

Summary
Lipid biology continues to emerge as an area of significant therapeutic interest, particularly as the result of an enhanced understanding of the wealth of signaling molecules with diverse physiological properties. This growth in knowledge is epitomized by lysophosphatidic acid (LPA), which functions through interactions with six cognate G protein-coupled receptors. Herein we present three crystal structures of LPA1 in complex with antagonist tool compounds selected and designed through structural and stability analysis. Structural analysis combined with molecular dynamics identified a basis for ligand access to the LPA1 binding pocket from the extracellular space contrasting with the proposed access for the sphingosine 1-phosphate receptor. Characteristics of the LPA1 binding pocket raise the possibility of promiscuous ligand recognition of phosphorylated endocannabinoids. Cell-based assays confirmed this hypothesis, linking the distinct receptor systems through metabolically related ligands with potential functional and therapeutic implications for treatment of disease.
Introduction
Lysophosphatidic acid (LPA) is a pleiotropic bioactive lipid produced from extracellular lysophospholipids by autotaxin (ATX) as well as derived from membrane glycerophospholipids by phospholipases (Aoki, 2004; Hishikawa et al., 2014; Perrakis and Moolenaar, 2014) to produce a range of chemical species with varied fatty acid chain length and saturation. LPA is present in nearly all cells, tissues and fluids of the body (Mirendil et al., 2013) and its effects are mediated by cognate cell-surface G protein-coupled receptors (GPCRs) consisting of six family members designated LPA1-LPA6 (Kihara et al., 2014) that couple to heterotrimeric G protein complexes to activate downstream signaling pathways. Targeted deletion of the LPA receptors has revealed physiological effects on every organ system examined thus far, with receptor dysregulation linked to a range of disease indications including hydrocephalus (Yung et al., 2011), infertility (Ye et al., 2005), fibrosis (Tager et al., 2008), pain (Inoue et al., 2004) and cancer (Mills and Moolenaar, 2003). Three of the six LPA receptors, including LPA1 (Kihara et al., 2014), are part of a larger lysophospholipid receptor family (the EDG family) that includes the sphingosine 1-phosphate (S1P) receptors, of which the structure of one member (S1P1) has been reported (Hanson et al., 2012). The closely related cannabinoid receptor CB1 (Hecht et al., 1996; Matsuda et al., 1990) interacts with endogenous ligands structurally related to LPA raising the question of potential polypharmacology between the two receptors, effectively connecting these signaling systems. Additional structural details for this family will improve our understanding of their associated function and physiology.
Results
Design of LPA1 Ligands for Structural Studies
A key for success in GPCR structure determination is the identification of a ligand that stabilizes the receptor into a single and stable conformation. To facilitate finding such a ligand, a stability assay based on analytical size exclusion chromatography (aSEC) was developed and used to screen a library of compounds (Figure S1). The compound used for initial studies of LPA1, ONO-9780307 (Table S1), was selected from this library based on the initial SEC stability assay data (Figure S1). This assay was used in place of the more traditional CPM assay (Alexandrov et al., 2008) to allow early access to stability data without the need for pure receptor protein. Using insights gained from the initial structure, several analogues of ONO-9780307 were designed by considering lipophilicity, structural diversity and potential metabolic stability. These analogues were synthesized, tested for stability induction and used to generate additional co-crystal structures (Table S1). The design feature of ONO-9910539 is an acetyl group for enhanced polar interactions and reduced lipophilicity. The design of ONO-3080573 replaced a methylene and the pyrrole ring with an ether and phenyl ring, respectively, to both reduce torsional strain and increase structural diversity and potential metabolic stability.
Structure determination of LPA1-ONO9780307, LPA1-ONO9910539 and LPA1-ONO3080573
To facilitate crystallization, a thermostabilized b562RIL (bRIL) (Chun et al., 2012), was inserted into the third intracellular loop (3IL) at Ballesteros index positions 5.66 (R233) and 6.24 (R247); (Ballesteros and Weinstein, 1995), and thirty-eight residues were truncated from the carboxyl terminus (C-terminus). With this engineered construct (LPA1-bRIL) in complex with compounds ONO-9780307 and ONO-9910539, crystals were produced in LCP supplemented with cholesterol that diffracted to a final resolution of 3.0 å and 2.9 Å respectively and supported structure solution (Figure 1A). Despite similar stability induction for ONO-3080573 (Figure S1) we were unable to crystallize this compound with the original LPA1-bRIL construct. In order to further improve stability and reduce conformational heterogeneity for crystallization in the presence of ONO-3080573, a series of disulfide bonds were engineered into various sites within the extracellular half of the transmembrane region of LPA1-bRIL, and one was selected based on superior expression and stability analysis (dsLPA1-bRIL) (Figure 2A and C). The effect of disulfide bond engineering at this site was evaluated using signaling in response to LPA concentration in a calcium mobilization assay (Figure 2B). Crystals of dsLPA1-bRIL in complex with ONO-3080573 were grown in a LCP cholesterol mixture and diffracted to 2.9Å (Figure 2D). While there are clear differences in experimental electron density for the ligands (Figure S2), aside from small conformational changes in and around the binding pocket, the receptor structures are highly analogous.
