

Significance
DNA is wrapped around nucleosomes, so that repairing double-strand breaks (DSBs) requires significant nucleosome reorganization. Nucleosome reorganization requires exchange of histone H2A.Z. This work reveals that H2A.Z only transiently accumulates at breaks and demonstrates that Anp32, a histone chaperone, is recruited to DNA breaks and removes H2A.Z from the nucleosomes. Further, removal of H2A.Z from the intact nucleosome by Anp32 promotes acetylation of histone H4, remodels the local chromatin, and facilitates DNA repair. The dynamic exchange and removal of H2A.Z by Anp32e is therefore critical for chromatin reorganization at DSBs. Further, modulation of the nucleosome surface by Anp32e directs histone modification, recruitment of repair proteins, and the processing and repair of DSBs.
Keywords: NHEJ, DSB repair, H2A.Z, Anp32e, genome instability
Abstract
The repair of DNA double-strand breaks (DSBs) requires open, flexible chromatin domains. The NuA4–Tip60 complex creates these flexible chromatin structures by exchanging histone H2A.Z onto nucleosomes and promoting acetylation of histone H4. Here, we demonstrate that the accumulation of H2A.Z on nucleosomes at DSBs is transient, and that rapid eviction of H2A.Z is required for DSB repair. Anp32e, an H2A.Z chaperone that interacts with the C-terminal docking domain of H2A.Z, is rapidly recruited to DSBs. Anp32e functions to remove H2A.Z from nucleosomes, so that H2A.Z levels return to basal within 10 min of DNA damage. Further, H2A.Z removal by Anp32e disrupts inhibitory interactions between the histone H4 tail and the nucleosome surface, facilitating increased acetylation of histone H4 following DNA damage. When H2A.Z removal by Anp32e is blocked, nucleosomes at DSBs retain elevated levels of H2A.Z, and assume a more stable, hypoacetylated conformation. Further, loss of Anp32e leads to increased CtIP-dependent end resection, accumulation of single-stranded DNA, and an increase in repair by the alternative nonhomologous end joining pathway. Exchange of H2A.Z onto the chromatin and subsequent rapid removal by Anp32e are therefore critical for creating open, acetylated nucleosome structures and for controlling end resection by CtIP. Dynamic modulation of H2A.Z exchange and removal by Anp32e reveals the importance of the nucleosome surface and nucleosome dynamics in processing the damaged chromatin template during DSB repair.
The repair of DNA double-strand breaks (DSBs), which cleave the DNA backbone, requires remodeling of the local chromatin architecture. This reorganization of the chromatin is important for promoting access to the site of damage, for creating a template for the repair machinery, and for repackaging the chromatin and resetting the epigenetic landscape following repair. Chromatin remodeling at DSBs is linked to changes in posttranslational modification of histones. DSBs activate the ataxia-telangiectasia mutated (ATM) and DNA–PKcs kinases, which phosphorylate multiple DNA repair proteins, including histone H2AX. Phosphorylated H2AX (γH2AX) provides a binding site for mdc1, which promotes spreading of γH2AX for hundreds of kilobases either side of the break (1–3). DSBs also promote complex patterns of chromatin ubiquitination, including ubiquitination of H2A/H2AX by the RNF8/RNF168 ubiquitin ligases, which, in turn, creates binding sites for repair proteins such as 53BP1 and brca1 (4–7). DSBs also lead to increased methylation of histone H3 on lysine 36, which can regulate DNA repair pathway choice (8, 9) and methylation of H3 on lysine 9 (10), which drives activation of the Tip60 acetyltransferase and the ATM kinase (11, 12). Further, the NuA4–Tip60 complex (5, 13–15) promotes acetylation of histone H4 at DSBs and drives the formation of open, flexible chromatin domains at DSBs (5, 13, 14). The repair of DSBs is therefore fundamentally a chromatin-driven process requiring dynamic changes in histone modification and chromatin reorganization, which directly promote recruitment of DSB repair proteins (16).
The NuA4–Tip60 remodeling complex plays a central role in nucleosome reorganization at DSBs (16). NuA4–Tip60 is a 16 subunit complex containing 2 key subunits—the p400 SWI/SNF ATPase and the Tip60 acetyltransferase. The p400 ATPase promotes exchange of H2A for the histone variant H2A.Z (13, 17). This increase in H2A.Z at DSBs then promotes acetylation of histone H4 by the Tip60, creating open, flexible chromatin at sites of DNA damage (5, 11, 14). Inactivation of NuA4–Tip60 blocks both H2A.Z exchange and H4 acetylation, leading to a reduction in chromatin mobility at DSBs. Consequently, loss of H2A.Z exchange leads to defective DSB repair, increased sensitivity to DNA damage, and genomic instability (13, 18, 19).
Here, we demonstrate that H2A.Z exchange at DSBs is dynamic, with H2A.Z accumulating at DSBs within minutes of damage, followed by rapid H2A.Z eviction. Further, we show that Anp32e, an H2A.Z-specific histone chaperone, binds specifically to the docking domain of H2A.Z and is required to remove H2A.Z from the damaged chromatin template. Failure to remove H2A.Z leads to defects in DSB repair, including a loss of H4 acetylation, defects in nonhomologous end joining (NHEJ), and increased end resection of DSBs.
