

Abstract
In solution, the Trp 59 indole ring at the base of the active site cleft in the FKBP domain protein FKBP12 is rotated by ~90° at a population level of 20%, relative to its canonical crystallographic orientation. NMR measurements on the homologous FK1 domains of human FKBP51 and FKBP52 indicate no observable indole ring flip conformation, while the V101I variant of FKBP12 decreases the population having a perpendicular indole orientation by 10-fold. A set of three parallel 400 ns CHARMM27 molecular simulations for both wild type FKBP12 and the V101I variant examined how this ring flip might be energetically coupled to a transition of the Glu 60 sidechain which interacts with the backbone of the 50’s loop located ~12 Å from the indole nitrogen. Analysis of the transition matrix for the local dynamics of the Glu 60 sidechain, the Trp 59 sidechain, and of the structurally interposed α-helix hydrogen bonding pattern yielded a statistical allosteric coupling of 10 kJ/mol with negligible concerted dynamical coupling for the transitions of the two sidechains.
Keywords: FKBP12, NMR, allostery, molecular simulation, statistical coupling, concerted transition
Introduction
The FK506-binding domain protein FKBP12 is best known for its role in mediating the immunosuppressive effects of FK506 and rapamycin. Among the fourteen FKBP domain proteins in the human genome [1], FKBP12.6 and the first FKBP domain of FKBP51 and FKBP52 exhibit the closest sequence homology to FKBP12 and they are believed to provide the largest functional overlap [2–5], although the significance of that overlap remains open to debate [6]. While each of these FKBP domains retain the cis-trans isomerization activity on model prolyl peptides found in their eubacterial homologs, genetic and cell biology studies indicate that the primary physiological roles for FKBP12 and FKBP12.6 as well as for FKBP51 and FKBP52 involve protein recognition interactions that contribute to the regulation of various signaling pathways, most notably the FKBP12/FKBP12.6 regulation of ryanodine receptor calcium channels [7–10] and the FKBP51/FKBP52 regulation of steroid receptor complexes [11–14]. Given the current challenges of structurally characterizing the regulatory conformational transitions within these large complexes, useful insights may be derived from a more detailed understanding of the energetically accessible conformational transitions of the isolated FKBP domains which may facilitate coupling to the larger scale transitions of the complex.
The long β4-β5 loop (or 80’s loop) is known to provide critical protein recognition interactions for various signaling functions of FKBP12 [15–18] in addition to its role in mediating the regulatory interactions of FKBP51 and FKBP52 with the steroid receptor [19]. In characterizing the structural basis for two distinct conformational transitions of FKBP12 centered in this loop which give rise to NMR resonance doubling (τ of 3.0 s at 43°C [20]) and conformational exchange linebroadening (t ~ 120 μs at 20°C [21–23]), respectively, we reported the crystal structure of the G89P variant [24].
Surprisingly, although the Cα atoms of Trp 59 and Gly 89 are separated by 21 Å, the indole ring in the G89P structure is rotated ~90° with respect to its canonical position in the crystal structure of the wild type FKBP12 (Graphical Abstract), resulting in the occlusion of most of the active site cleft. This sidechain transition, involving a χ1 torsional rotation from the gauche− to trans rotamer and a smaller shift in the χ2 torsion angle, had previously been reported in the 0.94 Å resolution X-ray structure for the E60Q variant of FKBP12 by Saven and colleagues [25]. Both the G89P and the E60Q crystal structures also exhibit a ~1 Å shift in the backbone of the central turn of the α-helix which enables the amide of Ala 64 to form a canonical hydrogen bond to the carbonyl oxygen of residue 60. In contrast to wild type FKBP12, the crystal structures of the highly homologous FKBP12.6 [26,27] exhibit an undistorted α-helix with canonical hydrogen bonding geometry between Glu 60 and Ala 64. This shift of the α-helix in FKBP12.6 has been proposed to be sterically accommodated due to its smaller Phe 59 sidechain at the base of the active site [28].
The E60Q substitution partially disrupts the hydrogen bonding interactions that occur between wild type Glu 60 sidechain and the backbone amides of the 50’s loop in the wild type structure. In the E60Q structure, a flip of the (ψ52, ϕ53) torsion angles brings the carbonyl oxygen of Lys 52 into a hydrogen bond with the sidechain amide of Gln 60. In a 12 ns CHARMM27 molecular simulation analysis by Park and Saven [29], they found that the Trp 59 indole ring in wild type FKBP12 flipped to a perpendicular orientation after ~8 ns. Although the absence of a return transition precluded any conclusions about the predicted equilibrium for the indole ring flip, their molecular dynamics study suggested the energetic accessibility of this transition.
In contrast to the crystal structure of the E60Q variant, the backbone geometry in the 50’s loop is not disrupted in the G89P structure. The sidechain carboxylate of Glu 60 in the G89P structure maintains hydrogen bonding interactions with the backbone amides of the 50’s loop, but in contrast to the gauche− χ1 rotamer seen in the crystal structure of wild type FKBP12, Glu 60 adopts a trans χ1 rotamer conformation in the G89P structure. This extended sidechain conformation enables the backbone of Glu 60 to shift toward the indole ring allowing for formation of a canonical α-helical hydrogen bonding geometry between its carbonyl oxygen and the amide of Ala 64.
Following up on our initial observation that the indole HN resonance of Trp 59 in wild type FKBP12 exhibits NOE crosspeaks to methyl resonances that are incompatible with the canonical crystal structure, we carried out quantitative NOE buildup analysis to show that the crosspeak pattern is consistent with a 20% population of the rotated indole conformation [24]. In the present study, we wished to establish whether this indole ring reorientation is suppressed in the FK1 domains of FKBP51 and FKBP52 since the marked alteration of the active site geometry offers an opportunity for design of selective inhibitors to facilitate discrimination among the FKBP domain proteins. Furthermore, the presence of trans χ1 rotamers for both Trp 59 and Glu 60 in the G89P structure, in contrast to the gauche− rotamers at both positions in the wild type crystal structure, is suggestive of an energetic coupling between the interactions of the 50’s loop backbone and the Trp 59 indole ring which might enable binding interactions at the 50’s loop to allosterically modulate the geometry of the active site.
