucose challenge (Fig. 1F and Fig. S3C). On the other hand, similar to the effects of urea in LLC-PK1 cells, both MIN6 cell volume and endosome motility recovered rapidly after glucose challenge but not after NaCl challenge (Fig. 2E and Fig. S3B).
Fig. 4.

Vesicle motility following NaCl-hypertonic challenge recovers well after cell volume is restored. (A, Left) LLC-PK1 cell volume was measured using calcein-AM under isosmotic conditions and after challenge (arrow) with NaCl (350 or 500 mOsmol/kg) or urea (500 mOsmol/kg). (Right) LLC-PK1 cell volume (solid lines) and FITC-dextran–loaded endosome motility (vesicle mvt, dotted lines) after challenge with NaCl or urea (500 mOsmol/kg), normalized to maximal variations for comparison of their recovery kinetics. (B) Projected FITC-dextran–loaded endosome motility in LLC-PK1 cells as a function of cell volume. (C) LLC-PK1 cells were challenged or not with an RVI inhibitor mixture (red arrow), and cell volume (Left) and FITC-dextran–loaded endosome motility (Right) were measured before and after (arrow) NaCl challenge (500 mOsmol/kg). (D) LLC-PK1 cell volume (Left) and FITC-dextran–loaded endosome motility (Right) before and after NaCl challenge (500 mOsmol/kg) and after osmolality returned to basal, isotonic conditions, either immediately after challenge or after a longer period (12 min), when near-maximal volume recovery was reached (blue and red arrows, respectively). Error bars show mean ± SEM from four independent experiments.
Recovery of cell volume after NaCl challenge relies on an increase in intracellular osmolality by a mechanism of regulatory volume increase (RVI) that itself relies on the influx of Na+ and Cl− ions. We examined the effects of RVI inhibition on FITC-dextran–loaded endosome motility by challenging LLC-PK1 cells with a mixture of Na+ transporter inhibitors (Materials and Methods). The recovery of endosome motility relied on the recovery of cell volume, as expected, because inhibition of RVI before NaCl challenge significantly delayed movement recovery (Fig. 4C). On the other hand, cell volume and motility decreased by NaCl challenge recovered quickly when extracellular osmolality was returned to isosmotic levels (Fig. 4D), when executed either immediately after NaCl challenge or after long periods of challenge (i.e., at a time when cell volume had recovered almost fully). In these latter experiments, the reduced steady-state endosome motility quickly returned to near baseline values. These data confirm previous work showing that molecular crowding contributes to deficient trafficking (14, 22). However, they also indicate that molecular crowding is not the only factor involved in decreased vesicle motility after hypertonic challenge.
Macromolecule Dynamics Are Reduced by an Isovolumetric Increase in [Cl−]i.
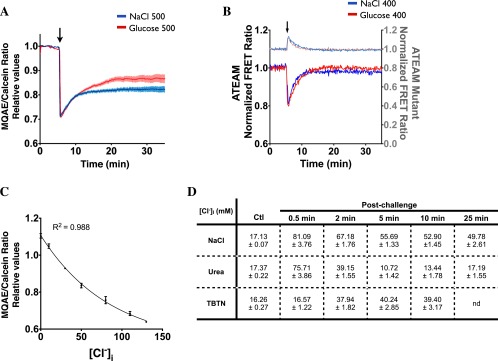
Loss of intracellular water upon hypertonic challenge increases Na+ and Cl− levels within cells. These levels remain elevated during RVI as the driving force for water reentry into cells. Consequently, hypertonicity induces an electrolyte imbalance and depolarizes the membrane of various cell types, including proximal tubule cells (54–56). We first tested whether membrane depolarization by either an acute isotonic increase of extracellular K+ concentration (to 72 mM) or K+ channel blockade by tetraethylammonium chloride (5 µM) had an effect on vesicle motility in LLC-PK1 cells. No differences were found, indicating that cell depolarization is not responsible for the observed defect in vesicle trafficking. As a model of ionic imbalance, we examined the effects of high levels of [Cl−]i on macromolecule motility under isovolumetric conditions by challenging cells with a previously described (57) combination of tributyltin and nigericin that already had been validated in LLC-PK1 cells (58). The Cl−/OH− antiporter tributyltin forms pores in the cell membrane that allow extracellular Cl− to equilibrate with intracellular Cl−. The H+/K+ ionophore nigericin is added to clamp intracellular pH. We confirmed that [Cl−]i was increased by tributyltin/nigericin ionophores (TBTN) by loading LLC-PK1 cells with N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE), a fluorescent dye that quenches in the presence of Cl− ions (59). To account for fluorescence changes induced by differences in cell volume, cells were loaded simultaneously with calcein-AM, and the ratio of MQAE/calcein fluorescence was recorded. MQAE fluorescence was quenched by TBTN, NaCl, and also by glucose challenge (500 mOsmol/kg) (Fig. 5 A and B and Fig. S7A). Similar to the effect of osmolyte challenge, fluorescence remained low for long periods of time in the presence of TBTN, which did not affect cell volume (Fig. 5A). As expected, contrary to the effects of TBTN and NaCl challenge, MQAE fluorescence was quenched only transiently by urea (Fig. 5B). We conclude that TBTN at least partly mimics ionic imbalance by NaCl challenge without the changes in cell volume that invariably accompany hypertonic challenge.
Fig. 5.

An isovolumetric increase in [Cl−]i decreases vesicle and microfilament motility. (A and B) LLC-PK1 cells were challenged (arrows) with the ionophores nigericin (5 µM)/tributyltin chloride (10 µM) (TBTN) (A), NaCl (500 mOsmol/kg), or urea (500 mOsmol/kg) (B). Error bars show mean ± SEM from three independent experiments. Relative changes of MQAE fluorescence were measured as the normalized emission ratio of MQAE (360 nm) to calcein (488 nm). MQAE quenching by KSCN (150 mM) confirms the probe was operating within its dynamic range. Inset in A shows changes in cell volume measured with calcein alone. (C) Decreased FITC-dextran–loaded endosome motility in LLC-PK1 cells after TBTN challenge (arrow). Error bars show mean ± SEM from four independent experiments. Curve fitting after TBTN challenge, indicated by the dotted line, was performed in two separate phases. (D and E) [Cl−]i as a function of cell volume (D) and projected FITC-dextran–loaded endosome motility (E) in LLC-PK1 cells. (F) Time-lapse pseudocolored images of GFP-α-tubulin and GFP-β-actin in LLC-PK1 cells before and after (arrow) TBTN challenge illustrating decreased but not abolished motility in both microfilaments after challenge. (Scale bar: 10 µm.) Representative images are shown on the left, and motility quantification is shown on the right. Error bars show mean ± SEM of three independent experiments. *P < 0.05 vs. control.
Fig. S7.