Results
Here, we examined the role of the histone chaperone Anp32e (20) in regulating H2A.Z exchange at DSBs. DNA damage created by laser microirradiation (“laser striping”) led to an increase in γH2AX and accumulation of Anp32e (Fig. 1A). No focal accumulation of Anp32e was seen in untreated cells (Fig. S1A). However, Anp32e accumulation was delayed relative to γH2AX formation, so that only 16% of γH2AX stripes contained Anp32e immediately after DNA damage (Fig. 1A). Despite this short delay, Anp32e was eventually loaded onto all of the γH2AX “stripes.” To further confirm that Anp32e is recruited to DSBs, we used the well-characterized p84-zinc finger nuclease (p84-ZFN) (13) to create a targeted DSB on chromosome 19 (Fig. S1C). ChIP demonstrated significant enrichment for Anp32e at the p84-ZFN DSB (Fig. 1B). Further, two siRNAs to Anp32e (which reduced Anp32e but not the cutting efficiency of p84-ZFN; Fig. S1 C and D) blocked Anp32e accumulation at the DSB (Fig. 1B). Anp32e is therefore rapidly loaded onto the chromatin at DSBs. Because Anp32e is an H2A.Z chaperone (20), we examined how Anp32e impacted the ability of the NuA4 complex to promote H2A.Z exchange (by the p400 ATPase) (5, 13) and H4 acetylation (H4Ac: by the Tip60 acetyltransferase) at DSBs. Loss of Anp32e reduced H4Ac at DSBs (Fig. 1C), indicating that Anp32e is required for H4Ac after DNA damage. We next examined if Anp32e contributed to H2A.Z exchange. H2A.Z rapidly accumulated at sites of laser damage (Fig. 1D). Surprisingly, H2A.Z was only transiently retained on the damaged chromatin, with essentially all of the H2A.Z removed within 10 min of damage (Fig. 1D). Further, depletion of Anp32e did not block accumulation of H2A.Z (Fig. 1D); rather, loss of Anp32e blocked removal of H2A.Z, leading to prolonged retention of H2A.Z on the chromatin at DSBs (Fig. 1D). The H2A.Z chaperone Anp32e therefore rapidly removes H2A.Z from sites of DNA damage. Further, because loss of Anp32e blocks H4Ac at DSBs (Fig. 1C), this implies that removal of H2A.Z by Anp32e may trigger H4Ac at DSBs.
Fig. 1.

Anp32e is rapidly recruited to DSBs. (A) U2OS cells expressing Flag-Anp32e were exposed to laser microirradiation and allowed to recover for the indicated times. γH2AX and Flag-Anp32e were detected by immunofluorescence. Percent of γH2AX stripes colocalizing with Anp32e is shown. (B and C) The 293T cells were incubated with nonspecific (siCon) or Anp32e-specific siRNAs (siAnp#3 or siAnp#5) for 48 h, followed by transfection with vector (−) or the p84-ZFN (+) to create DSBs. Cells were processed for ChIP 18 h later using Anp32e antibody (B) or H4Ac antibody (C), followed by real time-PCR (RT-qPCR) with primers located 500 bp to the right of the DSB. Results are ±SE (n = 3). (D) U2OS cells were incubated with nonspecific (siControl) or Anp32e-specific (siAnp#3) siRNA for 48 h, followed by laser striping. Cells were either fixed immediately (0 min) or allowed to recover for the indicated times, followed by immunofluorescent staining with γH2AX and H2A.Z antibodies.
Fig. S1.

Expression and analysis of H2AZ and Anp32e. (A) U2-OS cells were either mock irradiated (control) or exposed to laser microirradiation (laser) to create focused regions of DNA damage. Cells were then fixed and costained with DAPI (4′,6-diamidino-2-phenylindole) and antibodies to γH2AX (green) or Flag-Anp32e (red). (B) U2-OS cells were either mock irradiated (control) or exposed to laser microirradiation (laser) to create focused regions of DNA damage. Cells were then fixed and costained with DAPI and antibodies to γH2AX (green) or H2A.X (red). Conclusion from B and C is as follows: Bands of γH2AX, H2A.Z, and Anp32e are readily detected in the nucleus following laser microirradiation, with only diffuse staining detected in untreated, adjacent cells. (C) The 293T cells were transfected with the p84-ZFN and the percent of cells with a DSB was measured by PCR 18 h posttransfection. Results are ±SE (n = 3–5 biological replicates). The following cells were examined: 293T cells expressing HA-H2A.Z or HA-H2A.ZNG individually or in combination with LRS–GFP or LANA–GFP [containing the mutated (LRS) or wild-type (LANA) peptide sequences, respectively], control (untreated) 293T cells, or cells preincubated for 48 h with control siRNA (siCon) or siRNA Anp#3 (siAnp32e). Conclusions are as follows: Results indicate that depletion of Anp32e or expression of H2A.Z constructs did not significantly alter the ability of the p84-ZFN to create DSBs. (D) The 293T cells transiently transfected with either control siRNA (siCon) or two Anp32e-specific siRNAs (siAnp#3 and siAnp#5). Forty-eight hours later, cell extracts were prepared and Western blot analysis for Anp32e and cannexin (loading control) was carried out. Conclusion is as follows: siAnp#3 was chosen for use in experiments in Figs. 1–5. (E) The 293T cells were transfected with either control or Anp32e-specific shRNA and examined for Anp32e and Vinculin (loading control) by Western blot analysis.
The crystal structure of Anp32e bound to H2A.Z indicates that Anp32e makes multiple contacts with H2A.Z, including T103/I104 in the C-terminal docking domain (20, 21) (Fig. 2A). Although the Anp32e interacting residues are conserved in H2A, the extra glycine residue in H2A (blue circle; Fig. 2A) disrupts Anp32e–H2A interaction (20, 21), so that Anp32e only binds H2A.Z. We therefore replaced the Anp32e binding region of H2A.Z with the equivalent region from H2A, including the extra glycine, creating the chimeric H2A.Z protein H2A.ZNG (Fig. 2A). We then examined whether Anp32e–H2A.Z interaction was required for H2A.Z removal.
Fig. 2.

Anp32e uses H2A.Z’s docking domain to removes H2A.Z. (A) Sequence alignment of human H2A and H2A.Z, with conserved amino acids in blue. Residues required for Anp32e interaction (yellow circles) and the helix breaking glycine in H2A (blue circle) are indicated. Red lines delineate acidic domain. Sequence of chimeric H2A.ZNG is indicated. (B) The 293T cells expressing vector, HA-H2A.Z, or HA-H2A.ZNG were transfected with vector (−) or p84-ZFN (+) to create DSBs. Cells were processed for ChIP 18 h later using HA antibody and primers were located 500 bp to the right of the DSB. Results are ±SE (n = 3). (C) The 293T cells expressing HA-H2A.Z or HA-H2A.ZNG were transfected with vector (−) or p84-ZFN (+) to create DSBs. Cells were processed for ChIP 18 h later using H4Ac antibody and primers were located 500 bp to the right of the DSB. Results are ±SE (n = 3). (D) U2OS cells expressing HA-H2A.Z or HA-H2A.ZNG were exposed to laser striping and either fixed immediately (0 min) or recovered for 30 min. H2A.Z is detected with HA antibody. Percent of γH2AX stripes colocalizing with H2A.Z is shown.