Our 15N NMR relaxation analysis indicated that the indole ring flip may occur at a rate similar to that of the global molecular tumbling of the protein [24]. Since it appeared likely that the sidechain rotamer transition of Glu 60 and the shifting of the hydrogen bonding geometry in the central α-helix might occur at least as rapidly, we investigated whether molecular simulations on the μs timescale could usefully sample these three distinct local transitions to examine the degree of thermodynamic coupling between them and to analyze the degree to which this allosteric process arises from concerted transitions. The integration of thermodynamic analysis with the various mechanistic structural models of protein allostery continues to present a significant challenge for the field [30,31]. The role played by concerted conformational transitions has long remained a central aspect of the mechanistic analyses. Unfortunately, the kinetics underlying many of the best studied allosteric systems are too slow to allow for unbiased molecular dynamics simulations to provide estimates of the transition rates and equilibria. The comparatively rapid kinetics of the indole ring flip in FKBP12 provides an allosteric system that is amenable to such a molecular simulation analysis.
2. Materials and methods
2.1. Protein preparation
Genes for wild type [20] and the V101I variant of FKBP12 as well as the FK1 domains of FKBP51 and FKBP52 [32] were chemically synthesized (Genscript), with codon optimization for expression in Escherichia coli. The genes were cloned into the expression vector pET11a and then transformed into the BL21(DE3) strain (Novagen) for expression. The protein expression and purification procedure were carried out as described for FKBP12 [20,33] as well as for the FK1 domains of FKBP51 and FKBP52 [32]. For the selective 13C-methyl labeled samples, 85 mg/L of [3-2H, 4-13C] α-ketoisovalerate and 50 mg/L of [3-2H2, 4-13C] α-ketobutyrate [34] were supplemented into a medium for U-2H,15N enriched sample growths as previously described [24]. For back exchanging the amide hydrogens, 1 mM tris(2-carboxyethyl)phosphine and solid Tris base was added to a solution of the purified protein to obtain a pH value above 9, and the samples were incubated at 25°C for 3 hours and then neutralized with solid monobasic sodium phosphate. All protein samples were concentrated via centrifugal ultrafiltration and then equilibrated into a pH 6.50 buffer containing 25 mM sodium phosphate, 2 mM dithiothreitol and 2 mM tris(2-carboxyethyl)phosphine by a series of centrifugal concentration steps.
2.2. NMR spectroscopy
NMR data for the wild type and V101I variant of FKBP12 were collected on a Bruker Avance 600 MHz spectrometer and the NMR data for the FK1 domains of FKBP51 and FKBP52 were collected on a Bruker 800 MHz spectrometer at 25°C. Resonance assignments for the wild type FKBP12 (BMR Data Bank accession numbers 19240 and 19241 [20]) and FK1 domains of FKBP51 and FKBP52 (BMR Data Bank accession numbers 19787 and 19788 [32]) have previously been reported. The indole ring reorientation analyses were carried out using a 3D F1-filtered 1H-13C-1H NOESY experiment [35]. FELIX software (Felix NMR) was used for NMR data processing.
2.3. Molecular dynamics simulations
To form a model of the V101I variant, coordinates from the 0.92 Å resolution X-ray structure of FKBP12 (PDB code 2PPN [25]) were modified with CHIMERA [36] by adding a methyl group to Val 101 which positions this Cδ atom below the Trp 59 indole ring. Coordinates from the 1.00 Å resolution structure of the FK1 domain of FKBP51 (PDB code 3O5P [37]) were used for the simulations. Hydrogen atoms were added to the protein heavy atoms and crystallographically-defined water molecules with all carboxylate and aliphatic amine groups being charged using VMD [38]. VMD was then used to form a rectangular solvent box with no protein atoms within 10 Å of the boundary filled with TIP3P waters, as modified for CHARMM. Sodium and chloride ions added to establish electroneutrality and an ionic strength near 150 mM. For each protein, three parallel simulations were prepared, differing solely by independent placement of the added ions. MD simulations were carried out in NAMD2 [39] using the CHARMM27 force field [40] with Particle Mesh Ewald summation for long range electrostatics using a 1 Å grid spacing. Short range interactions utilized a switching distance of 10 Å and a cutoff of 12 Å. SHAKE constraints were applied for all bonds to hydrogen, and a step size of 1 fs was used. Steepest descent minimization was applied for 3,000 steps with 25 cal/mol/Å2 restraints applied to all protein heavy atoms and crystallographic water molecules, followed by a second minimization of 3,000 steps and a 30 ps warming protocol to 298 K in 25 degree increments with 5 kcal/mol/Å2 restraints. After 200 ps of NPT equilibration with constant temperature conditions enforced with a Langevin damping factor of 1.0 ps−1 and constant pressure conditions using Langevin piston pressure control, the heavy atom restraints were decreased from 5 kcal/mol/Å2 to 1 kcal/mol/Å2 at a rate of 5 ps per unit. The system was then allowed to stabilize under NPT conditions for 200 ps followed by another 200 ps with the heavy atom restraints reduced to 0.1 kcal/mol/Å2. The heavy atom restraints were then removed, and 400 ns of simulation were carried out under NVE conditions with the first 20 ns being treated as equilibration. Coordinate files were stored at 5 ps intervals. Temperature drift over the trajectory was minimal with the averages over consecutive 10 ns intervals remaining within 0.1 degrees of the overall average temperature.
