

Abstract
Recent progress in the development of single-nucleotide polymorphism (SNP) maps within genes and across the genome provides a valuable tool for fine-mapping and has led to the suggestion of genomewide association studies to search for susceptibility loci for complex traits. Test statistics for genome association studies that consider a single marker at a time, ignoring the linkage disequilibrium between markers, are inefficient. In this study, we present a generalized T2 statistic for association studies of complex traits, which can utilize multiple SNP markers simultaneously and considers the effects of multiple disease-susceptibility loci. This generalized T2 statistic is a corollary to that originally developed for multivariate analysis and has a close relationship to discriminant analysis and common measure of genetic distance. We evaluate the power of the generalized T2 statistic and show that power to be greater than or equal to those of the traditional χ2 test of association and a similar haplotype-test statistic. Finally, examples are given to evaluate the performance of the proposed T2 statistic for association studies using simulated and real data.
Introduction
Lack of tangible success of genetic linkage analyses for mapping of multifactorial trait loci with small-to-moderate effects, coupled with progress in the development of detailed SNP maps of the human genome (Gray et al. 2000), has led to the suggestion of population-based genomewide association studies (Risch and Merikangas 1996) that are based on linkage disequilibrium (LD). Traditional population-based association studies compare marker-allele frequencies between cases and control subjects, separately for each marker. However, when a collection of SNP markers is available, using only a single marker each time and ignoring the nonindependence among markers are inefficient. In addition, it is well known that complex diseases are influenced by multiple genes, requiring the development of statistical methods for evaluation of several trait loci collectively (Longmate 2001). Recently, discriminant analysis (Li et al. 2000), logistic regression (Czika et al. 2000), decision trees (Zhang and Bonney 2000), and neural networks (Bhat et al. 1999; Sherriff and Ott 2001) have been applied to genetic association studies using multiple marker loci. However, such methods provide only classification accuracy as a measure of significance—rather than P values, which are widely used to show significant evidence of association in the traditional context. Therefore, there is a need to describe the relationship between classification methods and traditional statistical testing.
In this article, we present, for population-based association studies of complex diseases, a generalized T2 test that simultaneously utilizes multiple SNP markers. The power of the generalized T2 statistic for the detection of a disease locus (or loci) will be evaluated, as will be comparability of the genotype T2 and haplotype T2 statistics.
In addition, we formulate the problem of identification of SNP markers or a combination of SNP markers, which make the largest contribution to disease risk, as a combinatorial optimization problem, and we develop efficient search algorithms. Finally, examples will be given to illustrate the applications of the proposed T2 statistic to association studies.
Test Statistic
Consider a design in which nA cases from an affected population and 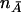 control subjects from a comparable unaffected population are sampled. Suppose that there are J markers that have been typed in the sample of cases and control subjects. The jth marker has alleles Bj and bj, with population frequencies PBj and Pbj, respectively. Define an indicator variable for the genotype of the jth marker for the ith individual from the affected population:
control subjects from a comparable unaffected population are sampled. Suppose that there are J markers that have been typed in the sample of cases and control subjects. The jth marker has alleles Bj and bj, with population frequencies PBj and Pbj, respectively. Define an indicator variable for the genotype of the jth marker for the ith individual from the affected population:
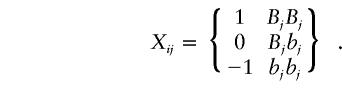 |
Similarly, we define an indicator variable, Yij, for an individual from the unaffected population. Let
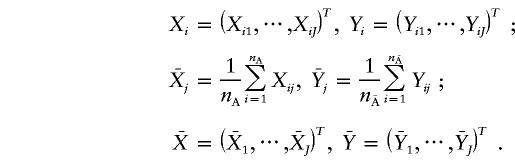 |
The pooled-sample variance-covariance matrix of the indicator variables for the marker genotypes is defined as
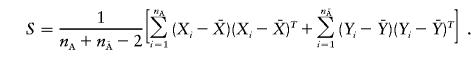 |
Hotelling’s (1931) T2 statistic is then defined as
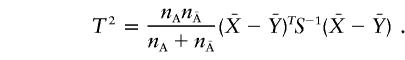 |
Under the null hypothesis that LD between any marker being tested and a disease locus does not exist, the covariance matrix of the indicator variables for the marker genotypes of the individuals from the affected population, ΣA=Cov(Xi,Xi), and the covariance matrix of indicator variables for the marker genotypes of the individuals from the unaffected population, 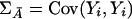 , are equal. Therefore, when the sample size is large enough to allow asymptotic theory to apply, under the null hypothesis,
, are equal. Therefore, when the sample size is large enough to allow asymptotic theory to apply, under the null hypothesis,
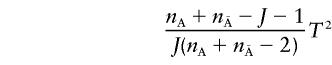 |
is asymptotically distributed as a central F distribution with J and 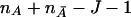 degrees of freedom. Under the alternative hypothesis that there is at least one marker showing LD with a disease locus, the covariance matrices ΣA and
degrees of freedom. Under the alternative hypothesis that there is at least one marker showing LD with a disease locus, the covariance matrices ΣA and 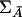 are no longer equal and
are no longer equal and
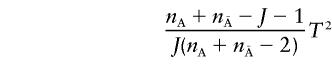 |
is not asymptotically distributed as a noncentral F distribution. In this case, it can be shown that T2 is asymptotically distributed as a χ2(J) distribution.