H2A.ZNG and H2AZ were efficiently expressed (Fig. S2A) and exchanged onto the chromatin at the p84-ZFN DSB (Fig. 2B). HA-H2A.Z accumulated at sites of laser DNA damage, and was removed by 30 min postirradiation, so that less than 5% of cells retained HA-H2A.Z (Fig. 2D). However, HA-H2A.ZNG was exchanged onto the damaged chromatin but was not removed (Fig. 2D), being retained for at least 30 min. This result is consistent with the ChIP data (Fig. 2B), where higher levels of H2A.ZNG accumulated at DSBs. Further, Anp32e was recruited to DSBs in the absence of H2A.Z (Fig. S2B), indicating that retention of H2A.ZNG was not due to failure to recruit Anp32e to the DSB. Binding of Anp32e to H2A.Z’s docking domain is therefore required to remove H2A.Z from DSBs, but is not for the initial exchange of H2A.Z onto the damaged chromatin. Further, retention of H2A.ZNG at DSBs blocked H4Ac after DNA damage (Fig. 2C). Thus, retention of H2A.Z on damaged chromatin through loss of Anp32e (Fig. 1C) or inactivation of the Anp32e binding site on H2A.Z (Fig. 2D) inhibits H4Ac at DSBs. We interpret this to mean that removal of H2A.Z by Anp32e from damaged chromatin facilitates histone H4 acetylation.
Fig. S2.
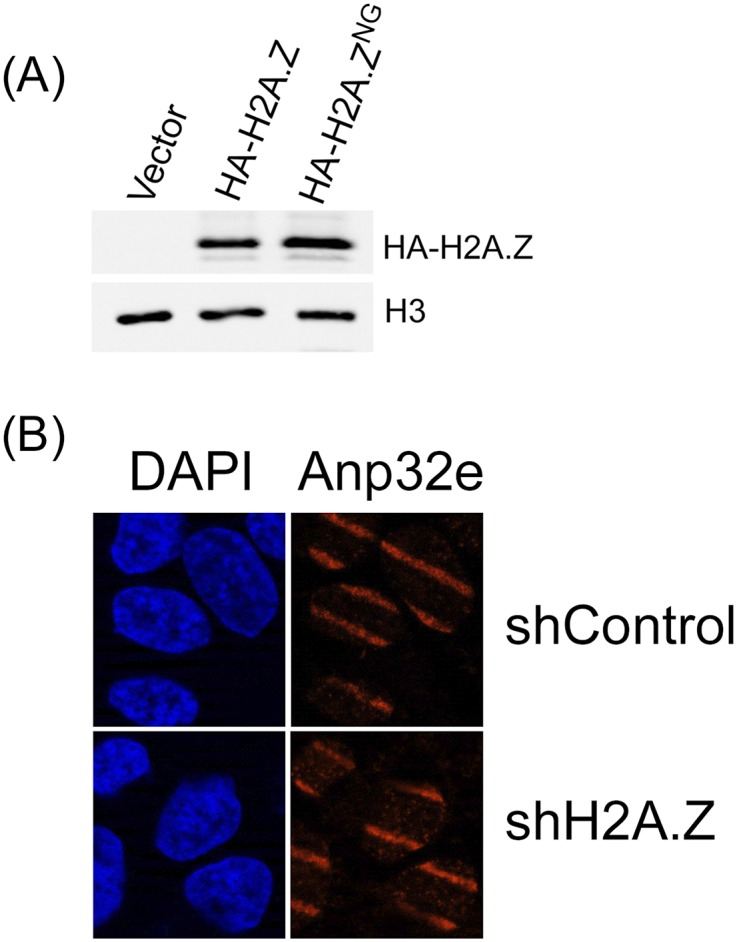
H2A.Z is not required to recruit Anp32e to sites of DNA damage. (A) The 293T cells expressing vector, HA-H2A.Z, or HA-H2A.ZNG were analyzed by Western blot using HA antibody or H3 antibody (loading control). (B) U2-OS cells expressing nonspecific shRNA or shRNA to H2A.Z were exposed to laser microirradiation to create DNA damage. Thirty minutes later, cells were fixed and incubated with antibodies to Anp32e or stained with DAPI. Conclusion is as follows: Recruitment of Anp32e to sites of DNA damage does not require H2A.Z, indicating that interaction between Anp32e and H2A.Z is not required to retain Anp32e at DSBs. Because Anp32e is a component of NuA4–Tip60, it is likely that Anp32e is recruited to DSBs as part of the NuA4–Tip60 complex.
Chromatin compaction involves binding of the H4 tail of one nucleosome to an acidic patch on the surface of an adjacent nucleosome. Acetylation of the H4 tails blocks this interaction and promotes unpacking of chromatin fibers (22–26). The acidic patch on the nucleosome surface is formed from a short stretch of acidic amino acids (the acidic domain) in the docking domain of H2A and H2A.Z (Fig. 2A). This acidic domain is adjacent to (and partially overlaps with) the Anp32e interaction domain on H2A.Z (20, 27–29) (Fig. 2A). Anp32e binding may therefore promote H4Ac by either displacing the H4 tail from the adjacent acidic domain or by removing H2A.Z and eliminating the entire H4 binding surface. To test this hypothesis, we used the LANA protein of Kaposi’s sarcoma herpes virus (27). LANA protein binds tightly to the acidic patch (30, 31), using the same acidic amino acids to which the H4 tail binds. The 23 amino acid domain of LANA, which binds to the acidic patch, and a control, in which an essential arginine was mutated to glycine (30), were fused to GFP and expressed in cells (Fig. S3A). GFP–LANA and GFP–Con had minimal impact on damage-induced H4Ac in control cells (Fig. 3A). However, GFP–LANA (but not GFP–Con) rescued H4Ac at DSBs in both H2A.ZNG cells (Fig. 3A) and Anp32e-depleted cells (Fig. 3B). This finding supports the idea that access to the H4 tail after DNA damage is restricted through interaction of H4 with the acidic patch on the nucleosome surface. Because the H4 tail and GFP–LANA both bind to this acidic patch (30, 31), displacement of the H4 tail, either by LANA peptide or Anp32e (which removes H2A.Z and eliminates the acidic patch) will promote H4Ac after DNA damage.