3. Results and discussion
3.1. NOESY analysis of indole ring flipping in the FK1 domains of FKBP51 and FKBP52
As in our previous analysis of ring flipping for Trp 59 of FKBP12 [24], samples of the FK1 domains were prepared in which the Cγ positions of valine, the Cδ positions of leucine and the Cδ position of isoleucine were substituted with 1H,13C while all other positions of the protein were uniformly labeled with 2H, 12C and 15N [34]. The amide and indole HN positions were back exchanged to yield a sample in which the NOESY interactions between the methyl and N-bound protons can be observed at high sensitivity with minimal effects from spin diffusion to complicate quantitative interpretation. In contrast to FKBP12, the active site Trp 90 of FKBP51 and FKBP52 have indole 1HNε1 resonances quite close to the solvent peak (5.05 ppm and 5.20 ppm, respectively). Exploiting the fact that no other N-bound protein resonance overlaps that 1H frequency, 3D F1-filtered 1H-13C-1H NOESY spectra were collected using a 200 ms mix time which we had previously found to yield approximately linear crosspeak buildups for FKBP12 [24]. For the F2-F3 plane at the indole 1HNε1 frequency, only crosspeaks for both FKBP51 (Fig. 1A) and FKBP52 (Fig. 1B) were observed for methyl groups that are near to the active site indole ring in the crystal structure. The relative intensities of those crosspeaks are consistent with the relative interproton distances derived from the crystal structure (to the inverse sixth power as expected for isolated spin pairs). Of particular note is the absence of crosspeaks to the methyl resonances of Val 55. The methyl groups of Val 55 are beyond 7 Å from the indole 1HNε1 in the crystal structures of FKBP51 and FKBP52 as are the analogous Val 24 methyl groups in the crystal structures of wild type FKBP12. However, strong NOE crosspeaks are observed for both methyls of Val 24 in FKBP12 (S/N of 50 for Cγ2), consistent with the flipped indole ring orientation seen in the crystal structure of the G89P variant [24]. Assuming a 20% population for the ring flipped FKBP12 conformation, the absence of the analogous crosspeaks to the Val 55 methyl groups in FKBP51 and FKBP52 indicates that the population of the flipped indole ring orientation must be less than ~0.5%.
Fig. 1.

NOESY crosspeaks between the active site Trp 90 HNε1 and nearby methyl groups in FKBP51 (A) and FKBP52 (B). The homologous position in FKBP12 is indicated in parentheses. The F2-F3 plane at the indole 1HNε1 frequency from a 3D F1-filtered 1H-13C-1H NOESY spectrum of collected using a 200 ms mix time (black) is superimposed upon the reference 2D 1H,13C HSQC spectrum containing all selectively enriched valine Cγ, leucine Cδ and isoleucine Cδ positions (green). The observable crosspeaks from the 3D NOESY spectrum have intensities that are consistent with the canonical orientation of the indole ring.
In addition to FKBP51 and FKBP52, crystal structures have been reported for two other active site Trp-containing FKBP domains of FKBP13 [41] and FKBP25 [42]. All of these four domains contain an aspartate residue at the position homologous to Glu 60. Indeed, during the evolution of FKBP12 this position is occupied by an aspartate throughout lower eukaryotes. The glutamate substitution did not appear until the evolution of bony fish at approximately the time that FKBP12 and FKBP12.6 began to diverge [43]. As illustrated for FKBP51, superpositioning onto the structure for the major conformer of the G89P variant of FKBP12 yields a close alignment for both the backbone conformation of the 50’s loop (Fig. 2A) and the residues which lie beneath the active site indole ring (Fig. 2B). The carboxyl oxygens of Asp 91 in FKBP51 and of Glu 60 in FKBP12 hydrogen bond to the backbone amides of residues 82/51 and 84/53 as well as to the highly conserved internal water at that site. On the other hand, not only does the orientation of the Trp indole ring differ between the structures of FKBP51 and the G89P variant of FKBP12, the backbone atoms of Trp 90 and Asp 91 in FKBP51 are shifted away from the canonical α-helical hydrogen bonding geometry (Fig. 2A), similar to what is observed in the wild type FKBP12 crystal structure. It would appear that the extended aspartate sidechain is too short to allow for shifting to the canonical α-helical hydrogen bonding geometry while still maintaining the hydrogen bonding interactions with the backbone of the 50’s loop.
Fig. 2.
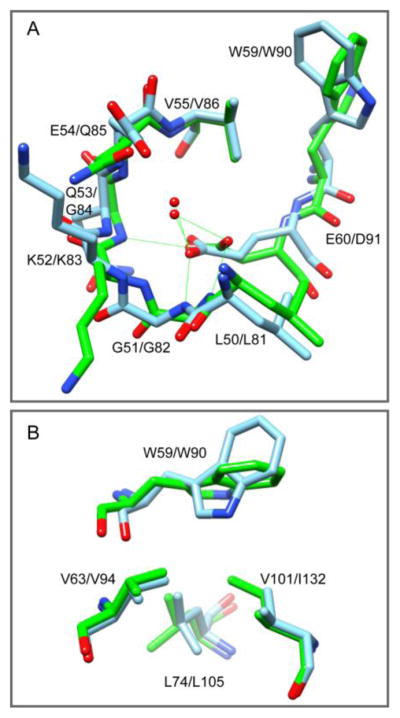
Superposition of the 0.96 Å resolution crystal structure for the first FKBP domain of FKBP51 (green) onto the G89P (blue) crystal structure. (A) The positioning of the 50’s loop backbone and the interactions of the Glu 60 / Asp 91 carboxylate groups are similar in the structures of the G89P variant [24] and the FK1 domain of FKBP51 [37]. However, the backbone segment connecting to the indole sidechain is shifted further away by ~1Å in the G89P crystal structure. (B) Underneath the indole ring of Trp 59, the Cδ1 of Ile 132 in FKBP51 projects into where a cavity is formed by the sidechains of Val 63, Leu 74 and Val 101 in the FKBP12 structure.