Figure 1.

The overall structure of LPA1 colored by region and compound structure in the bioactive conformation. (A) The ligand ONO-9780307, shown with green carbons, demarcates the binding pocket, which is partially occluded by an N-terminal alpha helix (red) packing closely against the extracellular loops, ECL1 and ECL2 (orange). There are three native disulfide bonds in the extracellular region, one of which constrains the N-terminal capping helix to ECL2. (B) The bioactive conformation of ONO-9780307 in green carbons with high torsional strain energy required for positioning the indane ring adjacent to the dimethoxy phenyl ring. Release of this strain with an optimal torsion angle would result in a clash with the binding pocket.
Figure 2.

Generation of the disulfide engineered LPA1 (dsLPA1-bRIL) crystallization construct. (A). Prediction of cysteine mutants replacing Asp2045.37 and Val2826.59 at the tip of TMV and VI respectively. (B). Functional analysis through a calcium mobilization assay measured in response to the LPA agonist dsLPA1 compared to full-length wild-type LPA1. The EC50 value of the stabilized receptor (994 nM) is about ten-fold higher compared to wild-type (153 nM). Data are presented as a mean (± SEM, n=3). (C). Thermal stability analysis of dsLPA1-bRIL compared to the non-engineered crystallization construct LPA1-bRIL. The engineered disulfide bond increases the apparent Tm by about 5°C in the presence of ONO-9780307, which was the ligand used for stability characterization of new constructs. (D). Crystal images for dsLPA1-bRIL in the presence of ONO-3080573. See also Figure S1.
Overall structural features of LPA1 in complex with ONO-9780307
The initial co-crystal structure of LPA1-bRIL in the antagonist state with the compound ONO-9780307 is used for a general description of the receptor. The main fold of the antagonist bound LPA1 receptor is similar to other inactive class A GPCRs, with a canonical seven-transmembrane α-helical bundle (Figure 1A). The N-terminus of LPA1 forms a six-turn alpha helix similar in length and orientation to the equivalent residues in S1P1, but with an additional disulfide bond linking the N-terminal capping helix to extracellular loop 2 (ECL2), resulting in additional fold stability. The N-terminus packs tightly against ECL1 and ECL2 and provides charged and polar amino acid side chains for interactions within the ligand-binding pocket. The ligand, ONO-9780307, positions its branching aromatic indane and dimethoxy phenyl rings adjacent to each other in the spherical binding pocket. This positioning requires a strained eclipsed conformation of the torsion angle on the bond adjacent to the indane ring, with a difference of 9.6 kcal/mol between the observed conformation and the local energy minimum estimated by gas-phase quantum mechanics calculations. The ligand conformation associated with the local energy minimum is not able to bind in the pocket due to projected clashes with TMVI (Figure 1B).
Structural comparison of the LPA1 and S1P1 receptors
The relative orientation of LPA1 helices is similar to the S1P1 receptor, which was expected given the similarity in both primary sequence (41% identical in transmembrane region) and recognition of lysophospholipid endogenous ligands (Chun et al., 2013). The primary point of divergence occurs at the tip of TMI, which is positioned 3 Å closer to TMVII compared to S1P1 (Figure 3A). This helical repositioning closes a gap between TMI and TMVII, which was postulated to enable access of hydrophobic ligands to the occluded extracellular binding pocket via the membrane for S1P1. The short segment between TMI and the N-terminal capping helix (ECL0) is helical in S1P1 but lacks secondary structure in LPA1. This change may open access to the ligand binding pocket from the extracellular space through increased flexibility and may also contribute to the inward shift of TMI through a release of secondary structure steric constraints. Interestingly, ECL1 and ECL2 are comparable between the two receptors, whereas the position of the third extracellular loop (ECL3) diverges from the S1P1 receptor by up to 8 Å at the furthest point, resulting in a loss of interactions between this loop and the rest of the extracellular region (Figure 3A). The overall result is the opening of a ligand access port into the extracellular milieu for LPA1, while access from the plasma membrane is closed (Figure 3A).