Power Evaluation
Noncentrality Parameter
To evaluate power, we need to calculate the noncentrality parameter of the χ2(J) distribution of the T2 statistic under the alternative hypothesis. We begin by computing the allele frequencies in the affected and unaffected populations. Consider a disease locus with alleles D and d. The alleles D and d have population frequencies PD and Pd, respectively. Let fDD, fDd, and fdd be the penetrance of the genotypes DD, Dd, and dd, respectively. Let PA denote the prevalence of the disease in the population. Then, PA is given by
Let PB(A) and 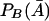 be the frequencies of marker allele B in the affected and unaffected populations, respectively. Let PBD, PBd, PbD and Pbd be the frequencies of haplotypes BD, Bd, bD, and bd, respectively. The frequency PB(A) is given by
be the frequencies of marker allele B in the affected and unaffected populations, respectively. Let PBD, PBd, PbD and Pbd be the frequencies of haplotypes BD, Bd, bD, and bd, respectively. The frequency PB(A) is given by
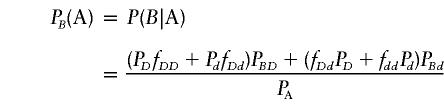 |
Similarly, we have
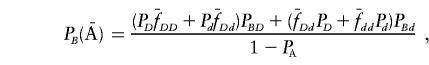 |
where 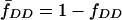 ,
, 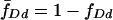 , and
, and 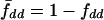 .
.
Consider the jth marker and the j′th marker. Let PBjBj′(A) and 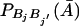 be the frequencies of haplotype BjBj′ in the affected and unaffected populations, respectively. If Hardy-Weinberg equilibrium is assumed, then it is easy to see that (see Appendix A)
be the frequencies of haplotype BjBj′ in the affected and unaffected populations, respectively. If Hardy-Weinberg equilibrium is assumed, then it is easy to see that (see Appendix A)
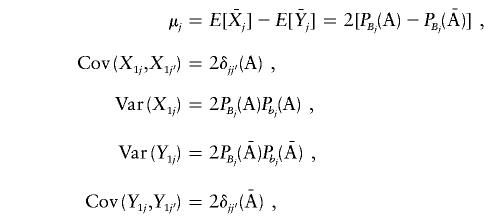 |
where
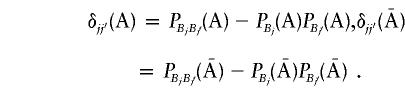 |
Define
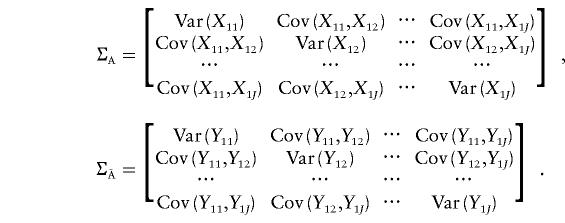 |
It is clear that the covariance matrices ΣA and 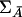 depend on the pairwise LD between the marker and trait loci. When
depend on the pairwise LD between the marker and trait loci. When 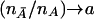 , the noncentrality parameter of the T2 statistic under the alternative hypothesis is given by
, the noncentrality parameter of the T2 statistic under the alternative hypothesis is given by
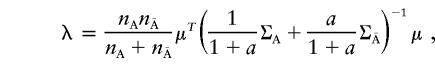 |
where μ=[μ1,…,μJ]T. Let
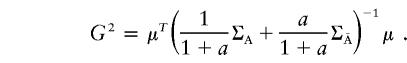 |
G2 can be considered to be a genetic-distance measure between two populations that is similar to that proposed by Balakrishnan and Sanghvi (1968). Intuitively, then, the noncentrality parameter λ can be expressed as a function of this genetic distance between the case and control populations;—that is,  . In the case in which all pairwise LD is equal to zero, G2 is reduced to
. In the case in which all pairwise LD is equal to zero, G2 is reduced to
 |
and the noncentrality parameter λ is
 |
For a single marker, we have
 |
and
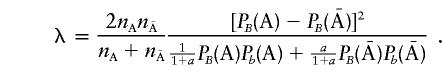 |
Therefore, the noncentrality parameter and power depend on the sample size and the genetic distance, which, in turn, are a function of allele frequencies and LD between the marker and trait loci.
The classic test statistic for a single-marker case-control study is given by (see Chapman and Wijsman 1998)
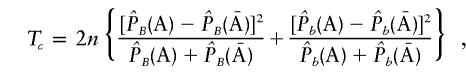 |
where 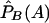 ,
, 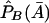 ,
, 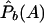 , and
, and 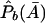 are the corresponding observed allele frequencies. Its noncentrality parameter, λc, is given by
are the corresponding observed allele frequencies. Its noncentrality parameter, λc, is given by
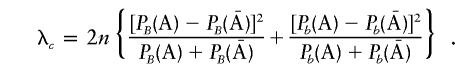 |
It can be shown (see Appendix B) that λ⩾λc. Therefore, for case-control association studies, the proposed T2 statistic has higher (or equivalent) power than does the classic Tc statistic. Figure 1 compares the power, for detection of a disease gene, of the T2 statistic and the classic χ2 statistic. From figure 1, we can see that, in all cases, the power of the T2 statistic is higher than that of the classic χ2 statistic. However, when the allele frequencies are small, the differences in the power of these two statistics are very small. It can be shown that, even in more complicated situations, such as multiple marker and trait loci, the T2 statistic has higher power than does the classic χ2 statistic (data not shown).