Fig. S3.

Retention of H2A.Z does not alter cell cycle distribution. (A) GFP–LANA, containing amino acids 1–23 of the LANA protein fused to GFP or GFP–Con, with an inactivating mutation in the LANA nucleosome binding domain, were expressed in 293T cells. The GFP tag was detected with GFP-specific antibody, with tubulin serving as loading control. (B) The 293T cells were transfected with control or CtIP-specific shRNA and examined for CtIP and tubulin (loading control) by Western blot analysis. (C) Cells were transfected with control siRNA (siControl) or siRNA to Anp32e (siAnp#3) for 48 h. Transfected cells and H2A.ZNG cells were collected, fixed, stained by propidium iodide, and analyzed by FACS for cell cycle distribution. Results are ±SE (n = 3 biological replicates). Conclusion is as follows: Depletion of Anp32e or expression of H2A.ZNG does not significantly perturb cell cycle distribution.
Fig. 3.

Binding of LANA to nucleosome surface rescues H4Ac in Anp32e-deficient cells. (A) The 293T cells expressing HA-H2A.Z, HA-H2A.ZNG, GFP–Con, or GFP–LANA were transfected with vector (−) or p84-ZFN (+). ChIP was performed 18 h later using H4Ac antibody and primers were located 500 bp to the right of the DSB. Results are ±SE (n = 3). (B) The 293T cells expressing GFP–Con or GFP–LANA were transiently transfected with siRNA to Anp32e (siAnp#3). Cells were transfected 48 h later with vector (−) or p84-ZFN (+) and processed for ChIP using H4Ac antibody. (C) Cells expressing HA-H2A.Z or HA-H2A.ZNG were untreated or exposed to bleomycin (7.5 μM) as indicated. Nuclei were extracted in 1.0 M NaCl, salt soluble proteins separated by SDS/PAGE, and H3 was detected by Western blot. Ponceau S staining indicates loading.
Increased H4 acetylation by NuA4–Tip60 at DSBs also facilitates the formation of open, relaxed chromatin at DSBs (13). Open chromatin at DSBs is associated with an increase in the sensitivity of histones to extraction in NaCl (5). When cells were exposed to bleomycin to create DSBs and extracted in 1.0 M NaCl, there was a rapid increase in the NaCl solubility of histone H3 (Fig. 3C), consistent with a reduction in nucleosome stability after DNA damage. Importantly, this increase in NaCl sensitivity was significantly reduced in cells expressing H2A.ZNG (Fig. 3C), indicating that retention of H2A.Z at DSBs tends to stabilize the nucleosomes. Because expression of H2A.ZNG also inhibits H4Ac (Fig. 3 A and B), we used the LANA peptide to restore H4Ac in H2A.ZNG cells. However, even though the LANA peptide can restore H4Ac (Fig. 3B), it only partially restored the increase in NaCl solubility of histone H3 caused by DNA damage in H2A.ZNG cells (Fig. 3C). Increased H4Ac, on its own, is therefore insufficient to decrease nucleosome stability. This result suggests that removal of H2A.Z, in combination with increased H4Ac, are required to decrease nucleosome stability at DSBs.
Previous studies implicated H2A.Z exchange in regulation of end processing of DSBs by CtIP (13). In fact, cells expressing H2A.ZNG or lacking Anp32e (Fig. 4A) exhibited a significant loss of Ku70 recruitment to laser stripes. In addition, ChIP analysis demonstrated an increase in the ssDNA binding protein RPA32 at DSBs in H2A.ZNG cells (Fig. 4B). This finding suggests that retention of H2A.Z at the DSB (by expressing H2A.ZNG or depleting Anp32e) directs processing of the DNA ends toward ssDNA production, favoring RPA binding over Ku70/80 binding. Further, GFP–LANA, which binds to the nucleosome surface and partially rescues H4Ac and formation of open chromatin in H2A.ZNG cells (Fig. 3), substantially restored Ku70/80 loading in both H2A.ZNG- and Anp32e-deficient cells (Fig. 4 C and D). Importantly, depletion of CtIP (Fig. S3B), which is essential for end resection, also restored Ku70 loading in the absence of Anp32e (Fig. 4E). Loss of Ku70 binding and increased ssDNA in Anp32e-depleted cells is therefore mediated through increased end resection of the break by CtIP. Because depletion of Anp32e blocks H2A.Z removal (Fig. 3) and H4Ac (Fig. 4), this result implies that dynamic H2A.Z exchange (by NuA4–Tip60) and removal by Anp32e, coupled with H4Ac, creates a chromatin conformation that functions to restrain CtIP-mediated end resection, retaining intact DSB ends and therefore favoring Ku70 binding.
Fig. 4.

Removal of H2A.Z by Anp32e is required for retention of Ku70 at DSBs. (A) U2OS cells expressing HA-H2A.Z or HA-H2A.ZNG or transfected with control or Anp32e siRNA siAnp#3 were exposed to laser striping and allowed to recover for 30 min, followed by immunofluorescent staining for γH2AX and Ku70. (B) The 293T cells expressing HA-H2A.Z or HA-H2A.ZNG were transfected with control or Anp32e siRNA siAnp#3 for 48 h followed by vector (−) or p84-ZFN (+) to create DSBs. ChIP using antibody to the RPA32 subunit of the RPA complex, with PCR primers located 100 bp to the right of the DSB, was then carried out. (C) The 293T cells expressing GFP–Con, GFP–LANA, HA-H2A.Z, or HA-H2A.ZNG were transfected with vector (−) or p84-ZFN (+) to create DSBs. ChIP, using Ku70/80-specific antibody, with primers located 100 bp to the right of the DSB, was then carried out. Results are ±SE (n = 3). (D) The 293T cells expressing GFP–Con or GFP–LANA were transfected with control (siCon) or Anp32e-specific (siAnp32e) siRNA. Cells were transfected 48 h later with vector (−) or p84-ZFN (+) to create DSBs, followed by ChIP using Ku70 antibody. Results are ±SE (n = 3). (E) U2OS cells were treated with control siRNA, siRNA to Anp32e (siAnp#3), or shRNA targeting CtIP. DNA damage was created by laser striping, and Ku70 was detected by immunofluorescence 30 min later.