Effects of NaCl and glucose on [Cl−]i and [ATP]i in LLC-PK1 cells and MQAE/calcein calibration curve for the determination of [Cl−]i. (A) Relative changes in [Cl−]i after NaCl or glucose challenge were measured as the normalized emission ratio of MQAE (360 nm) to calcein (488 nm). (B) [ATP]i was measured with ATP indicator ATeam before and after NaCl or urea challenge (400 mOsmol/kg). To estimate the actual relative loss of ATP, the wild-type ATeam FRET curves are normalized to the ATP-insensitive mutant probe FRET curves to account for volume-induced, ATP-independent FRET signals. Arrows indicate the time when cells were challenged. (C) Calibration curve relating relative MQAE/calcein ratios to known [Cl−] values. Error bars show mean ± SEM from three or four independent experiments. (D) Average [Cl−]i levels calculated using the calibration curve before or 0.5, 2, 5, 10, and 25 min after exposure to hypertonic NaCl and urea (500 mOsmol/kg) or TBTN.
TBTN rapidly decreased but did not abolish the motility of FITC-dextran–loaded endosomes (Fig. 5C). Interestingly, it decreased motility to an extent similar to the steady-state motility observed following strong (500 mOsmol/kg) NaCl challenge. We estimated actual [Cl−]i values in LLC-PK1 cells using a previously described calibration method performed in this same cell line (59). [Cl−]i was estimated to be 16.9 ± 0.6 mM under resting, isotonic conditions and increased strongly after NaCl, urea, or TBTN challenge (Fig. S7 C and D). The relationship among cell volume, [Cl−]i, and FITC-dextran–loaded endosome motility is depicted in Fig. 5 D and E. Endosome motility decreased steadily in response to increased [Cl−]i induced by TBTN, which does not alter cell volume, and then fell dramatically when [Cl−]i was increased by ≥40 mM. Endosome motility similarly was reduced at 50 mM after long periods of NaCl challenge (i.e., at a time when cell volume had recovered almost fully). Endosome motility then fell dramatically to near immobilization when [Cl−]i was increased above 50 mM immediately after either NaCl or urea challenge, likely reflecting the combined effects of increased [Cl−]i and reduced cell volume. These figures also indicate that, although urea influx allows fast recovery of cell volume and [Cl−]i, the intracellular accumulation of urea actually decreases endosome motility, although to a lesser extent than in response to elevated [Cl−]i. This finding may help explain the residual loss of endosome motility by long-term urea challenge (Fig. 1F) despite full recovery of both cell volume and [Cl−]i (Figs. 4A and 5B). Similar to endosome motility, microfilament motility was decreased by TBTN (Fig. 5F), suggesting that a sudden isovolumetric increase of [Cl−]i decreases microfilament motility as well.
High Levels of [Cl−]i Decrease Mitochondrial Function and Intracellular ATP Content.
Membrane trafficking is a tightly regulated process that relies on available energy. Perturbations of ATP production by hypertonicity could affect microfilament dynamics and vesicle motility. We examined this possibility by monitoring real-time changes in intracellular ATP concentration ([ATP]i) in LLC-PK1 cells using the recently developed FRET-based ATP indicator ATeam (60). We compared the effects of NaCl, urea, and the ATP synthase inhibitor oligomycin on YFP/CFP emission ratios of wild-type and mutant (AT1.03R122K/R126K) ATeam probes (Fig. 6A). As expected, the basal emission ratio of the mutant probe, which does not respond to [ATP]i (60), was lower than that of the wild-type probe (0.42 mutant FRET ratio compared with 1.3 wild-type FRET ratio). Unexpectedly, the emission ratio of the mutant probe was increased immediately by NaCl challenge (Fig. 6A). Equiosmolar urea produced a similar effect (Fig. 6A). Because equiosmolar NaCl and urea reduce cell volume to similar extents (Fig. 4A), this ATP-independent increase in the FRET emission ratio probably is caused by a conformational change in the probe's sensor domain that results from cell shrinkage-induced molecular crowding and/or high ionic strength. This effect increased dramatically with the magnitude of osmotic challenge and prevented us from conducting experiments with a challenge above 400 mOsmol/kg. Despite this drawback, the emission ratio of the wild-type probe was partly quenched immediately following NaCl challenge (400 mOsmol/kg) and, to a lesser extent, after urea challenge (400 mOsmol/kg), indicating that each osmolyte decreases [ATP]i but does so to different extents. Contrary to the high levels of [Cl−]i that are sustained by NaCl challenge (Fig. 5B), [ATP]i recovered almost completely after long periods of challenge (Fig. 6A). [ATP]i also decreased at first but then recovered completely after glucose challenge (Fig. S7B). As expected, a combination of oligomycin and the glycolysis inhibitor 2-deoxyglucose (2-DOG) decreased [ATP]i (Fig. 6A). Similar to our FRET data, analysis of [ATP]i by a fluorometric assay revealed that [ATP]i was decreased by NaCl challenge and, to a lesser extent, by urea challenge (500 mOsmol/kg), returned to basal levels after longer periods of challenge, and was decreased by oligomycin/2-DOG (Fig. 6B). FRET analysis of LLC-PK1 cells additionally revealed that [ATP]i was decreased initially by an isovolumetric increase in [Cl−]i induced by TBTN and recovered only partly after longer periods of challenge (Fig. 6C), possibly reflecting increased ATP consumption and/or loss.
Fig. 6.

Mitochondrial function is transiently decreased by hypertonic and Cl− ionophore challenge. (A and B) [ATP]i in LLC-PK1 cells was measured with ATP indicator ATeam (A, dark colors) or fluorometric analysis (B) before and after (arrow) NaCl or urea challenge [400 (A) or 500 (B) mOsmol/kg] and oligomycin (10 µg/mL)/2-DOG (10 mM). (C) [ATP]i in LLC-PK1 cells was measured by ATeam (dark tone) before and after the addition of TBTN followed by oligomycin/2-DOG. In A, to estimate the actual relative loss of ATP, the wild-type ATeam FRET curves are normalized to the ATP-insensitive mutant probe FRET curves to account for volume-induced, ATP-independent FRET signals. Mutant probe responses are illustrated in light tones (A and C) on the left axes. (D) Decreased FITC-dextran–loaded endosome motility kinetics (black, right axis) is similar to that of ATeam FRET signal loss (gray, left axis) in LLC-PK1 cells after addition of oligomycin/2-DOG (arrow). Error bars show mean ± SEM from four independent experiments. (E) Time-lapse pseudocolored images of GFP-α-tubulin and GFP-β-actin in LLC-PK1 cells before and after the addition of oligomycin/2-DOG illustrate decreased motility in both microfilaments after challenge. (Scale bars: 10 µm.) Representative images are shown on the left, and quantification of motility in three independent experiments is shown on the right. (F) Ratiometric imaging of the mitochondrial membrane potential dye JC-1 (7.7 µM) in control (Ctl) and LLC-PK1 cells challenged with NaCl (500 mOsmol/kg), Cl− ionophores (TBTN), or the positive controls CCCP (1 µM) or ArA (20 µM) for 15 min. Images show JC-1 fluorescence in control cells and after 10 min of NaCl challenge. (G) Analysis of mitochondrial fission in LLC-PK1 cells pretreated with MitoTracker Red (500 nM) and then challenged with NaCl (500 mOsmol/kg) or ionophores (TBTN, 10 min) or with CCCP or ArA (15 min). Error bars show mean ± SEM from four independent experiments. *P < 0.05 vs. control. Images show MitoTracker fluorescence in control cells and after 10 min of NaCl challenge. (Scale bars: 10 µm.)