Figure 3.

Comparison of S1P1 and LPA1 structural features. (A). Top-view of LPA1 with structurally divergent regions of S1P1 (PDB ID: 3V2Y) overlayed (cyan). The configuration of the extracellular region is constrained by two intraloop disulfide bonds in ECL2 and ECL3 and a disulfide bond connecting ECL2 with the N-terminus. Although the electron density for the ECL3 loop is poor compared to the rest of the structure, the approximate position could be properly determined and is further from the N-terminal helix compared to S1P1. In S1P1 Lys285 in this region forms a cation dipole interaction with the C-terminal end of the capping helix, an interaction that is missing in LPA1. TMI is shifted 3 Å closer to TMVII at the tip compared to S1P1 and ECL0 is a loop in LPA1. Neither ECL0 nor ECL3 are involved in crystal contacts. (B). The shape of the LPA1 binding pocket is spherical in nature (solid surface) compared to the more linear binding pocket of S1P1 (blue mesh surface). This difference is caused by three changes in the amino acid composition between the two receptors. The first at position 3.33 is an aspartate in LPA1 and a phenylalanine in S1P1. The second at position 5.43 is a tryptophan in LPA1 and a cysteine in S1P1. Finally at position 6.51 LPA1 has a glycine whereas S1P1 has a leucine. The reduction in side chain bulk at this position opens a sub-pocket occupied by the indane ring of the antagonist series.
These global changes combined with residue substitutions generate a binding pocket for LPA1 that is more spherical compared to S1P1 (Figure 3B). This characteristic supports the finding that LPA1 has the ability to recognize a more diverse repertoire of chemical species of endogenous ligands with acyl chains of different lengths and globular conformations beyond the commonly used LPA 18:1 sn1. Three sequence substitutions can be identified that alter both the shape and polarity of the LPA receptor binding pocket compared to S1P receptors. In LPA receptors, position 3.33 is occupied by an aspartate residue (Asp1293.33 in LPA1), whereas S1P receptors have a conserved phenylalanine that serves an important role in ligand recognition (van Loenen et al., 2011). At position 5.43, LPA receptors possess a tryptophan residue (Trp2105.43 in LPA1), contrasting with a cysteine residue at this position in most S1P receptors. Finally, LPA receptors have a glycine (Gly2746.51 in LPA1) at position 6.51, contrasting with a leucine or alanine that forms important van der Waals interactions with the S1P ligands (Figure 3B).
Molecular dynamics analysis supports ligand access from extracellular space
The divergent position of TMI in LPA1 compared to S1P1 is attributable to both enhanced polar interactions and a substantial reduction in volume at the interface of TMI and TMVII (Figure 4A and 4B). In order to further understand the mode of ligand access for both LPA1 and S1P1, comparative molecular dynamics simulations of both receptors were completed, with and without their respective ligands, and the distance between the TMI and TMVII helices was analyzed over the course of three independent 100ns simulations for each receptor both with and without ligand. Based on this analysis, it is clear that the distance between TMI and TMVII in LPA1 is indeed smaller and less variable compared to that observed for S1P1. The distances diverge most noticeably at the extracellular tip of the helices, with a difference of approximately 7 Å between LPA1 and S1P1 in the absence of ligand (Figure 4C). Overall, these results strengthen the conclusion that sequence differences between TMI and TMVII on LPA1 and S1P1 result in an altered ligand access path, whereby LPA1 preferentially receives ligands from the extracellular milieu compared to membrane access for S1P1 ligands.
Figure 4.