Finally, we examined how Anp32e contributes to DSB repair. Loss of Anp32e (Fig. 5A) or expression of H2A.ZNG (Fig. 5B) increased radiosensitivity, consistent with a key role for Anp32e and H2A.Z removal in DSB repair. Neither depletion of Anp32e or expression of H2A.ZNG impacted cell cycle kinetics (Fig. S3C). However, loss of Anp32e reduced NHEJ activity in cells (Fig. 5C), consistent with the loss of Ku70 at DSBs in the absence of Anp32e (Fig. 4E). Alternative-NHEJ (alt-NHEJ) is a backup pathway used when components of NHEJ, such as Ku70, are inactivated (32) and uses CtIP-mediated resection and microhomology to promote DSB repair (32, 33). Indeed, cells lacking Anp32e showed increased repair through the alt-NHEJ pathway (Fig. 5D), consistent with the increase in CtIP-dependent ssDNA and loss of Ku70 binding (Fig. 4). Finally, depletion of Anp32e led to a small but significant increase in repair by homologous recombination (HR) (Fig. 5E), suggesting that the increase in ssDNA following Anp32e depletion may also increase repair by the HR pathway.
Fig. 5.

Loss of Anp32e promotes repair by the alt-NHEJ pathway. (A) The 293T cells were transfected with control or Anp32e siRNA siAnp#3. Cells were irradiated 48 h later and surviving colonies counted. Results are ±SE (n = 3). (B) MCF-7 cells expressing HA-H2A.Z or HA-H2A.ZNG were irradiated and surviving colonies counted. Results are ±SE (n = 3). (C–E) Cells carrying either (C) a GFP–NHEJ reporter, (D) a GFP–alt-NHEJ reporter, or (E) a GFP–HR reporter were transfected with control or Anp32e-specific siRNA, followed by vector (Vec) or I-Sce1 to create DSBs. GFP positive cells were quantitated by FACS analysis. Results are ±SE (n = 4–6).
Discussion
We have demonstrated that the Anp32e histone chaperone is a DNA damage response protein which directs removal of H2A.Z from nucleosomes at DSBs. Previous work indicated that NuA4–Tip60 promotes both H2A.Z exchange and H4Ac on nucleosomes at DSBs, creating open chromatin domains that are required for DSB repair (Fig. 6) (11, 13, 14). Our results now extend this work to demonstrate that H2A.Z is only retained transiently at DSBs, and that rapid removal of H2A.Z by Anp32e is required to promote H4 acetylation and nucleosome reorganization at DSBs. The NuA4–Tip60 complex and Anp32e therefore function together to coordinate dynamic accumulation and removal of H2A.Z from nucleosomes during the repair of DSBs.
Fig. 6.

Control of nucleosome dynamics by Anp32e-directed removal of H2A.Z at DSBs. Unacetylated H4 tails bind to the acidic patch on adjacent nucleosomes. Exchange of H2A.Z by the p400 subunit of NuA4 creates a binding site for Anp32e on the nucleosome surface. Anp32e then removes the entire H2A.Z–H2B dimer, resulting in loss of the acidic patch. Loss of the acidic patch releases the H4 tail, allowing for acetylation by Tip60 and a shift to a more flexible chromatin structure. The resulting partial nucleosome may then be reconstituted by the addition of new H2A-H2B dimers or may contribute to the nucleosome-depleted region identified at DSBs.
We propose a model (Fig. 6) in which the initial exchange of H2A.Z by NuA4–Tip60 stabilizes nucleosomes at the break, limiting chromatin mobility and maintaining chromatin structure. Anp32e then removes the entire H2A.Z–H2B dimer, eliminating both H2A.Z and the acidic patch, and freeing the H4 tail for acetylation by Tip60 and processing of the DSB. How H2A.Z alters nucleosome function is complex and may depend on associated histone modifications and histone variants (29). For example, H2A.Z is associated with open chromatin (34–37), but is present in heterochromatin and can stabilize nucleosomes (38, 39). In addition, the acidic domain of H2A.Z is longer than that of H2A (Fig. 2A) and favors the formation of more compact chromatin (29, 39), presumably by increasing interaction between the H4 tail and the acidic patch. Further, we showed H2A.Z retention at DSBs blocked the shift to open chromatin, indicating that the initial exchange of H2A.Z stabilizes chromatin immediately after DNA damage. Other repressive factors, including HP1 and NuRD (10, 40–42), are also rapidly, but transiently recruited to DSBs. Transient loading of H2A.Z and other repressors may therefore temporarily “heterochromatinize” nucleosomes at breaks, stabilizing the damaged chromatin, and allowing the cell time to process the chromatin for DNA repair (Fig. 6).
H2A.Z contains a specific binding site for Anp32e (20, 21), which partially overlaps with the acidic domain to which the H4 tail binds (24–27). Anp32e binding to H2A.Z may displace the H4 tail, promoting H4 acetylation. The observation that the LANA protein, which binds the acidic domain (30), can rescue H4 acetylation in the absence of Anp32e supports this finding. However, Anp32e, in common with many histone exchange reactions, actually removes the entire H2A.Z–H2B dimer (20, 21), potentially creating tetrasomes (containing only H3–H4 dimers). Studies indicate that H2B is lost from the chromatin at DSBs (43), suggesting that removal of H2A.Z–H2B by Anp32e may create chromatin regions containing tetrasomes or possibly lacking any nucleosomes. Removal of the H2A.Z–H2B dimer will therefore eliminate the entire acidic patch from the nucleosome surface, releasing the H4 tail for acetylation by the Tip60 acetyltransferase (Fig. 6). However, although regions adjacent to the DSB may exhibit partial loss of nucleosomes (43), we have shown that regions more distal (>1 kb) to the DSB maintain intact nucleosomes (13, 16). Thus, removal of H2A.Z–H2B dimers by Anp32e is likely coupled to the addition of new H2A–H2B dimers by other remodeling ATPases implicated in DSB repair, such as Ino80 (44, 45). Further, because acetylation prevents the H4 tail from rebinding to the acidic patch (22–26), the combination of H2A.Z–H2B removal and H4Ac prevents interaction between nucleosomes and promotes the shift to open, flexible chromatin required for DSB repair (Fig. 6).