Regarding the residues that underlie the Trp 59 indole ring in the wild type FKBP12 crystal structure, Ile 132 in the FKBP51 structure is substituted for Val 101 (Fig. 2B). This added Cδ atom neatly fills the empty volume that lies directly underneath the indole ring in the wild type FKBP12 structure which becomes occupied following the indole reorientation that occurs in the G89P crystal structure. This apparent increase in steric hindrance to the reorientation of the indole ring combined with the restricted extension that results from the shorter Asp91 sidechain, suggests that such a reorientation of the indole ring may be significantly less energetically accessible for FKBP51 and is likely to be similarly inaccessible for the other active site tryptophan-containing FKBP domains.
3.2. NOESY analysis of indole ring flipping in the V101I variant of FKBP12
The additional methyl group arising from the V101I substitution would appear to be readily accommodated into the crystal structure of wild type FKBP12 with minimal perturbation. If that pre-existing cavity is energetically significant in allowing for the flipping of the indole ring, the V101I variant would be predicted to reduce the probability of that transition. In this regard, it should be noted that one of the high resolution crystal structures of this FKBP51 domain (PDB code 3O5P) published by Hausch and colleagues [37] has a minor (10%) conformer of the homologous Ile 132 sidechain in an alternate rotamer state which does not significantly occlude the cavity lying under the indole ring.
The F2-F3 plane at the Trp 59 indole 1HNε1 frequency from the F1-filtered 1H-13C-1H NOESY spectrum of the V101I variant (Fig. 3A) yields a simpler pattern of NOE crosspeaks to the neighboring methyl groups than what is seen for wild type FKBP12 (Fig. 3B). The Cδ crosspeaks of Ile 56 and Ile 76 dominate the NOESY interactions with the indole 1HNε1 of the V101I variant as predicted from the crystal structure of the wild type FKBP12. On the other hand, there are also a weak crosspeak for the Cγ2 resonance of Val 24 that is 10-fold less intense than the corresponding crosspeak in the wild type spectrum. This crosspeak from Val 24 would not be anticipated from the FKBP12 crystal structure conformation. As noted above, the structurally homologous Val 55 of FKBP51 and FKBP52 do not give rise to crosspeaks to the indole 1HNε1 resonance, even though the S/N ratio of these spectra, particularly for the FKBP52 data, is above that for the V101I variant. These results are consistent with the population of the flipped indole conformation being reduced 10-fold in the V101I variant, relative to wild type FKBP12.
Fig. 3.

NOESY crosspeaks between the active site Trp 59 HNε1 and nearby methyl groups in the V101I variant (A) and wild type FKBP12 (B) for the F2-F3 plane at the indole 1HNε1 frequency from a 3D F1-filtered 1H-13C-1H NOESY spectrum of collected using a 200 ms mix time (black) is superimposed upon the reference 2D 1H,13C HSQC spectrum containing all selectively enriched valine Cγ, leucine Cδ and isoleucine Cδ positions (green). The observable crosspeaks from the 3D NOESY spectrum of the V101I variant predominantly reflect intensities that are consistent with the canonical orientation of the indole ring. The crosspeak for Val 24 Cγ2 is 10-fold weaker in the V101I spectrum. This crosspeak would be unobservable for the canonical orientation of the indole ring and is consistent with a corresponding 10-fold decrease in the population of the perpendicular reoriented indole ring, relative to the wild type protein.
3.3. Molecular dynamics analysis of indole ring flipping in wild type and the V101I variant of FKBP12 and in the FK1 domain of FKBP51
For each FKBP domain, three parallel 400 ns simulations were carried out at 298K in NAMD2 [39] using the CHARMM27 force field [40]. As illustrated for one of the three 400 ns simulations of wild type FKBP12 (Fig. 4A), the perpendicular orientation of the Trp 59 Nε1-HNε1 bond vector markedly predominated over conformations similar to the initial frame in the canonical ring orientation. In contrast, the MD simulations for the V101I variant yielded a more equal distribution of the two indole ring orientations (Fig. 4C). Excluding the initial 5% of each MD run for equilibration, 1.14 μs of simulation for wild type FKBP12 predicted a canonical indole ring conformation population of only 7.9% which rose to 59.6% for the V101I variant. The predicted 17-fold (7.0 kJ/mol) shift in the indole ring flip equilibrium that is induced by the V101I substitution agrees reasonably well with the experimentally determined value despite the fact that the predicted populations for each state differ markedly from the experimental observations.
Fig. 4.

CHARMM27 simulations of the sidechain conformation for Trp 59 and Glu 60 in wild type (A and B) and the V101I variant (C and D) of FKBP12. Illustrated at 400 ps intervals, the orientation of the Trp 59 indole Nε1 – HNε1 bond vector is plotted relative to that of the initial frame (A and C). The χ1 sidechain torsion angle of Glu 60 is plotted at the same intervals (B and D) using a 0° to 360° scale to circumvent the discontinuity at 180°/−180°.
The orientation of the χ1 sidechain torsion angle for Glu 60 is predicted to be significantly correlated with the Trp 59 indole ring conformation for both wild type FKBP12 (Fig. 4B) and the V101I variant (Fig. 4D). In contrast to the crystal structures of wild type FKBP12, these simulations predict a high population for the trans χ1 rotamer of Glu 60. Given the correlation between the conformational states for the sidechains of Trp 59 and Glu 60, the errors in predicting the absolute population of the sidechain conformations may largely arise from inaccuracies in the modeling of the interactions between the Glu 60 sidechain and the backbone of the 50’s loop, including the bridging interactions of the conserved structural water molecule. Using the same force field in the NAMD2 simulation program, Park and Saven [29] observed that the predicted orientation of the Trp 59 ring is quite sensitive to the modeling of the interactions between the residue 60 sidechain and the 50’s backbone.