Our data indicate that increased [Cl−]i induced by NaCl and TBTN might contribute to decreased vesicle and microfilament motility partly because of decreased [ATP]i, at least during the early stages of challenge. This notion led us to examine the effects of ATP depletion by oligomycin/2-DOG on macromolecular dynamics in LLC-PK1 cells. Similar to the effect of TBTN, oligomycin/2-DOG decreased but did not abolish the motility of FITC-dextran–loaded endosomes (Fig. 6D) and reduced MT and actin motility (Fig. 6E). These observations indicate that a sudden increase in [Cl−]i transiently decreases [ATP]i, which in turn affects macromolecular dynamics.
The decreased [ATP]i induced by NaCl challenge could arise from perturbed mitochondrial function. Ratiometric imaging of the mitochondrial membrane potential (ΔΨ) dye JC-1 in LLC-PK1 cells revealed an F488/F568 signal ratio that increased significantly after NaCl challenge (Fig. 6F). Similar to recovered levels of [ATP]i after long periods of challenge (Fig. 6 A and B), ΔΨ gradually returned toward basal levels after longer periods of time (Fig. 6F). Cells challenged with TBTN displayed similar, transient mitochondrial dysfunction despite sustained elevated levels of [Cl−]i (Fig. 6F). Mitochondria exist as a network that is continuously shaped by fusion and fission events, and their excessive fragmentation is a common consequence of mitochondrial dysfunction (61). Both NaCl and TBTN challenge increased mitochondrial fission, and mitochondria remained fragmented even after long periods of time (Fig. 6G). Therefore, a sudden increase in [Cl−]i transiently decreases [ATP]i, at least partly as a consequence of mitochondrial dysfunction.
Discussion
The motility of various endosome/vesicle subpopulations was nearly abolished immediately after NaCl-hypertonic challenge, even when osmolality was increased only moderately (by 50 mOsmol/kg). This observation made in various mammalian cell types is similar to the decreased vesicle trafficking observed in yeast exposed to severe osmotic stress (3 M sorbitol; i.e., 3,000 mOsmol/kg) (14). Osmotic compression per se and accompanying molecular crowding undoubtedly play a large part in the vesicle immobilization that accompanies water efflux, because the intracellular space is filled to near capacity with macromolecules and organelles under basal conditions (62). An optimal protein density that maximizes biochemical reaction rates may exist in cells (22). By increasing macromolecule density, cell shrinkage hinders protein diffusion and biochemical reactions (14, 22). This idea is supported by the finding that eukaryotic cells under compressive stress behave as strong glass because of crowding-induced stiffening of the cytoplasm (21). Although vesicle motility certainly is affected by molecular crowding, our data suggest that other factors that arise as part of the hypertonic stress response may be involved, because volume recovery did not immediately relieve the depression in vesicular dynamics. Because RVI relies on electrolyte import, we hypothesized that electrolyte imbalance could be one of these additional factors. Although both the intracellular Na+ concentration [Na+]i and [Cl−]i increase in response to hypertonicity, our study focused on investigating how high levels of [Cl−]i may affect vesicle motility in conjunction with molecular crowding, taking advantage of tools for measuring and manipulating [Cl−]i already optimized in LLC-PK1 cells (58, 59). Similar to the effect of NaCl challenge, vesicle motility was abolished when cells were exposed to high concentrations (corresponding to 500 mOsmol/kg) of cell-permeable osmolytes (urea in LLC-PK1 cells and glucose in pancreatic β cells) but, unlike NaCl challenge, recovered quickly thereafter. Water is drawn back into the cell rapidly as cell-permeable osmolytes accumulate intracellularly. As a result, cell volume is restored quickly, and the accumulation of inorganic electrolytes inside the cell is minimized. The situation is very different in cells challenged with cell-impermeable osmolytes, such as NaCl and mannitol. In this case, cell volume is restored by RVI mechanisms that increase intracellular concentrations of inorganic electrolytes. After NaCl challenge, but not after urea challenge, vesicle motility recovers significantly more slowly than cell volume and remains low well after cell volume is restored. Motility is restored quickly by re-exposing cells to isotonic conditions, which restore intracellular electrolyte composition, but recovery is reduced by an isovolumetric increase of [Cl−]i by ionophores. These data demonstrate that vesicle motility is abated by high, but physiological, levels of [Cl−]i well after cell volume has recovered. In addition, along with molecular crowding, high levels of [Cl−]i probably contribute to reduced motility at the onset of hypertonic challenge, because water loss increases intracellular electrolyte concentration.
We show that a sudden increase of [Cl−]i transiently decreases ATP abundance and that chemical block of ATP synthesis reduces vesicle and microfilament motility. These observations indicate that attenuated levels of available ATP by high levels of intracellular salt may contribute to decreased macromolecule motility. ATP is consumed at approximately the same rate as it is synthesized. Our data indicate that high concentrations of [Cl−]i may impair mitochondrial function, thereby decreasing ATP synthesis, probably because protein crowding is even higher in mitochondria than in the cytosol (63) and because alterations in matrix volume strongly affect mitochondrial function (64, 65). Accordingly, hypertonicity initially decreases the volume of isolated mitochondria (64), and even mild hypertonic exposure (an increase of 60 mOsmol/kg) was reported to dissipate ΔΨ in cultured Vero cells within minutes of challenge (30). We show that ΔΨ is decreased by both hypertonic and TBTN challenge. We also show that, reflecting the recovery of cellular ATP abundance, ΔΨ returns to basal levels after long periods of challenge by either stimulus, although [Cl−]i levels remain elevated. Although our study focused on the effects of high [Cl−]i, imbalance of other ions (particularly Na+ but potentially others as well) is likely to be involved. Our data argue that an initial ionic imbalance temporarily impairs mitochondrial function but does not preclude mitochondria from adapting to their new milieu. We interpret our data as evidence that mitochondria are able to adapt and restore ATP production despite elevated intracellular ionic strength, but that this adaptation takes a few minutes. This adaptation may involve restored K+ equilibrium across the mitochondrial inner membrane, because matrix volume is controlled by the kinetic equilibrium between K+ entry and efflux (64, 65). Other mitochondrial ion transporters also may be involved in restoring mitochondrial ΔΨ. [ATP]i was decreased almost instantaneously upon NaCl challenge, faster than after oligomycin/2-DOG challenge, probably reflecting fast ΔΨ dissipation by the quasi-instantaneous cell shrinkage induced by NaCl as opposed to the slower effects of chemicals that need time to cross cell membranes. Increased ATP consumption after hypertonic challenge also might participate in decreasing [ATP]i. Altered protein structure by hypertonicity might lead to widespread and immediate hydrolysis of protein-bound ATP and/or to rapid ATP/ADP cycling, analogous to the so-called “catastrophe” events of MT depolymerization in which bound GTP is hydrolyzed immediately. Increased activity of ATP-consuming transporters that reestablish ionic balance and, notably, elevated Na+/K+-ATPase activity by high [K+]i also may contribute to decreased [ATP]i. Interestingly, our data show that although [ATP]i is reduced similarly immediately after NaCl or glucose challenge, it recovers completely after glucose challenge but not after NaCl challenge. This recovery after glucose challenge suggests that ATP synthesis initially is reduced by hypertonicity-induced cell shrinkage but that increased glucose availability helps restore [ATP]i. The combined effects of fast [Cl−]i recovery and increased ATP availability in glucose-challenged cells may help explain the higher steady-state levels of vesicle motility achieved in some cell types (LLC-PK1 and Huh7) and the fast recovery of motility in pancreatic β cells.