How NuA4–Tip60 and Anp32e precisely regulate the balance between exchange (by p400) and removal (by Anp32e) of H2A.Z is not clear. Anp32e was recently identified as a subunit of NuA4–Tip60 (20), suggesting that a single NuA4–Tip60/Anp32e complex can both rapidly add and remove H2A.Z from nucleosomes at DSBs. However, our results indicate that Anp32e recruitment to DSBs has different kinetics to that seen for other components of NuA4 (Fig. 1), suggesting that Anp32e functions separately from NuA4–Tip60 during DSB repair. Control of the balance between exchange and removal of H2A.Z may therefore be provided by other components of NuA4–Tip60, such as the ruvbl1 and ruvbl2 ATPases (46), modification of H2A.Z (e.g., through acetylation) or Anp32e, or through contributions of other motor ATPases implicated in DSB repair (15, 47).
A key finding was that retention of H2A.Z increased ssDNA at DSBs, and that these breaks were repaired mainly by the alt-NHEJ pathway. Importantly, this process was dependent on CtIP, suggesting that H2A.Z exchange and removal regulates CtIP activity. H2A.Z removal is required for several epigenetic modifications, including H4Ac (5, 13–15) and ubiquitination (5, 13, 14), which control nucleosome packing and loading of brca1 and 53BP1 at DSBs (5, 13, 14, 48). Consequently, retention of H2A.Z creates more compact nucleosomes lacking key histone modifications, such as H4Ac, at the site of damage. This shift in nucleosome conformation, combined with altered loading of DNA repair proteins such as 53BP1, may weaken the normal nucleosomal barrier to end resection (16). Interestingly, exo1 mediated end resection is increased on H2A.Z nucleosomes compared to H2A nucleosomes (45), supporting the idea that H2A.Z retention may favor end resection of DSBs. The failure to remove H2A.Z may therefore reduce the nucleosomal barrier to end resection by CtIP, leading to increased ssDNA and channeling of repair into the alt-NHEJ pathway. Dynamic H2A.Z exchange and subsequent removal by Anp32e represents a precise, ordered mechanism for regulating end processing during DSB repair.
The increase in alt-NHEJ repair when H2A.Z is retained at the DSB has parallels with repair of persistent DSBs. In yeast, persistent DSBs contain H2A.Z and are relocated to the nuclear membrane by a mechanism requiring H2A.Z-remodeling ATPases (49, 50). Further, DSBs tethered to the nuclear lamina can be repaired by the alt-NHEJ pathway (51). Failure to remove H2A.Z may create incorrectly processed chromatin at the DSB, which resembles a persistent, unrepaired DSB. These unrepaired DSBs are then processed through CtIP-dependent end resection and the alt-NHEJ pathway. H2A.Z exchange and removal by Anp32e are therefore important for ensuring accurate processing of the chromatin so that DSBs are channeled into the correct DSB repair pathway and therefore limit processing through the error-prone alt-NHEJ pathway.
In conclusion, we have shown that H2A.Z exchange and subsequent removal by Anp32e plays a critical role in the early chromatin processing events at DSBs, including promoting H4 acetylation. Further, the results indicate that the nucleosome surface plays a critical role in DSB repair, providing an essential binding site for key proteins involved in the earliest events of DSB repair. Finally, the requirement for rapid removal of H2A.Z by Anp32e demonstrates that the precise ordering and timing of epigenetic modifications and nucleosome reorganization at DSBs is required to suppress unwanted outcomes and promote efficient DSB repair.
Materials and Methods
Details of cell culture, DNA repair assays, transfection protocols, sequences of shRNA and siRNA, plasmid construction, Western blot analysis, nucleosome stability assay, and antibodies are in SI Materials and Methods.
Chromatin Immunoprecipitation.
ChIP assays used the SimpleChIP Chromatin IP Kit (Cell Signaling Technology). Cells were cross-linked with formaldehyde (5 min) and quenched with glycine. Cell pellets were solubilized in ChIP buffer (Cell Signaling Technology) and sonicated (Fisher BioRuptor U200). Part of the supernatant was digested with proteinase K (65 °C for 2 h), the DNA isolated by spin columns, and input DNA quantitated by real time PCR. Equivalent amounts of chromatin were incubated with primary antibody (overnight at 4 °C) followed by protein-G agarose beads precoated with sperm DNA. Immune complexes were washed in low and high salt ChIP buffers (Cell Signaling Technology), eluted and incubated in NaCl (65 °C for 2 h), and then digested with proteinase K. Purified DNA was quantitated by RT-qPCR using the Step One Plus real time PCR system (Applied Biosystems). Primer sequences, ChIP grade antibodies, and PCR conditions are in SI Materials and Methods.
Laser Microirradiation and Immunofluorescence.
Laser damage was produced using a 30-mW 405-nm diode laser focused through the 40×-Plan Apochromat/1.25 N.A. oil objective (Leica TCS SP5, Leica Microsystems). Laser output was 40–60% power to limit DNA damage to the laser path without noticeable cytotoxicity. Cells were preincubated with Hoechst 33258 (1 μg/mL for 5 min at 37 °C) before exposure to laser light. Cells were fixed with PBS/paraformaldehyde [4% (vol/vol)]. Time between laser exposure and fixation was 3 min. Cells were permeabilized in methanol, washed in PBS, and incubated in Triton X-100 (0.2% for 5 min). Cells were washed twice in PBS and blocked with FBS (10% for 20 min). For Ku70, cells were preextracted in buffer D (10 mM Pipes pH 7.0; 100 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 0.5% Triton X-100) before fixation. Slides were incubated with primary and secondary antibody with washing between each step, mounted with Fluoromount-G (Southern Biotech), and imaged with a Zeiss AxioImager Z1 microscope equipped with an Axiocam MRc Rev.3 color digital camera and Plan APO 63×/1.4 oil M27 lens (magnification 63×, aperture 1.4). Acquisition software and image processing used the Zeiss AxioVision software package (Zeiss Imaging).
SI Materials and Methods
Cell culture.