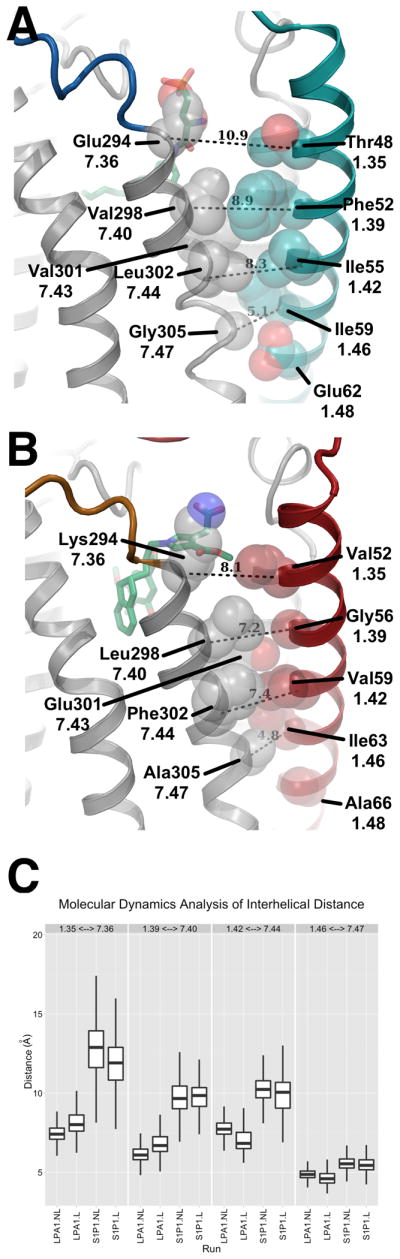
Analysis of the potential ligand entry for S1P1 and LPA1. (A). The position of TMI in S1P1 is shifted away from TMVII with a distance of 11 Å as measured between the Cα of Thr481.35 and Glu2947.36, this helical position allows access of ligand (ML056 shown in green carbon sticks) in the plasma membrane to the binding pocket of S1P1. (B). The position of TMI in LPA1 is closer to TMVII closing access of ligand (ONO-9780307 shown in green carbon sticks) in the plasma membrane to the binding pocket. The closer packing of TMI relative to S1P1 may be driven by the presence of Glu3017.43 which forms a hydrogen bond interaction with the backbone of Gly561.39 and Val591.42. (C). Molecular dynamics analysis of the distance between TMI and TMVII measured at four locations represented by a box plot for each measurement with the minimum and maximum indicated by vertical lines. The measurements correspond to the distances in shown in A monitored over the course of the simulation. The distance between the two helices is consistently larger and more variable for S1P1 than LPA1 both with and without ML056 and ONO-9780307 ligands respectively.
LPA1 ligand binding interactions with ONO-9780307, ONO-9910539 and ONO-3080573
Despite similar endogenous ligands, similar overall structural characteristics, and mutagenesis studies suggesting simple interchangeability between LPA1 and S1P1 (Wang et al., 2001), structural comparisons of the pocket shape and polarity revealed substantial divergence between the two receptors. At the top of polar region of the binding pocket, the carboxylic acid of ONO-9780307 interacts through polar and ionic bonds with residues His40, Lys39 and Tyr34, all of which are located on the N-terminal capping helix and ECL0 loop. Across the LPA family, both Lys39 and Tyr34 are highly conserved, however, His40 is unique to LPA1 and its protonation may be important for high affinity interactions with the carboxylic acid of ONO-9780307. Although direct experimental support is needed to draw definitive conclusions, calculations of binding energy between wild-type LPA1 with a protonated His40 and ONO-9780307 compared to a His40Ala in-silico mutation showed a greater than 1 kcal/mol difference in binding affinity supporting the hypothesis that this position could be partially responsible for the observed selectivity of this class of antagonists for LPA1 over LPA2 and LPA3 (Table S2)(Beard et al., 2013). The polar binding pocket continues on TMIII, where Arg1243.28 and Gln1253.29 form ionic and polar interactions with the carboxylic acid and chiral hydroxyl group of ONO-9780307, respectively. From TMVII, Glu2937.35 stabilizes the position of Lys39 but does not directly interact with the ligand (Figure 5A). The binding pocket surface formed by TMIII and TMV presents a significantly enhanced polar environment when compared to S1P1, consisting of an Asp1293.33 on TMIII and Trp2105.43 on TMV, which presents its indole nitrogen for hydrogen bonding interactions with the antagonist ligands (Figure 5A). Ligand design strategies for engaging this polarity while reducing lipophilicity were implemented with ONO-9910539, which features an acetyl group in the para position of the phenyl ring, placing a hydrogen bond acceptor for interaction with the indole nitrogen of Trp2105.43 (Figure 5B). The torsional strained bioactive conformation of ONO-9780307 helps to fill the large spherical binding pocket, placing the indane ring adjacent to Gly2746.51 on TMVI (Figure 1B and 5A). Ligand design strategies were implemented for ONO-3080573, which alleviates this torsional strain by replacing the methylene linker with an ether moiety (Figure 5B). This replacement was coupled with replacement of the metabolically labile pyrrole ring found in ONO-9780307 and ONO-9910439 with a phenyl ring which differentially positions the carboxylic acid, breaking interactions with Arg1243.28, His40 and Tyr34 but gaining an interaction with Lys2947.36.