The recovery of ATP abundance and ΔΨ recovery precede the recovery of vesicle and microfilament motility, indicating that the effects of [Cl−]i do not result from depleted ATP synthesis alone. Perturbed ionic balance within the vesicles themselves, which could alter vesicular shape and/or recruitment of microfilament tethering complexes, may contribute to the effects of [Cl−]i. Indeed, recent data examining defects in endosomal ClC-family channel mutants suggest that luminal Cl− content plays a role in endosomal trafficking beyond V-ATPase charge compensation (which facilitates endosomal acidification), which may include fission/fusion as well as endosomal volume regulation (3). We have shown recently that autophagy is induced by hypertonic stress (13). In this regard, it would be revealing to examine how vesicle motility that is reduced by ionic imbalance affects autophagy, because this process relies on autophagosome trafficking and fusion events with lysosomes/vacuoles (66). Vacuoles also contribute to protecting osmotically challenged cells by sequestrating Na+ and Cl−. This sequestration lowers intracellular osmolality and thereby helps avert the deleterious effects of cytoplasmic salt accumulation in plants and yeast (67). It remains to be determined whether a similar accumulation of salt in the vacuoles of at least some mammalian cell types may occur following hypertonic challenge and whether this mechanism participates in the mammalian cellular adaptation process.
Monovalent salts such as NaCl modify the ionic strength of solutions, such as the cellular cytoplasm, and can exert stabilizing or destabilizing effects on proteins, with the net effect depending on the nature of specific charge distribution and ionic interactions within the protein (68–70). Electrolyte concentration also is central to the screening of surface charge–charge interactions that influence protein stability, interaction, and function. Along with molecular crowding, increased ionic strength probably explains protein misfolding by hypertonicity (5, 12, 13, 71). Similarly, it might be expected to alter the functionality of at least a subset of proteins by affecting biomolecular electrostatic interactions. It has long been known that MT polymerization is decreased at high ionic strength in vitro (26, 72). This effect would help explain our observation that hypertonicity induces transient MT depolymerization in living cells. Actin filament assembly also is affected by hypertonicity, which increases their polymerization (4). Salt modulates actin assembly and its mechanical properties through nonspecific electrostatic effects (25). Occupancy of a distinct site by cations at concentrations similar to those occurring after strong hypertonic challenge recently was found also to stiffen actin filaments (73). Our data show that [Cl−]i decreases the motility of both actin filaments and MTs and that chemical agents that decrease microfilament motility also reduce vesicle motility. These data link microfilament dynamics reduced by [Cl−]i to decreased vesicle motility, which might arise from a combination of effects of salt bound to microfilaments, ATP loss, and/or modulated activities of any of dozens of proteins that bind to MTs and actin and that regulate microfilament assembly/disassembly dynamics.
Analogous to cell volume that is constrained within limits that maximize biochemical reaction rates (14, 22), intracellular electrolyte composition must be maintained to optimize cell function. Our results reveal the dramatic and broad effects of ionic imbalance on microfilament organization and membrane trafficking. By comparing time-dependent consequences of hypertonicity and isovolumetric elevation of [Cl−]i induced by TBTN, we were able to distinguish between the effects caused by compression-induced molecular crowding and those caused by high levels of [Cl−]i. Our data show that mammalian cells react to even a small increase in [Cl−]i, which triggers mitochondrial depolarization and microfilament remodeling and thereby slows vesicle trafficking. These effects persevere long after cell volume is restored. We propose that such change reflects the first of a series of events embodying altered membrane trafficking that allows cells to adapt to environmental change.
Materials and Methods
Reagents.
Chemicals were purchased from Sigma-Aldrich, and cell culturing reagents and dyes were from Life Technologies unless otherwise stated. ATeam and mutant ATeam probes were gifts from Hiromi Imamura, Kyoto University, Kyoto, and Nicolas Demaurex, University of Geneva, Geneva; LLC-PK1 cells expressing tubulin-GFP were a gift from Patricia Wadsworth, University of Massachusetts, Amherst, MA; human hepatoma Huh7 cells and primary rat hepatocytes were gifts from Michelangelo Foti, University of Geneva, Geneva; GFP-tagged Rab5 and GFP-tagged Rab7 were gifts from Christian Reinecker, Massachusetts General Hospital, Boston; Rab10 was a gift from Sebastian Schuck, Max Plank Institute, Heidelberg; and Rab11 was a gift from Jim Goldenring, Vanderbilt University, Nashville, TN. Please see SI Materials and Methods for commercial antibody sources and dilutions, and specifications of microscopes used.
Cell Cultures and Transfection.
Cells were cultured and transfected as previously described (41); please see SI Materials and Methods for details. Human monocytes were isolated from buffy coats collected from healthy volunteers according to the institutional guidelines of the Ethical Committee of the University of Geneva, using Lymphoprep (Axis-Shield). Isolated monocytes were differentiated into macrophages by culturing for 3 d with 100 ng/µL recombinant human macrophage colony-stimulating factor (Peprotech). Isosmotic medium (300 mOsmol/kg) was made hyperosmotic (350–500 mOsmol/kg) by adding 1,100 mOsmol/kg medium. Hyperosmotic medium (500 mOsmol/kg) was returned to isosmotic levels by the addition of 200 mOsmol/kg medium. To obtain isosmotic 72 mM KCl, 72 mM NaCl was replaced by isomolar KCl. Medium osmolality was verified using an osmometer.
Immunolabeling and Fluorescence Microscopy.
For GLUT2 and insulin analysis, cells grown on coverslips were fixed in methanol for 5 min at −20 °C; otherwise cells were fixed in 4% paraformaldehyde for 20 min. Dyes were applied at the following dilutions before fixation: JC-1 (Adipogen; 5 µg/mL, for 15 min), MitoTracker Red CMXRos (500 nM, for 15 min). Live-cell imaging was performed on cells grown on glass-bottomed dishes (World Precision Instruments).
Analysis of Endosome and Microfilament Motility.