U2OS, HeLa, and HEK293T cells (obtained from the American Type Culture Collection) were cultured according to the supplier’s instructions and certified mycoplasma and contaminant free. For clonogenic cell survival assays, cells were plated in triplicate at an appropriate dilution on six-well dishes and allowed to attach for 24 h. Cells were irradiated using a Cs137 irradiator and allowed to recover for 10–14 d. Cells were then fixed and stained with crystal violet, and colonies with >50 cells were scored visually as previously described (13).
HEK293 cells expressing the alt-NHEJ reporter (13), U2OS cells expressing the HR reporter, and HeLa cells expressing an NHEJ reporter (5, 13) were infected with vector or shRNA targeting Anp32e. Cells were transfected 72 h later with I-Sce I plasmid using Lipofectamine 2000 (Invitrogen). GFP positive cells were quantified 48 h later using the BD LSR II cell analyzer and data were analyzed with the BD Diva software package.
Real-Time Quantitative PCR Analysis.
PCR amplification protocol used an initial step of 95 °C for 5 min, followed by 33 cycles of: 30 sec at 95 °C/30 sec at 60 °C/30 sec at 72 °C, and a final extension step of 5 min at 72 °C. Serial dilutions of the starting material were used to determine the linear range of PCR amplification before use. Input genomic DNA was standardized against 18S rRNA genomic sequences. Standard controls included immunoprecipitation with IgG, which yielded essentially no signal. Relative fold enrichment was optimized by the input control and expressed as IP/input DNA.
For ChIP, the relative increase in signal after cutting by the p84-ZFN was calculated as [IPZFN/inputZFN]/[IPControl/inputControl]. All ChIP assays represent biological duplicates, with individual RT-qPCR reactions carried out in triplicate and the results presented ± SD.
Primers at p84-ZFN are as follows:
−0.5 kb: (TGGGTTCCCTTTTCCTTCTC and GTCCAGGCAAAGAAAGCAAG);
+0.5 kb: (ATGGTGCGTCCTAGGTGTTC and CCAAGGACTCAAACCCAGAA);
−100 bp: (AGAACCAGAGCCACATTAACC and CACTTCAGGACAGCATGTTTG)
+100 bp: (CTTAGAGGTTCTGGCAAGGAG and ACAGAAAAGCCCCATCCTTAG).
Additional primers are as follows:
18S primers: (CCCAGTAAGTGCGGGTCATA and GATCCGAGGGCCTCACTAAAC);
DSB primers for p84-ZFN: (CGGTTAATGTGGCTCTGGTT and ACAGGAGGTGGGGGTTAGAC).
Statistical Analysis.
All ChIP assays represent biological duplicates, with individual RT-qPCR reactions carried out in triplicate and the results presented are ±SD. Laser striping experiments were carried out with at least three biological replicates, with 30–55 cells striped per slide. Representative images, including at least seven cells, are shown in the figures in the main text. For quantitation of laser striping, γH2AX positive stripes with Anp32e or H2A.Z signal exceeding 1.25× background (calculated from signal intensity of stripe region divided by adjacent nonirradiated region within the same nucleus) were counted as colocalized. Results were averaged across 20–30 cells per field, with at least three biological replicates. For cell survival analysis, three biological replicates were carried out and results are expressed as mean ± SD.
Plasmids, shRNA, and siRNA.
shRNA to Anp32e was obtained from the Broad Institute collection (clone ID TRCN0000077936). Stable cell lines were derived by infecting HEK293T or U2OS cells with control or Anp32e shRNA followed by selection with puromycin (1 µg/mL) for 7 d. siRNA to Anp32e (Anp#3: TTAGTTCCACATTAGCCATAC and Anp#5: ATCCTCCTGATCAAATCCATC) was from Thermo Scientific Fisher. shRNA to CtIP used a previously validated shRNA (CCGGCGTCAGCCTTACAACGCAATACTCGAGTATTGCGTTGTAAGGCTGACGTTTTT) (13). H2A.Z cDNA was obtained from the DF/HCC DNA resource core (plasmid.med.harvard.edu/PLASMID). A HA-tag was added to the N-terminal by standard PCR protocols and the product was cloned into the pcDNA3.1 expression vector. H2A.ZNG was created by mutating the docking domain of H2A.Z (DSLIKA) to that of H2A (NKLLGKV), by site-directed mutagenesis (Agilent Technologies). GFP–Con and GFP–LANA were provided by Kenneth Kaye (Harvard Medical School, Boston) Anp32e by Ali Hamiche (Universite de Strasbourg, Strasbourg, France), and p84-ZFN by Fyodor Urnov (Sangamo BioSciences, Richmond, CA).
Transfection Protocols.
For p84-ZFN, 4 × 106 293T cells were plated on 10-cm dishes and allowed to grow for 36 h. Cells were transfected with p84-ZFN or control vectors using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. At 18 h posttransfection, cell pellets were collected and stored at −80 °C for ChIP analysis. For siRNA treatment, 293T cells were transiently transfected with Anp32e or control siRNAs using Lipofectamine RNAimax (Invitrogen) according to the manufacturer’s protocol. At 36 h posttransfection, cells were trypsinized, plated at 4 × 106 cells on 10-cm dishes, followed by p84-ZFN transfection as described above. GFP–control, GFP–LANA, and H2A.ZNG were transfected into 293T or U2OS cells using Lipofectamine 2000 and selected for 7–14 d with selection marker hygromycin 500 µg/mL or puromycin (1 µg/mL) as described previously by us (12).
Western Blot Analysis.
Cell extracts were prepared using RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Nonidet P-40, 1% sodium deoxycholate) containing protease inhibitor mixture (Roche). Protein concentration was measured using the Bio-Rad DC Protein assay kit. For Western blots, proteins were separated by SDS/PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk, incubated with primary antibodies for 1 h, washed in TBST (20 mM Tris, pH 7.5, 137 mM NaCl, 0.1% Tween 20), and then incubated with goat anti-mouse IR Dye-800CW or goat anti-rabbit IR Dye-680CW conjugated secondary antibodies (Li-Cor). Imaging was carried out using the Li-Cor Odyssey Near Infra Red System, and analysis was done with the Odyssey Software system 3.0 package (Li-Cor).
Nucleosome Stability Assay.