Figure 1.

Power curves of the T2 test and the χ2 test, with significance level α=0.0001, as a function of allele frequency, with penetrances assumed to be f11=0.4, f12=0.2, and f22=0.1 and with sample size 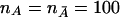 .
.
Two-Disease-Loci Model
To further evaluate the power of the T2 statistic, we consider two-locus disease models. Assume that there are two disease loci, D and d. Each disease locus has two alleles. The frequencies of the alleles D1 and D2 at disease locus D and of the alleles d1 and d2 at disease locus d can be denoted by PD1, PD2, Pd1, and Pd2, respectively. The frequencies of the genotypes DuDv and dkdl in the disease and normal populations are denoted by PDuDv and Pdkdl, respectively. The penetrance of the genotypes DuDvdkdl will be denoted by fuvkl. Then, the prevalence of the disease in the population is given by
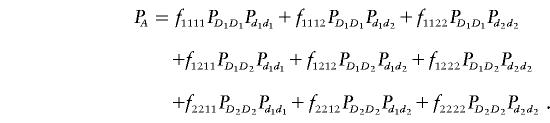 |
Denote the indicator variables for the genotypes of the first and second markers for the first individual from the affected population and for the first individual from the unaffected population by X11, X12, Y11, and Y12, respectively. Let
and let
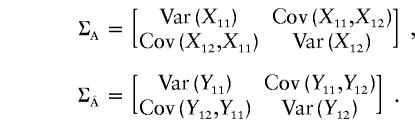 |
The elements of the vector μ and of the variance-covariance matrices ΣA and 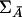 are given in Appendix C. The noncentrality parameter of the T2 statistic for the two-locus disease model is then given by
are given in Appendix C. The noncentrality parameter of the T2 statistic for the two-locus disease model is then given by
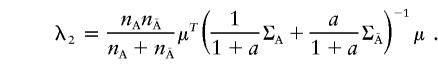 |
For convenience of presentation, we assume that the two disease loci are unlinked. Table 1 presents six types of two-locus disease models (Neuman and Rice 1992; Schork et al. 1993; Ott 1999). To illustrate the performance of the T2 statistic for the detection of disease loci, we plot figure 2, showing the power of the T2 statistic as a function of the allele frequency under the six types of two-locus–disease models in table 1.
Table 1.
Penetrance of Given Genotypes for Six Two-Locus Disease Models[Note]
|
Locus d |
|||
| Model and Locus D | d1d1 | d1d2 | d2d2 |
| Dom ∪ Dom: | |||
| D1D1 | f | f | f |
| D1D2 | f | f | f |
| D2D2 | f | f | 0 |
| Dom ∪ Rec: | |||
| D1D1 | f | f | f |
| D1D2 | f | f | f |
| D2D2 | f | 0 | 0 |
| Rec ∪ Rec: | |||
| D1D1 | f | f | f |
| D1D2 | f | 0 | 0 |
| D2D2 | f | 0 | 0 |
| Epistasis or Dom ∩ Dom: | |||
| D1D1 | f | f | 0 |
| D1D2 | f | f | 0 |
| D2D2 | 0 | 0 | 0 |
| Threshold: | |||
| D1D1 | f | f | 0 |
| D1D2 | f | 0 | 0 |
| D2D2 | 0 | 0 | 0 |
| Modifying: | |||
| D1D1 | f | f | f |
| D1D2 | f | 0 | 0 |
| D2D2 | 0 | 0 | 0 |
Note.— Adopted from Fan et al. (in press). f is a penetrance.
Figure 2.

Power curves of the T2 test, with significance level α=0.0001, as a function of allele frequency, in the case of Dom ∪ Dom, Dom ∪ Rec, Rec ∪ Rec, epistasis, threshold, and modifying models, when 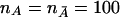 , PD1=Pd1, and f=0.6 are assumed.
, PD1=Pd1, and f=0.6 are assumed.
The Haplotype T2 Statistic
When haplotype information is available, we can define an indicator variable for the alleles of the jth marker on the ith chromosome from the affected population:
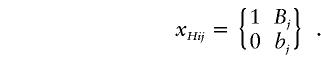 |
Similarly, we define an indicator variable yHij for the marker alleles located on the chromosomes from the unaffected population. Following the same development in the genotype T2 statistic, we can define the haplotype T2 statistic. Let
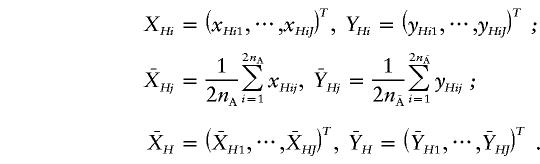 |
The covariance matrix is defined as
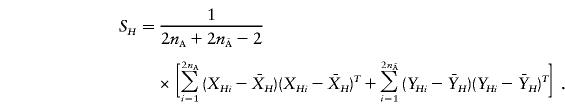 |
The haplotype T2 statistic is then defined as
 |
To compare the powers of the genotype T2 and haplotype T2H, we can compare their noncentrality parameters, because both T2 and T2H follow a χ2(J) distribution under the alternative hypothesis. It can be shown that the noncentrality parameter λ of the T2 statistic and the noncentrality parameter λH of the T2H statistic are equal (Appendix D).