Cells were loaded with FITC-dextran [1 µg/mL; 10,000 molecular weight (MW)] for 20 min at 37 °C. The motility of FITC-dextran–loaded endosomes, GFP-tagged Rab isoforms, and ssYFP was visualized at a single confocal plane and quantified using the Manual Tracking plug-in in ImageJ. The mean travel distance of 30 endosomes over a 2-min interval (10 frames) was measured. Mean velocity (travel distance/time) normalized to isotonic baseline (obtained during the first 12 s) is shown. Immobile or slow-moving (<0.3 µm/min) endosomes and endosomes that moved outside the plane of focus during the 2-min time interval were not considered. Endosome centroids were estimated manually (without centering), giving an estimated error of two or three pixels or 600–900 nm per measurement. Baseline and isotonic curves were fitted using a linear equation (Y = Slope*X + Intercept). Recovery phases were fitted using a Boltzmann sigmoidal model [Y = Bottom + (Remax − Bottom)/(1 + exp((Ret1/2 − X)/Slope))] with Bottom values constrained to >0. For washout experiments, motility values during treatment and washout phases were fitted separately. For experiments using tributyltin/nigericin ionophores, motility values during early and late phases of treatment were fitted separately, but both were fitted with Boltzmann sigmoidal equations, which gave the best fits. Kymographs were prepared using the Reslice function in ImageJ, and time-lapse pseudocolored images were prepared using Z Code Stack. To quantify microfilament motility, the STICS map jru v2 function of the Image Correlation Spectroscopy (ICS) ImageJ plug-in suite, developed by Jay Unruh (Stower's Institute for Medical Research, Kansas City, MO) (74) was used. Two to six subregions (90 × 90 pixels that avoided nuclei and areas of obvious whole-cell movements) per movie from three independent movies were analyzed per condition. Movie segments encompassing 10 frames (2-min timespans) were analyzed for each time point, starting 2 min before manipulation or at the indicated times. For each region and time point, 16 flow-velocity vectors were calculated by the program. To obtain the average movement of each subregion, the absolute value of the velocity vectors, defined as [absolute velocity = SQRT(x − velocity^2 + y − velocity^2)] was calculated for each vector, and the average of the 16 values was reported.
Cell Volume Analysis.
Cells were loaded with calcein-AM (5 µM) for 15 min at 37 °C. Regions of interest were drawn around the areas within cells that remained in focus. Photobleaching/dye leak correction was performed by extrapolating baseline fluorescence loss, estimated by fitting values from the first 6 min with a one-phase exponential decay function (Y = Span*exp(−K*X) + Plateau). Background-subtracted and photobleaching-corrected calcein fluorescence, which is inversely proportional to cell volume (75), was converted to cell-volume changes. Recovery phases were fitted with an exponential association function [Y = (Rvmax – Bottom)*(1 − exp(−X*ln (2)/Rvt1/2) + Bottom]. Results are expressed normalized to baseline. RVI was delayed by treating cells with benzamil (10 µM), bumetanide (50 µM), and gadolinium (100 µM).
Measurement of Relative Changes in [Cl−]i by MQAE Analysis.
Cells were loaded with 5 mM MQAE for 60 min at 37 °C, and 5 µM calcein-AM was added during the last 15 min. Cells were alternately excited at 360 nm/488 nm, and emission was collected at 530 nm to visualize MQAE or calcein, respectively. Photobleaching correction was performed as described above on MQAE and calcein signals separately, before the F360/F488 ratio was calculated. To transform ratio values into actual [Cl−]i values, three independent calibration curves were constructed as described previously (59) and averaged. Briefly, cells loaded with MQAE/calcein-AM were sequentially exposed to solutions containing TBTN and 0, 30, 50, 80, 110, or 130 mM Cl−, in which gluconate was used to replace Cl− as the counterion to maintain osmolarity, for 5 min each. The background-subtracted and photobleaching-corrected average ratio values within the middle of the 5-min exposure time frame then were plotted against the known Cl− concentration values of each calibration solution. The resulting curve then was fitted with a one-phase exponential decay function (which gave the best fit), and the equation was used to transform ratio values into mM [Cl−]i values.
Analysis of Cellular Glucose, ATP, and Mitochondrial Function.
To compare glucose-influx kinetics of LLC-PK1 and MIN6 cells, cells transfected with the FRET-based glucose sensor FLII12Pglu-700uDelta6 (76) were incubated in glucose-free Hank’s medium for 10 min before imaging. Images were collected using 440 nm excitation and alternate 485/535 nm emission as the solution was changed to an iso-osmolar Hanks’ solution containing 5 mM glucose, a change previously shown to lie within the middle of the dynamic range of the probe (76). Changes in cellular ATP levels were measured using a similar excitation/dual emission protocol, using the FRET-based ATeam probe (60) and by fluorometric analysis using the ATP Assay Kit (Abcam), in which the excitation/emission fluorescence (535/587 nm) was measured using a SpectraMax Paradigm microplate reader (Molecular Devices). Mitochondrial depolarization was measured as an increase in the background-subtracted 488/568 nm JC-1 ratio. Mitochondrial morphology was quantified using MitoTracker Red CMXRos as described (77). Data from at least 100 cells per condition were collected in each experiment. For experiments performed with oligomycin, glucose was replaced by 2-DOG (10 mM).
Western Blot Analysis and F-Actin/G-Actin Assay.
Cell lysates were prepared and quantified as described previously (78). Actin polymerization was quantified by determining the Triton X-100–soluble (G-actin)/TritonX-100–insoluble (F-actin) ratio. (See SI Materials and Methods for details) or by a TRITC-phalloidin methanol extraction method (79) adapted to 96-well format. F-actin content values are expressed as background-subtracted fluorescence values normalized to nontreated controls.
Statistics.
Results are given as the mean ± SEM from n independent experiments. Each experiment was performed on cells from the same passage. All experiments were performed at least three times. Statistical differences were assessed by one-way ANOVA, and curve fitting was performed using GraphPad Prism software. *P ≤ 0.05.
SI Materials and Methods
Antibody Sources and Dilutions.
Mouse anti-α-actin (1:10000), Sigma-Aldrich; rabbit anti-GLUT2 (1:50), Ventrex Labs; guinea pig anti-insulin (1:200), Ventrex Labs; goat anti-mouse HRP (1:5000), BD Pharmingen; donkey anti-rabbit Alexa Fluor 488 (1:1000) Life Technologies; sheep anti-guinea pig FITC (1:500) P.A.R.I.S (Compiègne, France).
Cell Cultures and Transfection.
Porcine LLC-PK1 cells were cultured as previously described (18). Plasmids were transiently transfected into LLC-PK1 cells using Lipofectamine 2000 (Life Technologies) at a either 0.5:1 (ATeam and ATeam mutant probes) or 1:1 (all others) µg DNA:µg Lipofectamine ratio per 500 µL serum-free culture medium. Lipofectamine was removed and serum re-added 24 h after transfection and 1--24 h prior to performing experiments. Mouse cortical collecting duct mCCDcl1 cells (80), mouse insulinoma MIN6 cells (44) and human hepatoma Huh7 cells (81) were cultured as previously described. Primary mouse pancreatic β cells and rat hepatocytes were isolated and cultured as previously described (82, 83). Cells were grown at 37 °C in a humidified atmosphere containing 5% CO2 to 80% confluency prior to performing experiments.
Microscopy.