In contrast to the MD simulations for both the wild type and V101I variant of FKBP12, the FK1 domain of FKBP51 was predicted to overwhelmingly adopt the canonical active site indole ring orientation for Trp 90 with the Asp 91 sidechain being in a trans χ1 rotamer state. Alternate conformations for these residues were populated on the order of 0.1%.
The issue of correlated conformational transitions for the Trp 59 and Glu 60 sidechains and the hydrogen bond geometry for the α-helical backbone was analyzed by examination of the transition matrix for the 23 states of the trans (120° to 240°) or gauche− (240° to 360°) χ1 rotamers of Trp 59 and Glu 60 and the presence or absence of a Glu 60 O – Ala 64 HN hydrogen bond, defined at the van der Waals separation of 2.50 Å [44]. The diagonal values in the transition matrix calculated from the simulations of wild type FKBP12 correspond to conformations that remain in the same state after a 5 ps sampling interval across 1.14 μs of simulation (Fig. 5). The ordering of the states is such that the rapidly interchanging hydrogen bond breakage/formation transitions lie within 2×2 submatrices, while the much slower torsion angle transitions of Glu 60 and Trp59 lie further from the diagonal. The value of each off-diagonal element is generally similar to that of the symmetrical element, indicating reasonably complete statistical sampling of most transitions in both directions.
Fig. 5.
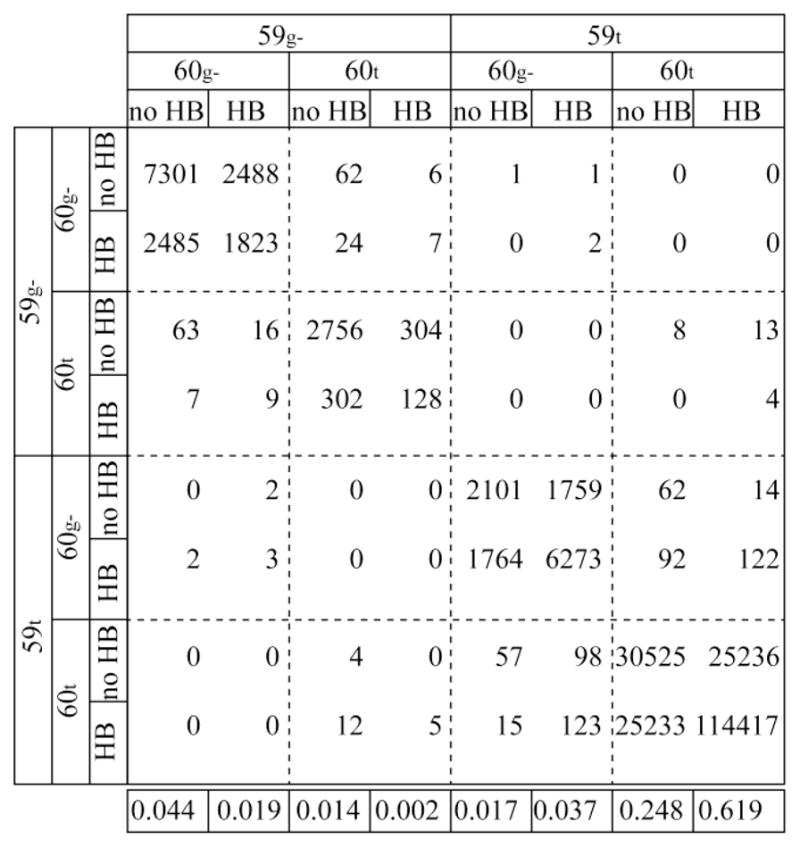
Transition matrix for wild type FKBP12 characterizing the trans-to-gauche− χ1 rotamer transitions for the sidechains of Trp 59 and Glu 60 and the hydrogen bonding status between Glu 60 O and Ala 64 HN (> or < 2.5 Å). The 1.14 μs of molecular simulation was sampled at 5 ps intervals. A small set of transitions of the χ1 rotamers with residence times less than 15 ps were disregarded. These generally corresponded to conformations that move only slightly beyond the torsional barrier maximum at 240° separating the trans and gauche− rotamers. The 59g− denotes Trp 59 sidechains with a gauche− χ1 rotamer and a Nε1-HNε1 angle (relative to the initial frame) < 50°, while 59t denotes the trans χ1 rotamer with a Nε1-HNε1 angle > 50°. The population average for each state is listed along the bottom.
Most striking is the absence of transitions lying within the 2×2 blocks along the anti-diagonal. Despite the marked statistical correlation between the sidechain conformations of Trp 59 and Glu 60 (Fig. 4A and 4B), this result indicates that throughout the entire 1.14 μs of simulation not a single concerted transition occurs for these two sidechains. There did occur 57 χ1 rotamer transitions for Trp 59 during this time period (63 such transitions for V101I – Fig. S1) indicating an average residence time of 20 ns. A similar calculation yields an average residence time of 1.5 ns for the Glu 60 χ1 rotamer states.
The allosteric coupling between the transitions of the Trp 59 and Glu 60 sidechains is determined by how the population ratio for the 59g− and 59t states depends upon the state of the Glu 60 sidechain (i.e., [(59g−/59t)|60g−]/[(59g−/59t)|60t]) which by symmetry equals [(60g−/60t)|59g−]/[(60g−/60t)|59t]. The predicted coupling ratio of 63 for wild type FKBP12 corresponds to a coupling free energy of 10.3 kJ/mol. Since the predicted coupling free energy is larger than the free energy difference between any pair of Trp 59 and Glu 60 sidechain states, the molecular dynamics-derived energy level diagram deviates from the generic diamond pattern typically assumed (Fig. 6).