Therefore, the power of the multilocus T2 statistic is the same as that of the haplotype T2 statistic. Equivalence of the two statistics is important, because unequivocal haplotypes are usually not available in the majority of case-control studies. Intuitively, this equivalence can be attributed to the fact that the multilocus T2 statistic contains the same pairwise LD information in the covariance matrices—that is, ΣA and  —that is contained in the haplotypes.
—that is contained in the haplotypes.
Search Algorithm
To identify SNP markers (or the combination of SNP markers) that make the greatest contribution to disease risk and drug response, search algorithms are fundamental. In this study, we use a heuristic algorithm that seeks the best combination of SNP markers for risk assessment. The algorithm is based on the sequence-forward floating-selection (SFFS) algorithm of Pudil et al. (1994), which is easy to implement and which requires minimal computation. The SFFS algorithm is based on a sequence-forward–selection algorithm (SFS). The procedures for sequential-forward selection are as follows:
-
1.
Compute the desired criterion value for each of the markers, and select the marker with the best value;
-
2.
Form all possible two-dimensional vectors that contain the winner from the previous step, and compute the criterion value for each of them and then select the best one;
-
3.
Form all three-dimensional vectors expanded from the two-dimensional winners, and select the best one; continue this process until the prespecified dimension of the feature vector—say, l—is reached.
The SFS algorithm requires less computational burden than do other search algorithms, but it suffers from the so-called nesting effect—that is, once a marker is chosen, there is no way for it to be discarded in later steps. To overcome this problem, the SFFS algorithm was proposed. The SFFS algorithm balances the required computational time and overall optimality. (For details, interested readers are referred to Pudil et al. [1994] and Xiong et al. [2001].)
Examples
The proposed T2 test was applied to a simulated data set from Genetic Analysis Workshop 12 (GAW12) (Almasy et al. 2001). Simulated data were provided for an isolated population founded ∼20 generations ago by 100 individuals from the general population. Unrelated cases and control subjects ( ) were obtained by selection of founders and their spouses from 23 extended pedigrees. Sequence data are available for a major gene, MG6 on chromosome 6, that directly influences affection status of the individuals. MG6 is known to account for 25.3% of disease liability, and, in the GAW12 data, the sequence data are labeled “GENE 1.” Site 557 was identified as the SNP closely related to disease liability. The P values of the T2 statistic and of the classic χ2 statistic, for testing the association between SNP markers and affection status that are included within GENE 1 are summarized in table 2. We can see from table 2 that both the T2 test and the χ2 test identified a common set of SNP markers showing significant association with affection status but that, in all cases, the T2 test had smaller P values than did the χ2 test. The T2 test identified site 557 as having the smallest P value. Since strong LD exists between many SNP markers within GENE 1 (Czika et al. 2000; Huang et al. 2001), both the T2 test and the χ2 test identified a number of SNP markers that had small P values.
) were obtained by selection of founders and their spouses from 23 extended pedigrees. Sequence data are available for a major gene, MG6 on chromosome 6, that directly influences affection status of the individuals. MG6 is known to account for 25.3% of disease liability, and, in the GAW12 data, the sequence data are labeled “GENE 1.” Site 557 was identified as the SNP closely related to disease liability. The P values of the T2 statistic and of the classic χ2 statistic, for testing the association between SNP markers and affection status that are included within GENE 1 are summarized in table 2. We can see from table 2 that both the T2 test and the χ2 test identified a common set of SNP markers showing significant association with affection status but that, in all cases, the T2 test had smaller P values than did the χ2 test. The T2 test identified site 557 as having the smallest P value. Since strong LD exists between many SNP markers within GENE 1 (Czika et al. 2000; Huang et al. 2001), both the T2 test and the χ2 test identified a number of SNP markers that had small P values.
Table 2.