Widefield fluorescence images were captured with an Axioskop 2 microscope (Carl Zeiss AG, Germany) equipped with a 40X 1.3 NA Plan-neofluar oil objective, using AxioVision software. Confocal imaging was performed on a Leica TCS SP5 MP confocal microscope system equipped with a 63× 1.4 NA Plan-apo HCS oil objective, using Leica Application Suite software (Leica Microsystems AG, Germany). Live-cell imaging was performed on cells grown to 50% confluence and rinsed with Hanks solution, pH 7.4, supplemented with 11 mM glucose, 25 mM NaHCO3 and 20 mM Hepes. Visualization of macromolecule motility and cell volume was performed on a QLC100 spinning-disk confocal system equipped with a 63× 1.45 NA Apochromat oil immersion objective (Visitech International), maintained at 37 °C by a microscope temperature control system (Life Imaging Services) using Metamorph/Metafluor 5.0 software (Molecular Devices) or on a Nikon A1R inverted confocal microscope system equipped with a resonant scanner, Perfect Focus, 60X 1.4 NA CFI Plan-Apo oil immersion objective (Nikon, Tokyo), and maintained at 37 °C by a microscope temperature control system (Life Imaging Services). MQAE and ATeam FRET analysis were performed on a Zeiss Axiovert S100TV microscope equipped with a 40X Fluar 1.3 NA oil immersion lens, Visitron Systems monochromator illumination system and stage heater to maintain cells at 37 °C. Cells were alternately excited with 360 nm and 488 nm light and emission collected at 530 nm using a 430 DCLP dichroic, 510WB40 emission filter and MetaFluor software, to visualize MQAE and calcein, respectively. Cells were alternately excited with 440 nm and 488 nm light and emission collected at 530 nm using a 505 DCXR dichroic, and 510WB40 emission filter to visualize ATeam and mutant-ATeam probes. Frames were acquired either every 5 s (calcein, MQAE, ATeam) or 12 s (all others).
F-Actin/G-Actin Assay.
Actin polymerization was quantified by Western blot by determining the Triton X-100-soluble (G-actin)/TritonX-100-insoluble (F-actin) ratio. Cells grown in plastic 6-well plates were scraped and spun at 6,000 × g for 3 min. Cells were lysed for 10 min with 100 µL of ice-cold lysis buffer (84) and spun at 8,500 × g for 5 min. The supernatant was resuspended in 2× sample buffer while the pellet was resuspended in 300 µL of lysis buffer and spun at 8,500 × g for 5 min. The pellet was then resuspended in 100 µL of lysis buffer and 100 µL 2× sample buffer. Equal amounts of protein (10 µg) were loaded onto gels and proteins were separated by NuPage 4–12% BisTris gel (Life Technologies) electrophoresis and detected using anti-α-actin. Bands from at least 3 independent experiments were quantified using ImageJ. F-actin was also quantified by TRITC-phalloidin fluorescence by adapting a previously described method (79) to 96-well format. Briefly, cells were fixed in 50 mL of TRITC-phalloidin binding buffer [4% paraformaldehyde, 0.1% TritonX-100, 125 nM TRITC-phalloidin in 20 mM K2HPO4 10 mM PIPES, 5mM EGTA, 2 mM MgCl2] for 1 h. Wells were washed 3 times in PBS and incubated overnight at −20 °C in 100 mL of methanol. The extracted TRITC-phalloidin fluorescence (590 nm) was read on a FLUOstar (BMG Labtech) microplate reader. Background values were determined from wells extracted with binding buffer lacking phalloidin. F-actin content values are expressed as background-subtracted fluorescence values normalized to nontreated controls.
Supplementary Material
Acknowledgments
This work was supported by Swiss National Science Foundation Grant 31003A_138408/1 and a grant from the Fondation Schmidheiny (to U.H.), as well as a Young Researcher Subsidy from The Sir Jules Thorn Charitable Overseas Trust (to P.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421290112/-/DCSupplemental.
References
- 1.Willmer P, Stone G, Johnston I. Environmental Physiology of Animals. Blackwell Publishing; Malden, MA: 2005. [Google Scholar]
- 2.Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89(1):193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 3.Stauber T, Jentsch TJ. Chloride in vesicular trafficking and function. Annu Rev Physiol. 2013;75:453–477. doi: 10.1146/annurev-physiol-030212-183702. [DOI] [PubMed] [Google Scholar]
- 4.Burg MB, Ferraris JD, Dmitrieva NI. Cellular response to hyperosmotic stresses. Physiol Rev. 2007;87(4):1441–1474. doi: 10.1152/physrev.00056.2006. [DOI] [PubMed] [Google Scholar]
- 5.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu Rev Biophy. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28(1-4):155–159. doi: 10.1152/advan.00034.2004. [DOI] [PubMed] [Google Scholar]
- 7.Houpt TR. Patterns of duodenal osmolality in young pigs fed solid food. Am J Physiol. 1991;261(3 Pt 2):R569–R575. doi: 10.1152/ajpregu.1991.261.3.R569. [DOI] [PubMed] [Google Scholar]
- 8.Anjaria DJ, Mohr AM, Deitch EA. Haemorrhagic shock therapy. Expert Opin Pharmacother. 2008;9(6):901–911. doi: 10.1517/14656566.9.6.901. [DOI] [PubMed] [Google Scholar]
- 9.Daviskas E, Anderson SD. Hyperosmolar agents and clearance of mucus in the diseased airway. J Aerosol Med. 2006;19(1):100–109. doi: 10.1089/jam.2006.19.100. [DOI] [PubMed] [Google Scholar]
- 10.Soupart A, Decaux G. Therapeutic recommendations for management of severe hyponatremia: Current concepts on pathogenesis and prevention of neurologic complications. Clin Nephrol. 1996;46(3):149–169. [PubMed] [Google Scholar]
- 11.Dmitrieva NI, Burg MB, Ferraris JD. DNA damage and osmotic regulation in the kidney. Am J Physiol Renal Physiol. 2005;289(1):F2–F7. doi: 10.1152/ajprenal.00041.2005. [DOI] [PubMed] [Google Scholar]
- 12.Burkewitz K, Choe K, Strange K. Hypertonic stress induces rapid and widespread protein damage in C. elegans. Am J Physiol Cell Physiol. 2011;301(3):C566–C576. doi: 10.1152/ajpcell.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nunes P, et al. Hypertonic stress promotes autophagy and microtubule-dependent autophagosomal clusters. Autophagy. 2013;9(4):550–567. doi: 10.4161/auto.23662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miermont A, et al. Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc Natl Acad Sci USA. 2013;110(14):5725–5730. doi: 10.1073/pnas.1215367110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TH, Linstedt AD. Osmotically induced cell volume changes alter anterograde and retrograde transport, Golgi structure, and COPI dissociation. Mol Biol Cell. 1999;10(5):1445–1462. doi: 10.1091/mbc.10.5.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carpentier JL, et al. Potassium depletion and hypertonic medium reduce “non-coated” and clathrin-coated pit formation, as well as endocytosis through these two gates. J Cell Physiol. 1989;138(3):519–526. doi: 10.1002/jcp.1041380311. [DOI] [PubMed] [Google Scholar]