Fig. 6.

The free energy diagram (kJ/mol) for the gauche− and trans χ1 rotamer states of Trp 59 and Glu 60 predicted from 1.14 μs of simulation for wild type FKBP12.
As illustrated in the four 2×2 blocks along the diagonal of the transition matrix (Fig. 5), the kinetics of hydrogen bond formation and rupture for the Glu 60 O – Ala 64 HN interaction occurs on the timescale of the 5 ps sampling interval. Assuming exponential kinetics for the hydrogen bond dynamics yields time constants ranging from 3 ps to 5 ps for the four states of the Trp 59 and Glu 60 sidechains. The population ratios for the hydrogen bonded and the non-hydrogen bonded states vary over a 17.6-fold range from the (59g−,60t) state to the (59t,60t) state. Expressing these hydrogen bonding population ratios in terms of an energy level diagram strongly indicate that the conformational shift underlying the Glu 60 O – Ala 64 HN interaction provides a substantial component of the coupling energetics linking the sidechain conformations of Trp 59 and Glu 60, although other contributions are clearly required (Fig. 7). Shifting the hydrogen bond distance cutoff to either 2.30 Å or 2.70 Å had minimal effects on the derived energy level diagram (Fig. S2).
Fig. 7.

The differential in free energy corresponding to the shift in hydrogen bond formation between Glu 60 O and Ala 64 HN as a function of the χ1 rotamer state for both Trp 59 and Glu 60 in wild type FKBP12 assuming hydrogen bond formation at van der Waals distance (2.50 Å).
Inspection of the transition matrix also provides mechanistic insight into how the transition of the Glu 60 O – Ala 64 HN hydrogen bonding facilitates transitions in the two sidechains. Focusing on the lower right hand quadrant of the transition matrix (Fig. 5) for which the Trp 59 sidechain remains in a trans χ1 rotamer state, the transfer from HB(60g−) to noHB(60t) is more populated (92) than would be expected for the sequence HB(60g−) → HB(60t) → noHB(60t) since by that pathway the off-diagonal block population ratio for noHB(60t)/ HB(60t) could not be more than the corresponding diagonal block ratio (25233/114417). This implies that the dominant pathway for this transition is HB(60g−) → noHB(60g−) → noHB(60t). A similar argument applies for the nominally reverse transition noHB(60t) → HB(60t) → HB(60g−). Although the sampling statistics for the Trp 59 χ1 transitions are limited, preferred kinetic pathways in the Glu 60 trans state appear to be present for the noHB(59g−) → HB(59g−) → HB(59t) and the HB(59t) → noHB(59t) → noHB(59g−) transitions.
If the 1.14 μs of molecular simulation for the V101I variant of FKBP12 were to model a pure modulation of the Trp 59 – Glu 60 sidechain allostery, the transition matrix (Fig. S1) would yield a free energy diagram in which the two 59t states would be shifted upward 7.0 kJ/mol (Fig. 8A), relative to that of the wild type FKBP12 simulation (Fig. 6), to reflect the difference in population for the 59t state in these simulations (Fig. 4A and 4C). However, the MD-derived energy level diagram for the V101I variant differs appreciably, particularly with regards to the lower free energy of the (59t,60g−) state (Fig. 8B). One potential source for the resultant decrease in modeled allosteric coupling is the high population (96.6%) predicted for the alternate χ1 rotamer of Ile 101 which corresponds to the minor (10%) rotamer in the 3O5P structure of FKBP51. At least in part, this strong biasing against the initially assigned χ1 rotamer state for Ile 101 likely reflects limitations in the force field as the corresponding simulations for the FKBP51 domain predict a 70% population for the χ1 rotamer of Ile 132 that is present at only 10% in one high resolution crystal structure and undetected in several other crystal forms of comparable resolution [37]. As a result of the altered sidechain packing arrangement of Ile 101 in FKBP12, the predicted orientation of the flipped indole ring of Trp 59 in this variant is slightly rotated away from its position in the wild type protein, as indicated by the center of the Nε1-HNε1 vector distribution being shifted up to ~110° relative to the initial ring orientation in the simulation (Fig. 4D).
Fig. 8.

Free energy diagrams (kJ/mol) for the gauche− and trans χ1 rotamer states of Trp 59 and Glu 60 in the V101I variant of FKBP12. If the V101I variant were to act as a pure allosteric modulator, the energy levels for the states containing the trans χ1 rotamer of Trp 59 would be shifted 7.0 kJ/mol (A) from the free energy diagram predicted for wild type FKBP12 to reflect the 17-fold predicted shift in population for this state in the V101I variant (Fig. 4C). The energy levels for this variant were then predicted directly from its 1.14 μs of simulation (B).
4. Conclusion
The conformational transition of the Trp 59 indole ring in FKBP12 is highly sensitive to the sidechain transitions of the adjacent glutamate residue. Given the structural constraints imposed by the shorter asparate sidechain found in the FK1 domains of FKBP51 and FKBP52 and several other human FKBP domains containing an active site tryptophan, it appears likely that among these proteins only FKBP12 significantly populates a perpendicular orientation of the indole ring which largely occludes the active site cleft. Molecular simulations have provided a structural interpretation of how this ring flip transition is linked to interactions in the backbone of the 50’s loop with a predicted allosteric coupling of 10 kJ/mol that could provide a basis for how binding interactions at this distal site might modulate the geometry of the catalytic site.