Results of the T2 Test and the Classic χ2 Test, When Applied to Simulated Data within Gene 1 from GAW12[Note]
|
P, by Type of Test |
|||
| Position | T2 | χ2 | r2 |
| 557 | 6.6 × 10 −14 | 3.04 × 10−11 | |
| 1553 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 2619 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 3456 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 3573 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 3742 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 3835 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 3853 | 6.6 × 10−14 | 3.04 × 10−11 | .97 |
| 76 | 2.66 × 10−13 | 9.67 × 10−11 | .99 |
| 11180 | 2.86 × 10−13 | 3.04 × 10−11 | |
| 2923 | 6.41 × 10−13 | 2.44 × 10−11 | .86 |
| 5757 | 8.019 × 10−13 | 2.12 × 10−11 | .93 |
| 7281 | 8.019 × 10−13 | 2.12 × 10−11 | .93 |
| 1478 | 1.199 × 10−12 | 4.59 × 10−11 | .85 |
| 4471 | 1.2832 × 10−11 | 1.43 × 10−10 | .86 |
| 4752 | 1.2832 × 10−11 | 1.43 × 10−10 | .86 |
| 2942 | 1.5017 × 10−11 | 3.93 × 10−10 | .91 |
| 3534 | 1.16706 × 10−8 | 5.41 × 10−8 | .73 |
| 3653 | 1.16706 × 10−8 | 5.41 × 10−8 | .73 |
| 5094 | 1.16706 × 10−8 | 5.41 × 10−8 | .73 |
| 5244 | 1.16706 × 10−8 | 5.41 × 10−8 | .73 |
| 2732 | 1.42422 × 10−8 | 3.02 × 10−8 | .66 |
| 2853 | 1.42422 × 10−8 | 3.02 × 10−8 | .66 |
| 596 | 2.28199 × 10−8 | 5.41 × 10−8 | .73 |
| 5542 | 4.81874 × 10−8 | 1.36 × 10−7 | .71 |
| 189 | 6.05394 × 10−8 | 1.40 × 10−7 | .72 |
| 12185 | 8.64 × 10−8 | 3.54 × 10−7 | |
| 13074 | 8.64 × 10−8 | 4.54 × 10−7 | |
| 5688 | 8.73906 × 10−8 | 1.36 × 10−7 | .71 |
| 4602 | 9.36796 × 10−8 | 2.12 × 10−7 | .66 |
| 4688 | 9.36796 × 10−8 | 2.12 × 10−7 | .66 |
Note.— Data are from Almasy et al. (2001).
Table 3 shows (1) the results of the T2 test for 17 two-SNP combinations that have P values <10−14 and (2), of all possible three-SNP combinations, the top 15 that have the smallest P value. Two features are evident from table 3: first, the P values of the optimal combination of two or three SNPs are smaller than that of each single SNP in the combination; second, an individual SNP may have a large P value, but its combinations with other SNPs may have a very small P value.
Table 3.
Results of the T2 Test Applied to Simulated Data within GENE 1 from GAW12, When Two or Three SNPs Are Used[Note]
| 1st SNP |
2d SNP |
3d SNP |
||||
| Position | P | Position | P | Position | P | P for 1st and 2d SNPs or for 1st, 2d, and 3d SNPs |
| 557 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−16 | ||
| 76 | 2.66 × 10−13 | 9150 | 3.55 × 10−7 | 2.00 × 10−15 | ||
| 1553 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 2619 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 3456 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 3573 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 3742 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 3835 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 3853 | 6.60 × 10−14 | 9150 | 3.55 × 10−7 | 5.55 × 10−15 | ||
| 557 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 1553 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 2619 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 3456 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 3573 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 3742 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 3835 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 3853 | 6.60 × 10−14 | 4315 | 1.83 × 10−6 | 6.99 × 10−15 | ||
| 76 | 2.66 × 10−13 | 4315 | 1.83 × 10−6 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 5654 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 5688 | 8.74 × 10−8 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 5721 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 5922 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 6912 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 7577 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 7654 | 3.76 × 10−2 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 7890 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 9341 | 1.24 × 10−7 | 9150 | 1.40 × 10−5 | 1.11 × 10−16 |
| 1553 | 6.60 × 10−14 | 5757 | 8.02 × 10−12 | 7890 | 3.76 × 10−2 | 1.11 × 10−16 |
| 1553 | 6.60 × 10−14 | 7281 | 8.02 × 10−12 | 7890 | 3.76 × 10−2 | 1.11 × 10−16 |
| 557 | 6.60 × 10−14 | 1407 | 5.94 × 10−1 | 9150 | 1.40 × 10−5 | 2.22 × 10−16 |
Note.— Data are from Almasy et al. (2001).
The proposed T2 test was also applied to a real data set of cases of scleroderma, or systemic sclerosis (SSC) (X. Zhou, F. K. Tan, J. D. Reveille, C. Ahn, A. Wang, and F. C. Arnett, personal communication). SSC is a multisystem disease of unknown etiology and is characterized by cutaneous and visceral fibrosis, small-blood-vessel damage, and autoimmune features (Medsger 1997; X. Zhou, F. K. Tan, J. D. Reveille, C. Ahn, A. Wang, and F. C. Arnett, personal communication). Three SNP markers—SPARC 998, SPARC 1551, and SPARC 1992—were genotyped in 20 unrelated patients with SSC and in 75 normal control subjects from the Oklahoma Choctaw population. It has been reported that, in this population, (a) the clinical disease pattern is relatively homogeneous and (b) the prevalence of SSC in this population is high (X. Zhou, F. K. Tan, J. D. Reveille, C. Ahn, A. Wang, and F. C. Arnett, personal communication). To further evaluate the performance of the T2 test, both the T2 test and the χ2 test were applied to the samples from the Oklahoma Choctaw, to examine association between the SPARC gene and SSC. Table 4 presents the results. It is evident from table 4 that marker SPARC 998 has a T2-associated P value that is much smaller than that associated with the classic χ2 test. It has been reported that expression of the SPARC gene is ∼2.5-fold and ∼5-fold increased, respectively, when cDNA microarrays and western-blot analysis are used, in case-control comparisons for SSC (X. Zhou, F. K. Tan, J. D. Reveille, C. Ahn, A. Wang, and F. C. Arnett, personal communication). The SPARC gene is being increasingly recognized as playing a variety of roles in tissue development, remodeling, and fibrosis (Motamed 1999).