Figure 5.

Analysis of the ligand binding pocket of LPA1. (A). Detailed binding interactions for ONO-9780307 in the LPA1 pocket. Polar interactions are represented by dashed yellow lines, and calculated positions of polar hydrogens are shown to more accurately represent the hydrogen bonding network. The indane binding pocket is formed by Gly2746.51 and Leu2756.52 which are represented by van Der Waals spheres. Areas for improving ligand binding are indicated in red text, and red spheres indicate positions for additional polar interactions. (B). Binding pocket interactions for additional co-crystal structures of ONO-9910539 and ONO-3080573. ONO-9910539 has improved interactions due to the introduction of an acetyl group in the para position of the phenyl ring which interacts with Trp2105.43. ONO-3080573 reduces the torsional strain induced by the indane position in ONO-9780307 through the replacement of a methylene linker with an ether linkage. In addition replacement of the pyrrole ring with a para substituted phenyl ring alters the interactions of the acid group away from the phosphate binding pocket toward Lys2947.36. See also Figure S2, Table S1 and Table S2.
Intersecting endocannabinoid pharmacology
The additional polarity provided by Asp1293.33 and Trp2105.43 in the hydrophobic binding pocket may have implications for specifying the preference of LPA receptors for long unsaturated acyl chains, serve as a trigger for agonist induced conformational changes, and is interesting in the context of GPCR phylogenetic evolution. The presence of a tryptophan in this position only occurs in 1% of all class A receptors and is unique to the lysophospholipid and cannabinoid receptors (Van Durme et al., 2006). The most abundant endogenous agonist for the cannabinoid receptor CB1 is 2-arachidonyl glycerol (2-AG; 1,3-Dihydroxy-2-propanyl (5Z,8Z,11Z,14Z)-5,8,11,14-eicosatetraenoate) (Stella et al., 1997; Sugiura et al., 1997). Despite binding at a much lower affinity than the other identified endocannabinoid, anandamide, its higher relative abundance makes 2-AG a major signaling molecule for cannabinoid receptors (Stella et al., 1997). Interestingly, 2-AG is phosphorylated and converted to 2-arachidonyl phosphatidic acid (2-ALPA) through the action of monacylglycerol (MAG) kinase/acyl glycerol kinase (AGK) (Kanoh et al., 1986), producing a potential signaling molecule accommodated by the spherical LPA1 binding pocket, although the relative abundance of 2-ALPA has not been investigated. Furthermore, an alternative biosynthetic pathway has been reported for the production of anandamide that progresses through a lysophospholipid analog (pAEA), adding support for the idea of metabolic cross-talk between LPA and cannabinoid receptor systems (Figure 6A) (Liu et al., 2006).
Figure 6.

Pharmacologically pleitropic signaling lipids overlap through common acyl chain binding pockets in the LPA and cannabinoid receptors and enzymatic interconversion of polar head group. (A). The LPA, S1P and cannabinoid receptors are grouped in the same clade based on a sequence alignment of the transmembrane region. LPA and cannabinoid receptors are proposed to overlap in ligand specificity when binding polyunsaturated acyl chains delimited by different head group requirements. The biosynthetic map details the conversion of standard phospholipids to both LPA and CB receptor agonists through common pathways. Both known agonists for the cannabinoid system (2-AG and AEA) are one enzymatic step away from LPA ligands via phosphorylation of the head group (2-ALPA and pAEA). (B). Molecular modeling of the endogenous agonist lysophosphatidic acid with an 18 carbon acyl chain (LPA). In the presence of agonist, both Trp2716.48 and Trp2105.43 are predicted to change rotameric states to increase the volume of the binding pocket and direct the respective aromatic π clouds toward the bound ligand. The predicted agonist binding pocket is capable of docking 2-ALPA and pAEA which are generated through enzymatic action from endogenous cannabinoids. (C). Conformational root mean square fluctuation of LPA over the course of a 100 ns molecular dynamics simulation. The orientation of the ligand is consistent throughout the calculation indicating a reasonable starting point for modeling agonist interactions. (D). Analysis of intracellular signaling and cellular functions in B103 cells heterologously expressing full-length wild-type LPA1. The data are presented as mean normalized values (± SEM) to maximum responses induced by 3 μM 1-oleoyl-LPA (n =3) for Ca2+ mobilization assay, and by 10 μM forskolin (n =3) for cAMP assay. For the Ca2+ mobilization assay EC50 values for LPA 18:1 sn1 (n=3), 1-ALPA (n=3), 2-ALPA (n=2) and pAEA (n=2) are 146 ± 74 nM, 1125 ± 1484 nM, 98 ± 143 nM and 2120 ± 3885 nM respectively. For the cAMP assay EC50 values for LPA 18:1 sn1 (n=2), 2-ALPA (n=2) and pAEA (n=2) are 0.345 ± 0.202 nM, 0.302 ± 0.54 nM and 1.50 ± 2.04 nM respectively. Neurite retraction was quantified under microscopic observation. Filamentous (F)-actin stress fibers, produced by Rho GTPase activation like neurite retraction, were also produced by pAEA stimulation. The representative cell morphologies were shown (bars, 50 μm). The F-actin stress fiber formation was observed after 15 min treatment with LPA and pAEA (bars, 10 μm).