This allosteric interaction involves a negligible contribution from concerted dynamical transitions. At least three distinct local conformational transitions with markedly differing characteristic time constants participate in this allosteric coupling: the active site Trp 59 sidechain reorientation, the sidechain transition of Glu 60 interacting with the 50’s loop backbone, and the shift in hydrogen bonding geometry for the backbone atoms of those two residues which provides a significant component of the structural mechanism for the coupling between the first two transitions. The conformational equilibria for both the sidechains of Trp 59 and Glu 60 are determined by sets of interactions with the surrounding protein structure, and it is the overlap between these two sets of interactions that provide the energetic coupling of these sidechain transitions.
Viewed in terms of two statistically coupled but dynamically decoupled transitions, allostery can be expected to be the rule rather than the exception for such nearby transition sites as those considered herein. Long range concerted intramolecular motion, as with the helical shift in the aspartate receptor of chemotaxis [45,46], can modulate the conformational equilibria surrounding the extracellular aspartate binding site as well as the cytoplasmic binding site of the HAMP domain over a distance of 100 Å. Yet allostery need not require that this helical shift to be dynamically coupled to the transitions underlying the conformational equilibria at either binding site of the aspartate receptor. Although transitions such as the helical piston model of the aspartate receptor offer an efficient mechanism to transmit the modulation of interactions at the two distant binding sites, it is unnecessarily restrictive to assume that, in general, the coupling mechanism within an allosteric process must be dynamically concerted [47]. In the ongoing efforts to identify large scale collective transitions in proteins, the FKBP12 system offers a valuable cautionary example. The presence of μs-ms conformational exchange NMR linebroadening throughout much of that protein led several groups to propose a single collective transition [21–23], an assumption later shown to be severely in error [20]. Analyzing how nature might exploit the dynamical heterogeneity of protein conformational transitions may provide a more fruitful path for understanding the structural basis of allostery.
Supplementary Material
HIGHLIGHTS.
Unlike FKBP12, the active site Trp 90 ring of FKBP51 and FKBP52 does not flip
The V101I substitution decreases the indole ring flipping in FKBP12 by 10-fold
Transitions of the Trp 59 and Glu 60 sidechains are strongly correlated
1.14 μs of CHARMM27 simulation indicate no concerted transitions for these residues
Allosteric analysis of statistically coupled but dynamically uncoupled transitions
Acknowledgments
This work was supported in part by National Institutes of Health grant GM 088214. We acknowledge the use of the NMR facility and Molecular Genetics cores at the Wadsworth Center, the NMR facility at the New York Structural Biology Center, and the use of the IBM Intelligent Cluster at Union College.
Abbreviations
- PDB
protein data bank
- FKBP
FK506 binding protein
- NOESY
nuclear Overhauser enhancement spectroscopy
Footnotes
Performance site: Wadsworth Center, New York State Department of Health, Empire State Plaza, Albany, New York, 12201
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Galat A. Functional drift of sequence attributes in the FK506-binding proteins (FKBPs) J Chem Inf Model. 2008;48:1118–1130. doi: 10.1021/ci700429n. [DOI] [PubMed] [Google Scholar]
- 2.Weiwad M, Edlich F, Kilka S, Erdmann F, Jarczowski F, Dorn M, Moutty MC, Fischer G. Comparative Analysis of Calcineurin Inhibition by Complexes of Immunosuppressive Drugs with Human FK506 Binding Proteins. Biochemistry. 2006;45:15776–15784. doi: 10.1021/bi061616p. [DOI] [PubMed] [Google Scholar]
- 3.Marz AM, Fabian AK, Kozany C, Bracher A, Hausch F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol Cell Biol. 2013;33:1357–1367. doi: 10.1128/MCB.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Farmer JJD, Lane WS, Friedman J, Wiessman I, Schreiber SL. Calcineurin is a common target for cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 5.Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, Abraham RT, O’Keefe SJ, O’Neill EA, Wiederrecht GJ. A novel FK506 protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J Biol Chem. 1995;270:26511–26522. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- 6.Xu X, Su B, Barndt RJ, Chen H, Xin H, Yan G, Chen L, Cheng D, Heitman J, Zhuang Y, Fleischer S, Shou W. FKBP12 is the only FK506 binding protein mediating T-cell inhibition by the immunosuppressant FK506. Transplantation. 2002;73:1835–1838. doi: 10.1097/00007890-200206150-00023. [DOI] [PubMed] [Google Scholar]
- 7.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, Fleischer S. The calcium release channel of sacroplasmic reticulum is modulated by FK506-binding protein. J Biol Chem. 1993;268:22992–22929. [PubMed] [Google Scholar]
- 8.Timerman AP, Onoue H, Xin HB, Barq S, Copello J, Wiederrecht G, Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 9.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 10.Maruyama M, Li BY, Chen H, Xu X, Song LS, Guatimosim S, Zhu W, Yong W, Zhang W, Bu G, Lin SF, Fishbein MC, Lederer WJ, Schild JH, Field LJ, Rubart M, Chen PS, Shou W. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ Res. 2011;108:1042–1052. doi: 10.1161/CIRCRESAHA.110.237867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T, Gaber R, Picard D, Smith DF. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158–1167. doi: 10.1093/emboj/cdg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tai PK, Maeda Y, Nakao K, Wakim NG, Duhring JL, Faber LE. A 59-kilodalton protein associated with progestin, estrogen, androgen and glucocorticoid receptors. Biochemistry. 1986;25:5269–5275. doi: 10.1021/bi00366a043. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez ER. HSP56: a novel heat shock protein associated with untransformed steroid receptor complexes. J Biol Chem. 1990;265:22067–22070. [PubMed] [Google Scholar]