Table 4.
Test for Association between SPARC SNP Markers and SSC in Oklahoma Choctaw, by the T2 Test and the χ2 Test[Note]
|
P, by Type of Test |
||
| Marker | T2 | χ2 |
| SPARC 998 | .003886 | .01108 |
| SPARC 1551 | .04859 | .1198 |
| SPARC 1922 | .1889 | .1915 |
Note.— Data are from X. Zhou, F. K. Tan, J. D. Reveille, C. Ahn, A. Wang, and F. C. Arnett (personal communication).
Discussion
In this study, we have proposed a generalized T2 statistic to relate DNA sequence variations to the occurrence of disease. We show that the noncentrality parameter of the T2 statistic is larger than that of the well-known χ2 statistic, indicating that the T2 statistic has greater power than does the χ2 statistic. In addition, simulation studies and examples with real data demonstrate that, for case-control studies, the P value of the T2 test is smaller than that of the χ2 test.
The proposed generalized T2 statistic has utility in three areas of contemporary human genomic analysis. First, there is general interest in using a dense set of SNPs spanning each of the chromosomes, to localize genes via genomewide association analyses. For such association studies to be effective, it is not necessary that the SNPs be the disease-susceptibility loci; rather, the SNPs may aid in identification of the location of a disease-susceptibility locus, on the basis of LD between the SNP marker loci and nearby disease-susceptibility loci. The magnitude of LD among SNPs (including disease-susceptibility loci) is largely determined by the recombination rates among loci and by stochastic sampling variation, including genetic drift, migration, and sampling. Therefore, association between an SNP (or SNPs) and a trait of interest is generally attributable to LD between the SNP (or SNPs) and a disease-susceptibility locus. Such association may indicate proximity of the inferred disease-susceptibility locus to the SNP marker locus.
The second area in which the proposed statistic has utility is in analysis of the spectrum of variation within a gene, to identify sites or combinations of sites influencing the trait of interest. Variations at these sites are candidates for further experimental and functional studies. An association-mapping perspective is of little utility in this situation, because the recombination rate among sites is practically zero. In this case, one should first identify the complete menu of variable sites within the gene and then consider the ability of these sites to predict levels or prevalence rates of the phenotype of interest. Recent studies (e.g., see Horikawa et al. 2000) have indicated that there are not sufficient data to predict a priori which sites (e.g., cSNPs) are likely to predict and which are likely not to predict.
The third application of the T2 analysis is to the development of a more comprehensive vision of the genetic architecture (see Boerwinkle et al. 1986) of a trait. It is widely accepted that risk to a common disease is influenced by multiple genes and that these genes are interacting both among themselves and with environmental factors. One of the goals of studying the genetics of common diseases is to identify the contributing genes and mutations and to characterize their interaction as they combine with other agents to influence disease risk. To achieve this goal, it is necessary to have methods that evaluate multiple loci—and their interactions—simultaneously. The proposed T2 statistic, by virtue of the fact that it simultaneously considers the effects of multiple loci and does not assume additivity among those effects, is an important step in this direction. Future developments will include (1) extension of the T2 method to quantitative traits and (2) stepwise site-selection procedures. One aspect of considerable interest in the area of genome association studies is the use of haplotype information. Recently, some have argued that haplotypes may be the relevant functional unit in the consideration of genotype-phenotype relationships (Drysdale et al. 2000). In addition, haplotype information can facilitate a cladistic approach to genotype-phenotype relationships (Templeton et al. 1987). In the case of the T2 test, haplotype information has here been shown not to lend additional information about genotype-phenotype relationships, relative to multilocus genotype information. Initially, this result was surprising. However, on further investigation it was realized that the sample variance-covariance matrix, S, contains the pairwise relationships among loci. Therefore, the T2 statistic captures the pairwise-association information found in haplotypes. Higher-order associations, however, may not be included in the regular T2 statistic, indicating that the T2H statistic may have advantages in those situations.
LD analyses and association mapping are powerful tools for contemporary human genetics. Efforts to build a collection of SNP markers in all genes of the human genome (e.g., see The International SNP Map Working Group 2001 ) and advances in genotyping technologies bode well for large-scale applications in the near future. Such undertakings are not without complications, however. The cost-per-locus test for SNPs remains high. The pattern of LD may vary considerably between populations. And there is a further need to develop, evaluate, and apply novel methods for relating the considerable genomic information to risk of disease—methods such as the T2 test proposed here.
Acknowledgments
M.X. and J.Z. are supported by NIH grants GM56515 and HL 5448, and E.B. is supported by NIH grant HL 5448.