- 17.Hansen SH, Sandvig K, van Deurs B. Clathrin and HA2 adaptors: Effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol. 1993;121(1):61–72. doi: 10.1083/jcb.121.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasler U, et al. Acute hypertonicity alters aquaporin-2 trafficking and induces a MAPK-dependent accumulation at the plasma membrane of renal epithelial cells. J Biol Chem. 2008;283(39):26643–26661. doi: 10.1074/jbc.M801071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108(2):389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garner MM, Burg MB. Macromolecular crowding and confinement in cells exposed to hypertonicity. Am J Physiol. 1994;266(4 Pt 1):C877–C892. doi: 10.1152/ajpcell.1994.266.4.C877. [DOI] [PubMed] [Google Scholar]
- 21.Zhou EH, et al. Universal behavior of the osmotically compressed cell and its analogy to the colloidal glass transition. Proc Natl Acad Sci USA. 2009;106(26):10632–10637. doi: 10.1073/pnas.0901462106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dill KA, Ghosh K, Schmit JD. Physical limits of cells and proteomes. Proc Natl Acad Sci USA. 2011;108(44):17876–17882. doi: 10.1073/pnas.1114477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-Boulan E, Kreitzer G, Müsch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005;6(3):233–247. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- 24.Galjart N. Plus-end-tracking proteins and their interactions at microtubule ends. Curr Biol. 2010;20(12):R528–537. doi: 10.1016/j.cub.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 25.Kang H, Bradley MJ, Elam WA, De La Cruz EM. Regulation of actin by ion-linked equilibria. Biophys J. 2013;105(12):2621–2628. doi: 10.1016/j.bpj.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olmsted JB, Borisy GG. Ionic and nucleotide requirements for microtubule polymerization in vitro. Biochemistry. 1975;14(13):2996–3005. doi: 10.1021/bi00684a032. [DOI] [PubMed] [Google Scholar]
- 27.Devin A, Espié P, Guérin B, Rigoulet M. Energetics of swelling in isolated hepatocytes: A comprehensive study. Mol Cell Biochem. 1998;184(1-2):107–121. [PubMed] [Google Scholar]
- 28.Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim Biophys Acta. 1989;973(3):355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- 29.Bazil JN, Buzzard GT, Rundell AE. Modeling mitochondrial bioenergetics with integrated volume dynamics. PLOS Comput Biol. 2010;6(1):e1000632. doi: 10.1371/journal.pcbi.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Copp J, Wiley S, Ward MW, van der Geer P. Hypertonic shock inhibits growth factor receptor signaling, induces caspase-3 activation, and causes reversible fragmentation of the mitochondrial network. Am J Physiol Cell Physiol. 2005;288(2):C403–C415. doi: 10.1152/ajpcell.00095.2004. [DOI] [PubMed] [Google Scholar]
- 31.Michea L, Combs C, Andrews P, Dmitrieva N, Burg MB. Mitochondrial dysfunction is an early event in high-NaCl-induced apoptosis of mIMCD3 cells. Am J Physiol Renal Physiol. 2002;282(6):F981–F990. doi: 10.1152/ajprenal.00301.2001. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Chen D, Chen Z, Moeckel GW. Hypertonicity-induced mitochondrial membrane permeability in renal medullary interstitial cells: Protective role of osmolytes. Cell Physiol Biochem. 2010;25(6):753–760. doi: 10.1159/000315095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holzbaur EL, Goldman YE. Coordination of molecular motors: From in vitro assays to intracellular dynamics. Curr Opin Cell Biol. 2010;22(1):4–13. doi: 10.1016/j.ceb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42(1):39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallee RB, Wall JS, Paschal BM, Shpetner HS. Microtubule-associated protein 1C from brain is a two-headed cytosolic dynein. Nature. 1988;332(6164):561–563. doi: 10.1038/332561a0. [DOI] [PubMed] [Google Scholar]
- 36.Liang ZY, et al. The transport of alpha(1A)-adrenergic receptor with 33-nm step size in live cells. Biochem Biophys Res Commun. 2007;353(2):231–237. doi: 10.1016/j.bbrc.2006.11.116. [DOI] [PubMed] [Google Scholar]
- 37.Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401(6753):613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 38.Mehta AD, et al. Myosin-V is a processive actin-based motor. Nature. 1999;400(6744):590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 39.Rock RS, et al. Myosin VI is a processive motor with a large step size. Proc Natl Acad Sci USA. 2001;98(24):13655–13659. doi: 10.1073/pnas.191512398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357(6375):257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- 41.Nunes P, et al. A fluorimetry-based ssYFP secretion assay to monitor vasopressin-induced exocytosis in LLC-PK1 cells expressing aquaporin-2. Am J Physiol Cell Physiol. 2008;295(6):C1476–C1487. doi: 10.1152/ajpcell.00344.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42(9):3858–3863. [PubMed] [Google Scholar]
- 43.Calabrese A, et al. Connexin 36 controls synchronization of Ca2+ oscillations and insulin secretion in MIN6 cells. Diabetes. 2003;52(2):417–424. doi: 10.2337/diabetes.52.2.417. [DOI] [PubMed] [Google Scholar]
- 44.Miyazaki J, et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127(1):126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 45.Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58(2):221–232. doi: 10.1007/s00125-014-3451-1. [DOI] [PubMed] [Google Scholar]
- 46.Machacek M, Danuser G. Morphodynamic profiling of protrusion phenotypes. Biophys J. 2006;90(4):1439–1452. doi: 10.1529/biophysj.105.070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hebert B, Costantino S, Wiseman PW. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys J. 2005;88(5):3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol. 1996;6(10):1279–1289. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- 49.Danowski BA. Fibroblast contractility and actin organization are stimulated by microtubule inhibitors. J Cell Sci. 1989;93(Pt 2):255–266. doi: 10.1242/jcs.93.2.255. [DOI] [PubMed] [Google Scholar]
- 50.Foe VE, Field CM, Odell GM. Microtubules and mitotic cycle phase modulate spatiotemporal distributions of F-actin and myosin II in Drosophila syncytial blastoderm embryos. Development. 2000;127(9):1767–1787. doi: 10.1242/dev.127.9.1767. [DOI] [PubMed] [Google Scholar]
- 51.Grzanka A, Grzanka D, Zuryń A, Grzanka AA, Safiejko-Mroczka B. Reorganization of actin in K-562 and HL-60 cells treated with taxol. Neoplasma. 2006;53(1):56–61. [PubMed] [Google Scholar]
- 52.Jung HI, Shin I, Park YM, Kang KW, Ha KS. Colchicine activates actin polymerization by microtubule depolymerization. Mol Cells. 1997;7(3):431–437. [PubMed] [Google Scholar]
- 53.Rosenblum MD, Shivers RR. 'Rings' of F-actin form around the nucleus in cultured human MCF7 adenocarcinoma cells upon exposure to both taxol and taxotere. Comp Biochem PhysiolC Toxicol Pharmacol. 2000;125(1):121–131. doi: 10.1016/s0742-8413(99)00101-2. [DOI] [PubMed] [Google Scholar]