The nucleosome stability assay is described in ref. 5. Briefly, cells were washed twice in ice-cold PBS and resuspended in 500 μL buffer A (20 mM Hepes, pH 7.9, 0.5 mM DTT, 1 mM PMSF, 1.5 mM MgCl 2, 0.1% Triton) containing 1.0 M NaCl for 40 min at 4 °C with constant agitation. Cells were collected by centrifugation at 100,000 × g (Beckmann Ultracentrifuge) for 20 min, and the supernatant was retained for Western blot analysis.
Antibodies.
The following antibodies were used: acetylated H4 (Millipore), ChIP grade antibodies to H3 and H4 (Abcam), H2A.Z (Active Motive and Cell Signaling Technology), RPA32 (Abcam), Ku70/80 (Neomarker), γH2AX (2577S, Cell Signaling) and (05-636, Millipore), HA antibodies (Abcam), sc-7392 (Santa Cruz), GFP (Abcam), Flag (Sigma), Anp32e (Sigma), and Vinculin and Calnexin (Sigma). CtIP antibody was a gift from Richard Baer, Columbia University, New York.
Supplementary Material
Acknowledgments
We thank Ali Hamiche for Anp32e cDNA, Ken Kaye for LANA constructs, and Sangamo Biosciences for p84-ZFN. We also thank Jessica Tyler for sharing unpublished data and for insightful discussions and Timur Yusufzai for advice. This work was supported by NIH Grants CA93602, CA64585, and CA177884.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504868112/-/DCSupplemental.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonner WM, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8(12):957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 4.Mattiroli F, et al. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150(6):1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, et al. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol. 2010;191(1):31–43. doi: 10.1083/jcb.201001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fradet-Turcotte A, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499(7456):50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49(5):795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 8.Fnu S, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci USA. 2011;108(2):540–545. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pai CC, et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat Commun. 2014;5:4091. doi: 10.1038/ncomms5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci USA. 2014;111(25):9169–9174. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11(11):1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. 2005;102(37):13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, et al. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48(5):723–733. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murr R, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8(1):91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 15.Downs JA, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16(6):979–990. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152(6):1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishibuchi I, et al. Reorganization of damaged chromatin by the exchange of histone variant H2A.Z-2. Int J Radiat Oncol Biol Phys. 2014;89(4):736–744. doi: 10.1016/j.ijrobp.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 18.Morillo-Huesca M, Clemente-Ruiz M, Andújar E, Prado F. The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS ONE. 2010;5(8):e12143. doi: 10.1371/journal.pone.0012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011;144(2):200–213. doi: 10.1016/j.cell.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obri A, et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. 2014;505(7485):648–653. doi: 10.1038/nature12922. [DOI] [PubMed] [Google Scholar]
- 21.Mao Z, et al. Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Res. 2014;24(4):389–399. doi: 10.1038/cr.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyrick JJ, Kyriss MN, Davis WB. Ascending the nucleosome face: Recognition and function of structured domains in the histone H2A-H2B dimer. Biochim Biophys Acta. 2012;1819(8):892–901. doi: 10.1016/j.bbagrm.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potoyan DA, Papoian GA. Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc Natl Acad Sci USA. 2012;109(44):17857–17862. doi: 10.1073/pnas.1201805109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha D, Shogren-Knaak MA. Role of direct interactions between the histone H4 Tail and the H2A core in long range nucleosome contacts. J Biol Chem. 2010;285(22):16572–16581. doi: 10.1074/jbc.M109.091298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou J, Fan JY, Rangasamy D, Tremethick DJ. The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nat Struct Mol Biol. 2007;14(11):1070–1076. doi: 10.1038/nsmb1323. [DOI] [PubMed] [Google Scholar]
- 26.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 27.Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, Hansen JC. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface. 2013;10(82):20121022. doi: 10.1098/rsif.2012.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat Rev Mol Cell Biol. 2012;13(7):436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zlatanova J, Thakar A. H2A.Z: View from the top. Structure. 2008;16(2):166–179. doi: 10.1016/j.str.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Barbera AJ, et al. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311(5762):856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 31.Chodaparambil JV, et al. A charged and contoured surface on the nucleosome regulates chromatin compaction. Nat Struct Mol Biol. 2007;14(11):1105–1107. doi: 10.1038/nsmb1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4(6):e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18(1):75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber CM, Henikoff JG, Henikoff S. H2A.Z nucleosomes enriched over active genes are homotypic. Nat Struct Mol Biol. 2010;17(12):1500–1507. doi: 10.1038/nsmb.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2009;19(3):460–469. doi: 10.1101/gr.087619.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21(12):1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Roberts DN, Cairns BR. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123(2):219–231. doi: 10.1016/j.cell.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park YJ, Dyer PN, Tremethick DJ, Luger K. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J Biol Chem. 2004;279(23):24274–24282. doi: 10.1074/jbc.M313152200. [DOI] [PubMed] [Google Scholar]
- 39.Fan JY, Rangasamy D, Luger K, Tremethick DJ. H2A.Z alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol Cell. 2004;16(4):655–661. doi: 10.1016/j.molcel.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 40.Chou DM, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA. 2010;107(43):18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luijsterburg MS, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185(4):577–586. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol. 2011;193(1):81–95. doi: 10.1083/jcb.201101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldstein M, Derheimer FA, Tait-Mulder J, Kastan MB. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc Natl Acad Sci USA. 2013;110(42):16874–16879. doi: 10.1073/pnas.1306160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Symington LS. Overcoming the chromatin barrier to end resection. Cell Res. 2013;23(3):317–319. doi: 10.1038/cr.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adkins NL, Niu H, Sung P, Peterson CL. Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol. 2013;20(7):836–842. doi: 10.1038/nsmb.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi J, Heo K, An W. Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 2009;37(18):5993–6007. doi: 10.1093/nar/gkp660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34(2):71–77. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 48.Tang J, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20(3):317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalocsay M, Hiller NJ, Jentsch S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell. 2009;33(3):335–343. doi: 10.1016/j.molcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 50.Horigome C, et al. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell. 2014;55(4):626–639. doi: 10.1016/j.molcel.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 51.Lemaître C, et al. Nuclear position dictates DNA repair pathway choice. Genes Dev. 2014;28(22):2450–2463. doi: 10.1101/gad.248369.114. [DOI] [PMC free article] [PubMed] [Google Scholar]