Appendix A :
Assuming Hardy-Weinberg equilibrium, we can calculate  and
and 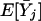 as follows:
as follows:
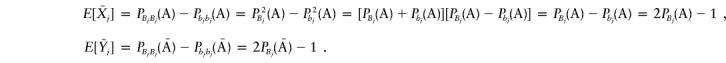 |
Therefore, we have
Next, we calculate the variance-covariances, Var(Xj) and Cov(Xj,Xj′). Note that
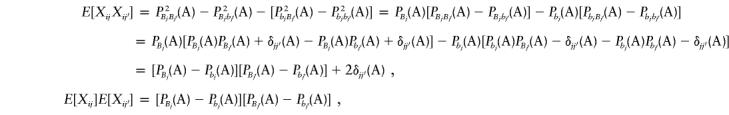 |
where δjj′(A)=PBjBj′(A)-PBj(A)PBj′(A).
Combining the above equations yields Cov(Xij,Xij′)=E[XijXij′]-E[Xij]E[Xij′]=2δjj′(A). It is not difficult to see that Var(Xij)=E[X2ij]-(E[Xij])2=P2Bj(A)+P2bj(A)-[PBj(A)-Pbj(A)]2=2PBj(A)Pbj(A). Similarly, we have 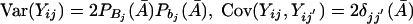 .
.
Appendix B :
Note that Pb(A)=1-PB(A) and 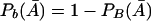 . Thus,
. Thus,
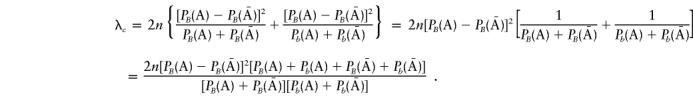 |
However, 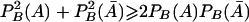 , which implies that
, which implies that
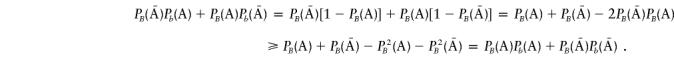 |
Therefore, we have
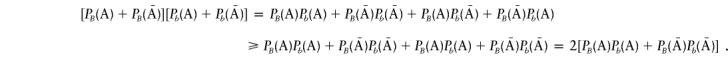 |
It follows that
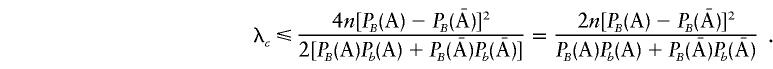 |
But, when 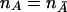 , λ is reduced to
, λ is reduced to
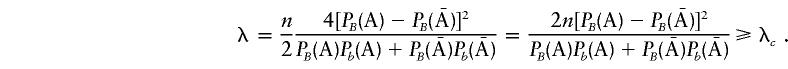 |
Appendix C:
To calculate the noncentrality parameter, λ2, we begin with the calculation of the frequencies of the genotypes at the two disease loci, in the affected population and in the control populations. It follows from the definition of the genotype frequencies in the disease population that
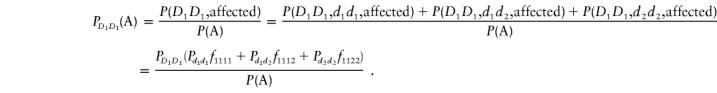 |
Similarly, we have
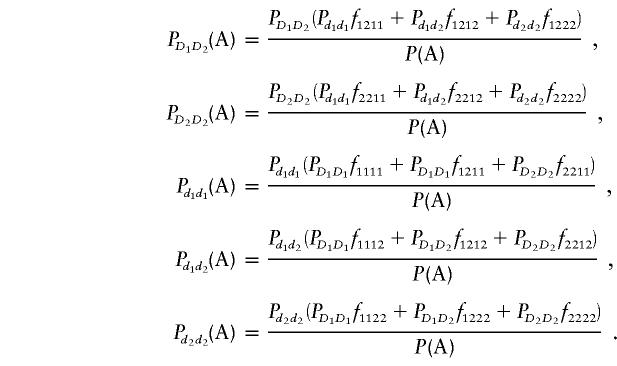 |
Let 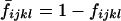 be the probability that an individual with genotypes DiDj and dkdl is unaffected. By an argument similar to that used above, we obtain
be the probability that an individual with genotypes DiDj and dkdl is unaffected. By an argument similar to that used above, we obtain
 |
Now we calculate the expectation of the indicator variables X11 and Y11. Using the definition of the indicator variable, we have
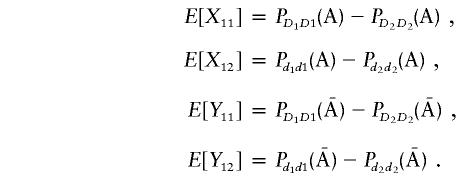 |
Thus, the vector μ can be calculated by μ=(E[X11]-E[Y11],E[X12]-E[Y12])T. Next we calculate the variance-covariance matrix ΣA. It is easy to see that
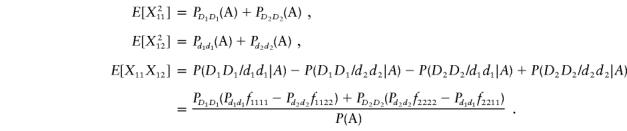 |
Therefore, we obtain
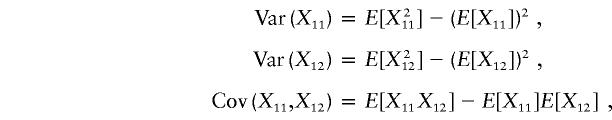 |
and
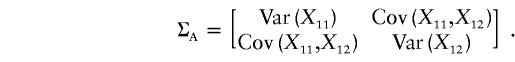 |
Appendix D :
If we assume Hardy-Weinberg equilibrium, we find that it is not difficult to show that
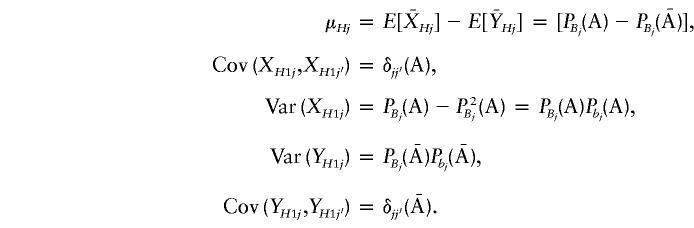 |
Thus, μH=(μH1,…,μHK)T; ΣHA=(1/2)ΣA and 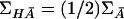 . The noncentrality parameter λH is then given by
. The noncentrality parameter λH is then given by
References
- Almasy L, Terwilliger JD, Nielsen D, Dyer TD, Zaykin D, Blangero J (2001) GAW12: simulated genome scan, sequence, and family data for a common disease. Genet Epidemiol 21:S332–S338 [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Sanghvi LD (1968) Distance between populations on the basis of attribute data. Biometrics 24:859–865 [Google Scholar]
- Bhat A, Lucek PR, Ott J (1999) Analysis of complex traits using neural networks. Genet Epidemiol 17 Suppl 1:S503–507 [DOI] [PubMed] [Google Scholar]
- Boerwinkle E, Chakraborty R, Sing CF (1986) The use of measured genotype information in the analysis of quantitative phenotypes in man. I. Models and analytical methods. Ann Hum Genet 50:181–194 [DOI] [PubMed] [Google Scholar]