The ability of lysophosphatidic acid derivatives of cannabinoid ligands to bind and activate LPA1 was tested both with 2-ALPA and pAEA using molecular modeling (Figure 6B and C) and in receptor signaling assays previously used to define LPA1 as an LPA receptor (Figure 6D). A model for lipid agonist binding generated through molecular modeling was used to dock the endogenous ligands into the LPA1 binding pocket. The slight expansion of the binding pocket through rotameric shifts of Trp2105.43 and Trp2716.48, and the exposure of the π clouds of their respective indole rings enabled favorable interactions with the double bonds of the phosphorylated cannabinoid ligands (Figure 6B). The ability of both 2-ALPA and pAEA to bind in the modeled agonist pocket without significant ligand strain, supports the notion that the hydrophobic binding pockets of LPA1 and CB1 can indeed bind the same, poly-unsaturated acyl chains with metabolically interconvertible head groups.
The molecular modeling provided sufficient rationale for testing both 2-ALPA and pAEA in LPA1 signaling assays. Remarkably, all phosphorylated ligands produced robust signaling as judged by calcium response and cAMP inhibition in stable cell lines heterologously expressing LPA1 (Fukushima et al., 1998). Further, a neurite retraction assay which measures cell morphology changes mediated by LPA1 showed robust activation by pAEA (Figure 6D). Neurite retraction involves activation of the small GTPase Rho that can also activate F-actin stress fibers in adherent cells, and this classic response was also activated by pAEA. Together these results highlight the selectivity of the phosphate headgroup on LPA1 in addition to the promiscuity of the binding site for acyl chain geometry and degree of saturation, wherein chemically distinct ligands that are capable of accessing this space can produce common responses of receptor activation and down-stream signaling.
Discussion
The growing family of GPCRs mediating membrane-derived lysophospholipid signals (Kihara et al., 2014) is remarkable for its ability to respond to diverse changes in the cellular milieu through multiple signaling mechanisms, and to discriminate structurally similar molecules. The ability of antagonist structures to inform receptor agonist binding is atypical, yet may reflect the evolutionary selection of binding pockets for lipid ligands that are produced from precursor reservoirs within which the receptors are embedded, which may further contribute to constitutive or inverse agonist activities (Bond and Ijzerman, 2006). A somewhat surprising result was the structural preference of LPA1 for ligand access from the extracellular milieu, rather than via the membrane as was reported for S1P1. Nevertheless, this structural prediction is supported by the biological existence and routine experimental use of albumin-bound LPA (Postma et al., 1996) to activate LPA1 (Fukushima et al., 1998), and is further consistent with the proposed delivery of LPA by its major extracellular biosynthetic enzyme, autotaxin, via a hydrophobic channel identified in its crystal structure (Nishimasu et al., 2011). The structure of LPA1 identified binding interactions between antagonist and His40, having a calculated pKa of 6.8 and 4.5 in the presence and absence of ligand respectively. Although speculative, computational analysis indicates that full protonation of His40 increases ligand binding affinity by more than 1kcal/mol (Beard et al., 2013). If substantiated through direct experimental observation, this could further support the hypothesis that the neoplastic activity of LPA1 is enhanced (Hama et al., 2004; Hausmann et al., 2011; Mills and Moolenaar, 2003; Perrakis and Moolenaar, 2014) in the acidic environment associated with hypoxic tumors(Herr et al., 2011; Justus et al., 2013; Kim et al., 2006; Tannock and Rotin, 1989; Wojtkowiak et al., 2011). Structural analyses predicts a functional complementarity and/or redundancy between CB1 and LPA1 depending on the balance of phosphorylation states for AEA and 2-AG in particular since it is a favored substrate of MAG kinase, at least in the brain (Kanoh et al., 1986). The interplay of these receptor-ligand systems may contribute to phenotypic variability seen amongst LPA1 knock-out strains and variable null-mutant survival within susceptible strains that are attributable to brain phenotypes (Contos et al., 2000; Matas-Rico et al., 2008), since brain expression of both receptors, along with ligand metabolism, is widespread and can overlap as occurs in oligodendrocytes (Benito et al., 2007; Weiner et al., 1998). Complementary access to LPA1 through phosphorylated CB1 ligands - and vice versa - represent examples of a single molecular species that could serve as both a primary receptor modulator and simultaneous prodrug for a different receptor, as represented by the endocannabinoids acting at their receptors, with phosphorylation producing selective LPA receptor ligands. Currently, compounds developed to target LPA receptors have entered Phase II clinical trials for pulmonary fibrosis and systemic sclerosis. Compounds exhibiting antagonist and/or agonist activity for both the CB1 and LPA1 receptors have yet to be tested but may be superior for certain indications.