- 14.Smith DF, Faber LE, Toft DO. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J Biol Chem. 1990;265:3996–4003. [PubMed] [Google Scholar]
- 15.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 16.Liang J, Choi J, Clardy J. Refined structure of the FKBP12-rapamycin-FRB ternary complex at 2.2 A resolution. Acta Cryst. 1999;D55:736–744. doi: 10.1107/s0907444998014747. [DOI] [PubMed] [Google Scholar]
- 17.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF-beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 18.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boegermann JH, Triffitt JT, vonDelft F, Knapp S, Knaus P, Bullock AN. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem. 2012;287:36990–36998. doi: 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riggs DL, Cox MB, Tardif HL, Hessling M, Buchner J, Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol. 2007;27:8658–8669. doi: 10.1128/MCB.00985-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustafi SM, Chen H, Li H, LeMaster DM, Hernández G. Analyzing the visible conformational substates of the FK506-binding protein FKBP12. Biochem J. 2013;453:371–380. doi: 10.1042/BJ20130276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brath U, Akke M, Yang D, Kay LE, Mulder FAA. Functional dynamics of human FKBP12 revealed by methyl 13C rotating frame relaxation dispersion NMR spectroscopy. J Am Chem Soc. 2006;128:5718–5727. doi: 10.1021/ja0570279. [DOI] [PubMed] [Google Scholar]
- 22.Brath U, Akke M. Differential responses of the backbone and side-chain conformational dynamics in FKBP12 upon binding the transition-state analog FK506: Implications for transition-state stabilization and target protein recognition. J Mol Biol. 2009;387:233–244. doi: 10.1016/j.jmb.2009.01.047. [DOI] [PubMed] [Google Scholar]
- 23.Sapienza PJ, Mauldin RV, Lee AL. Multi-timescale dynamics study of FKBP12 along the rapamycin-mTOR binding coordinate. J Mol Biol. 2011;405:378–394. doi: 10.1016/j.jmb.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 24.Mustafi SM, Brecher M, Zhang J, Li H, LeMaster DM, Hernández G. Structural Basis of Conformational Transitions in the Active Site and 80’s loop in the FK506-binding Protein FKBP12. Biochem J. 2014;458:525–536. doi: 10.1042/BJ20131429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szep S, Park S, Boder ET, VanDuyne GD, Saven JG. Structural coupling between FKBP12 and buried water. Proteins. 2009;74:603–611. doi: 10.1002/prot.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deivanayagam CC, Carson M, Thotakura A, Narayana SV, Chodavarapu RS. Structure of FKBP12.6 in complex with rapamycin. Acta Cryst D. 2000;56:266–271. doi: 10.1107/s0907444999016571. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Mustafi SM, LeMaster DM, Li Z, Héroux A, Li H, Hernández G. Crystal structure and conformational flexibility of the unligated FK506-binding protein FKBP12.6. Acta Cryst. 2014;D70:636–646. doi: 10.1107/S1399004713032112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fulton KF, Jackson SE, Buckle AM. Energetic and structural analysis of the role of tryptophan 59 in FKBP12. Biochemistry. 2003;42:2364–2372. doi: 10.1021/bi020564a. [DOI] [PubMed] [Google Scholar]
- 29.Park S, Saven JG. Statistical and molecular dynamics studies of buried waters in globular proteins. Proteins. 2005;60:450–463. doi: 10.1002/prot.20511. [DOI] [PubMed] [Google Scholar]
- 30.Cui Q, Karplus M. Allostery and cooperativity revisited. Prot Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Mustafi SM, LeMaster DM, Hernández G. Differential conformational dynamics in the closely homologous FK506-binding domains of FKBP51 and FKBP52. Biochem J. 2014;458:525–536. doi: 10.1042/BJ20140232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernández G, Anderson JS, LeMaster DM. Polarization and polarizability assessed by protein amide acidity. Biochemistry. 2009;48:6482–6494. doi: 10.1021/bi900526z. [DOI] [PubMed] [Google Scholar]
- 34.Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile(δ1) methyl-protonated 15N, 13C, 2H labeled proteins. J Biomolec NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- 35.Ogura K, Teresawa H, Inagaki F. An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. J Biomol NMR. 1996;8:492–498. doi: 10.1007/BF00228150. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Bracher A, Kozany C, Thost AK, Hausch F. Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Cryst D. 2011;67:549–559. doi: 10.1107/S0907444911013862. [DOI] [PubMed] [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Taikhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultz LW, Martin PK, Liang J, Schreiber SL, Clardy J. Atomic structure of the immunophilin FKBP13-FK506 complex: Insights into the composite binding surface for calcineurin. J Am Chem Soc. 1994;116:3129–3130. [Google Scholar]
- 42.Liang J, Hung DT, Schreiber SL, Clardy J. Structure of the human 25 kDa FK506 binding protein complexed with rapamycin. J Am Chem Soc. 1996;118:1231–1232. [Google Scholar]
- 43.Somarelli JA, Herrara RJ. Evolution of the 12 kDa FK506-binding protein gene. Biol Cell. 2007;99:311–321. doi: 10.1042/BC20060125. [DOI] [PubMed] [Google Scholar]
- 44.Rashin AA. Buried surface area, conformational entropy, and protein stability. Biopolymers. 1984;23:1605–1620. doi: 10.1002/bip.360230813. [DOI] [PubMed] [Google Scholar]
- 45.Otteman KM, Xiao W, Shin YK, Koshland JDE. A piston model for transmembrane signaling of the aspartate receptor. Science. 1999;285:1751–1753. doi: 10.1126/science.285.5434.1751. [DOI] [PubMed] [Google Scholar]
- 46.Yu EW, Koshland JDE. Propagating conformational changes over long (and short) distances in proteins. Proc Natl Acad Sci U S A. 2001;98:9517–9520. doi: 10.1073/pnas.161239298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daily MD, Phillips JGN, Cui Q. Many local motions cooperate to produce the adenylate kinase conformational transition. J Mol Biol. 2010;400:618–631. doi: 10.1016/j.jmb.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.