- Chapman NH, Wijsman EM (1998) Genome screens using linkage disequilibrium tests: optimal marker characteristics and feasibility. Am J Hum Genet 63:1872–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czika WA, Weir BS, Edwards SR, Thompson RW, Nielsen DM, Brocklebank JC, Zinkus C, Martin ER, Hobler KE (2001) Applying data mining techniques to the mapping of complex disease genes. Genet Epidemiol 21 Suppl 1:S435–S440 [DOI] [PubMed] [Google Scholar]
- Drysdale CM, McGraw DW. Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R, Floros J, Xiong MM. Transmission disequilibrium test of two unlinked disease loci: application to respiratory distress syndrome. Adv Appl Stat (in press) [Google Scholar]
- Gray IC, Campbell DA, Spurr NK (2000) Single nucleotide polymorphisms as tools in human genetics. Hum Mol Genet 9:2403–2408 [DOI] [PubMed] [Google Scholar]
- Hotelling H (1931) The generalization of student’s ratio. Ann Math Stat 2:360–378 [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PEH, et al (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 [DOI] [PubMed] [Google Scholar]
- Huang QQ, Marrison AC, Boerwinkle E (2001) Linkage disequilibrium structure and its impact on the localization of a candidate functional mutation. Genet Epidemiol 21: S620–S625 [DOI] [PubMed] [Google Scholar]
- International SNP Map Working Group, The (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 [DOI] [PubMed] [Google Scholar]
- Li X, Rao S, Elston RC, Olson JM, Moser KL, Zhang T, Guo Z (2001) Locating the genes underlying a simulated complex disease by discriminant analysis. Genet Epidemiol 21 Suppl 1:S516–521 [DOI] [PubMed] [Google Scholar]
- Longmate JA (2001) Complexity and power in case-control association studies. Am J Hum Genet 68:1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medsger TA Jr (1997) Systemic sclerosis (scleroderma): clinical aspects. In: Koopman WJ (ed) Arthritis and allied conditions: a textbook of rheumatology. Williams & Wilkins, Baltimore 1433-1464 [Google Scholar]
- Motamed K (1999) SPARC (osteonectin/BM-40). Int J Biochem Cell Biol 31:1363–1366 [DOI] [PubMed] [Google Scholar]
- Neuman RJ, Rice JP (1992) Two-locus models of diseases. Genet Epidemiol 9:347–365 [DOI] [PubMed] [Google Scholar]
- Ott J (1999) Analysis of human genetic linkage, 3d ed. Johns Hopkins University Press, Baltimore [Google Scholar]
- Pudil P, Novovicova J, Kittler J (1994) Floating search methods in feature selection. Pattern Recognition Lett 15:1119–1125 [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Schork NJ, Boehnke M, Terwilliger JD, Ott J (1993) Two-trait-locus linkage analysis: a powerful strategy for mapping complex genetic traits. Am J Hum Genet 53:1127–1136 [PMC free article] [PubMed] [Google Scholar]
- Sherriff A, Ott J (2001) Applications of neural networks for gene finding. Adv Genet 42:287–297 [DOI] [PubMed] [Google Scholar]
- Templeton AR, Boerwinkle E, Sing C (1987) A Cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in drosophila. Genetics 117:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong MM, Fang XZ, Zhao JY (2001) Biomarker identification by feature wrappers. Genome Res 11:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HP, Bonney G (2000) Use of classification trees for association studies. Genet Epidemiol 19:323–332 [DOI] [PubMed] [Google Scholar]