Experimental Procedures
Ligand selection optimization and stability analysis
Compounds based on a 3-pyrrolylpropionic acid core were selected from a library of previously developed LPA1 antagonists based on superior receptor stability induction properties. From this initial screen, ONO-9780307 was selected for structural determination. Analysis of the binding pocket revealed three regions for improved interactions. ONO-9910539 was designed to improve polar interactions with Trp2105.43 through the introduction of an acetyl moiety on the phenyl ring, and ONO-3080573 was designed to reduce torsional strain associated with the bioactive conformation of the scaffold through the introduction of an ether linkage to the indane ring system (Figure 5B and Table S1).
To measure the effect of compounds on the stability of the receptor, each compound was tested using a fluorescence based SEC stability assay. Briefly, after heating the GFP-tagged protein in the presence of ligand, an SEC profile of each data point was analyzed for fraction of folded protein which was plotted as a function of temperature or time to determine approximate melting temperature or half-life respectively (see also Extended experimental procedures and Figure S1).
Crystallization and diffraction analysis of LPA1-bRIL and dsLPA1-bRIL
Synthetic complementary DNA encoding the full length LPA1 receptor was generated with a stabilized apocytochrome c (bRIL) protein inserted within the third intracellular loop at positions V232 and R248. Baculovirus directed expression in Spodoptera frugiperda (Sf9) insect cells was driven by the ie1GP64 promoter with a cleavable HA signal sequence on the N-terminus of the receptor and a cleavable purification tag consisting of a 10x poly histidine tag upstream of a FLAGTM epitope tag. The resulting construct (LPA1-bRIL) was expressed in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac baculovirus expression system (Invitrogen) at a yield of ∼1mg/L and purified using IMAC chromatography (see also Extended experimental procedures). Protein samples of LPA1-bRIL and dsLPA1-bRIL were reconstituted into lipidic cubic phase (LCP) and crystallization set-ups were performed at room temperature (∼20 °C). Data-collection quality crystals were obtained over a period of 30 days from precipitant condition containing 0.1 M sodium citrate (pH 5.5), 34 - 38% (v/v) PEG400 and 200 mM ammonium acetate (Figure 2D). Crystals were harvested directly from LCP matrix and flash frozen in liquid nitrogen for diffraction analysis. X-ray diffraction data were collected on the 23ID-D/B beamline (GM/CA-CAT) at the Advanced Photon Source (Argonne, IL) using a 10 μm diameter minibeam with a MarMosaic 300 CCD detector. A rastering and data-collection strategy was followed as previously described (Cherezov et al., 2009; Hanson et al., 2012). Unattenuated exposures of 5-10 seconds per degree were required to observe diffraction to ∼3 Å resolution. Due to the limited amount of data collected per crystal, a protocol similar to that developed for S1P1 diffraction analysis was utilized to assemble the data. After data integration and scaling the structures were solved using molecular replacement followed by multiple cycles of refinement and rebuilding in order to generate the final model (see Extended experimental procedures). Statistics are presented in the crystallographic summary table (Table 1).
Table 1.