- 54.Cabado AG, Vieytes MR, Botana LM. Effect of ion composition on the changes in membrane potential induced with several stimuli in rat mast cells. J Cell Physiol. 1994;158(2):309–316. doi: 10.1002/jcp.1041580213. [DOI] [PubMed] [Google Scholar]
- 55.Geukes Foppen RJ, van Mil HG, van Heukelom JS. Effects of chloride transport on bistable behaviour of the membrane potential in mouse skeletal muscle. J Physiol. 2002;542(Pt 1):181–191. doi: 10.1113/jphysiol.2001.013298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macri P, Breton S, Marsolais M, Lapointe J, Laprade R. Hypertonicity decreases basolateral K+ and Cl- conductances in rabbit proximal convoluted tubule. J Membr Biol. 1997;155(3):229–237. doi: 10.1007/s002329900175. [DOI] [PubMed] [Google Scholar]
- 57.Krapf R, Berry CA, Verkman AS. Estimation of intracellular chloride activity in isolated perfused rabbit proximal convoluted tubules using a fluorescent indicator. Biophys J. 1988;53(6):955–962. doi: 10.1016/S0006-3495(88)83176-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chao AC, Dix JA, Sellers MC, Verkman AS. Fluorescence measurement of chloride transport in monolayer cultured cells. Mechanisms of chloride transport in fibroblasts. Biophys J. 1989;56(6):1071–1081. doi: 10.1016/S0006-3495(89)82755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verkman AS, Sellers MC, Chao AC, Leung T, Ketcham R. Synthesis and characterization of improved chloride-sensitive fluorescent indicators for biological applications. Anal Biochem. 1989;178(2):355–361. doi: 10.1016/0003-2697(89)90652-0. [DOI] [PubMed] [Google Scholar]
- 60.Imamura H, et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA. 2009;106(37):15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meeusen SL, Nunnari J. How mitochondria fuse. Curr Opin Cell Biol. 2005;17(4):389–394. doi: 10.1016/j.ceb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 62.Ellis RJ, Minton AP. Cell biology: Join the crowd. Nature. 2003;425(6953):27–28. doi: 10.1038/425027a. [DOI] [PubMed] [Google Scholar]
- 63.Srere PA. Complexes of sequential metabolic enzymes. Annu Rev Biochem. 1987;56:89–124. doi: 10.1146/annurev.bi.56.070187.000513. [DOI] [PubMed] [Google Scholar]
- 64.Devin A, Guérin B, Rigoulet M. Response of isolated rat liver mitochondria to variation of external osmolarity in KCl medium: Regulation of matrix volume and oxidative phosphorylation. J Bioenerg Biomembr. 1997;29(6):579–590. doi: 10.1023/a:1022435102552. [DOI] [PubMed] [Google Scholar]
- 65.Kaasik A, Safiulina D, Zharkovsky A, Veksler V. Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol. 2007;292(1):C157–C163. doi: 10.1152/ajpcell.00272.2006. [DOI] [PubMed] [Google Scholar]
- 66.Yorimitsu T, Klionsky DJ. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blumwald E. Sodium transport and salt tolerance in plants. Curr Opin Cell Biol. 2000;12(4):431–434. doi: 10.1016/s0955-0674(00)00112-5. [DOI] [PubMed] [Google Scholar]
- 68.Record MT, Jr, Zhang W, Anderson CF. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: A practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv Protein Chem. 1998;51:281–353. doi: 10.1016/s0065-3233(08)60655-5. [DOI] [PubMed] [Google Scholar]
- 69.Dominy BN, Perl D, Schmid FX, Brooks CL., 3rd The effects of ionic strength on protein stability: The cold shock protein family. J Mol Biol. 2002;319(2):541–554. doi: 10.1016/S0022-2836(02)00259-0. [DOI] [PubMed] [Google Scholar]
- 70.Kohn WD, Kay CM, Hodges RS. Salt effects on protein stability: Two-stranded alpha-helical coiled-coils containing inter- or intrahelical ion pairs. J Mol Biol. 1997;267(4):1039–1052. doi: 10.1006/jmbi.1997.0930. [DOI] [PubMed] [Google Scholar]
- 71.Munishkina LA, Ahmad A, Fink AL, Uversky VN. Guiding protein aggregation with macromolecular crowding. Biochemistry. 2008;47(34):8993–9006. doi: 10.1021/bi8008399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haga T, Abe T, Kurokawa M. Polymerization and depolymerization of microtubules in vitro as studied by flow birefringence. FEBS Lett. 1974;39(3):291–295. doi: 10.1016/0014-5793(74)80133-x. [DOI] [PubMed] [Google Scholar]
- 73.Kang H, et al. Identification of cation-binding sites on actin that drive polymerization and modulate bending stiffness. Proc Natl Acad Sci USA. 2012;109(42):16923–16927. doi: 10.1073/pnas.1211078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yi K, et al. Dynamic maintenance of asymmetric meiotic spindle position through Arp2/3-complex-driven cytoplasmic streaming in mouse oocytes. Nat Cell Biol. 2011;13(10):1252–1258. doi: 10.1038/ncb2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zelenina M, Brismar H. Osmotic water permeability measurements using confocal laser scanning microscopy. Eur Biophys J. 2000;29(3):165–171. doi: 10.1007/pl00006645. [DOI] [PubMed] [Google Scholar]
- 76.Takanaga H, Chaudhuri B, Frommer WB. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta. 2008;1778(4):1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cogliati S, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155(1):160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hasler U, Vinciguerra M, Vandewalle A, Martin P-Y, Féraille E. Dual effects of hypertonicity on aquaporin-2 expression in cultured renal collecting duct principal cells. J Am Soc Nephrol. 2005;16(6):1571–1582. doi: 10.1681/ASN.2004110930. [DOI] [PubMed] [Google Scholar]
- 79.Peracino B, et al. G protein beta subunit-null mutants are impaired in phagocytosis and chemotaxis due to inappropriate regulation of the actin cytoskeleton. J Cell Biol. 1998;141(7):1529–1537. doi: 10.1083/jcb.141.7.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaeggeler HP, et al. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16(4):878–891. doi: 10.1681/ASN.2004121110. [DOI] [PubMed] [Google Scholar]
- 81.Emerson SU, et al. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J Virol. 2004;78(9):4838–4846. doi: 10.1128/JVI.78.9.4838-4846.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meda P, et al. Rapid and reversible secretion changes during uncoupling of rat insulin-producing cells. J Clin Invest. 1990;86(3):759–768. doi: 10.1172/JCI114772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramadori P, Sheikh N, Ahmad G, Dudas J, Ramadori G. Hepatic changes of erythropoietin gene expression in a rat model of acute-phase response. Liver Int. 2010;30(1):55–64. doi: 10.1111/j.1478-3231.2009.02131.x. [DOI] [PubMed] [Google Scholar]
- 84.Bustamante M, et al. Regulatory volume increase is associated with p38 kinase-dependent actin cytoskeleton remodeling in rat kidney MTAL. Am J Physiol Renal Physiol. 2003;285(2):F336–F347. doi: 10.1152/ajprenal.00003.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.