Crystallographic data collection and refinement statistics. Data for high resolution shells is shown in parenthesis where applicable.
| Data Collection and Refinement Statistics | |||
|---|---|---|---|
| Ligand | ONO-9780307 | ONO-9910539 | ONO-3080573 |
| Resolution Cutoff (Å) | 3 | 2.9 | 2.9 |
| Number of Crystals | 86 | 53 | 39 |
| Data Collection Statistics | |||
| Space Group | P212121 | P212121 | P212121 |
| Cell Dimensions a,b,c (Å) | 34.3, 112.2, 154.6 | 34.6, 112.4, 155.7 | 34.4, 111.9, 154.0 |
| Number of Reflections Measured | 42,304 | 27,843 | 69,138 |
| Number of Unique Reflections | 10,599 | 10,883 | 12,701 |
| Resolution (Å) | 45 - 3.0(3.16 - 3.0) | 47 - 2.9(3.1 - 2.9) | 47 - 2.9(3.1 - 2.9) |
| Rmerge | 0.19 (0.46) | 0.14 (0.49) | 0.13 (0.59) |
| Mean I/σ(I) | 4.7 (1.4) | 4.0 (1.0) | 7.2 (1.3) |
| Completeness | 85 (75) | 78 (62) | 92 (80) |
| Redundancy | 4.0 (2.1) | 2.6 (1.7) | 5.4 (2.3) |
| CC1/2 | 0.99(0.27) | 0.99(0.38) | 1.0(0.56) |
| Refinement Statistics | |||
| Resolution (Å) | 30 - 3.0 | 30 - 2.9 | 30 - 2.9 |
| Number of Reflections (test set) | 10,576 (531) | 11,627 (584) | 12,660 (625) |
| Rwork/Rfree (%) | 25.4/28.1 | 26.5/27.9 | 27.1/28.5 |
| Number of Atoms | |||
| Protein | 2,991 | 2,977 | 3,015 |
| Ligand | 38 | 41 | 38 |
| Lipid and other | 18 | 21 | 21 |
| Average B Factor (Å2) | |||
| Protein | 73.2 | 95.1 | 88.4 |
| bRIL | 95.5 | 114.8 | 120.2 |
| Ligand | 48.6 | 70.9 | 65.4 |
| Lipid and other | 62.3 | 82.2 | 71.3 |
| R.M.S. Deviation | |||
| Bond Lengths (Å) | 0.01 | 0.009 | 0.009 |
| Bond Angles (°) | 1.08 | 0.92 | 0.87 |
| Ramachandran Plot Statistics (%) | |||
| Favored Regions | 96.3 | 96.8 | 97.3 |
| Allowed Regions | 3.7 | 3.2 | 2.7 |
| Disallowed Regions | 0 | 0 | 0 |
Stability engineering using disulfide bond addition
Candidates for disulfide bond engineering were generated computationally using algorithms implemented in BioLuminate (version 1.0, Schrödinger, LLC, New York, NY, 2012) and similar to previously described procedures (see also Extended experimental procedures)(Salam et al., 2014). Five potential candidates were identified on the extracellular half of the receptor consisting of two cysteine mutations per site to facilitate the formation of an additional disulfide bond. After generation of the mutant constructs and testing for expression, the double mutant D204C and V282C was selected for crystallization studies. This candidate was selected due to the enhanced expression profile as compared to the other four constructs, which showed little to no expression, as well as an enhanced thermal stability profile as characterized by the SEC thermal stability assay using highly purified receptor without a GFP tag (see also Extended experimental procedures).
Molecular modeling and dynamics studies
Crystal structures of the S1P1 (PDB ID: 3V2Y) and LPA1 were prepared using the Schrödinger Protein Preparation Wizard (Sastry et al., 2013) and embedded into a pre-equilibrated POPC (1-palmytoil-2-oleoylsn-glycero-3-phosphatidylcholine) lipid bilayer and solvated with water using the Desmond System Builder. Chloride ions were added to neutralize the system and the ionic strength was set to 0.15 M. The OPLS 2.1 force field (“OPLS2.1, Schrödinger, Inc., New York, NY, 2014,”), (Shivakumar et al., 2012) was utilized for all calculations and systems were relaxed using the default membrane relaxation protocol as implemented within the Schrödinger package. Molecular Dynamics simulations were performed in the NPT ensemble at 300K and 1 atm pressure using Desmond (“Desmond Molecular Dynamics System, version 3.9, D. E. Shaw Research, New York, NY, 2014. Maestro-Desmond Interoperability Tools, version 3.9, Schrödinger, New York, NY, 2014.,”). The two receptors were analyzed both with and without ligands with Van der Waals and short range electrostatic interactions cut off at 9 Å and smooth particle mesh Ewald (PME) (Essmann et al., 1995) method employed for calculation of long range electrostatic interactions. The reversible reference system propagation algorithm multiple time step approach was used with a time step of 2 ps and long-ranged electrostatic interactions were computed every 6 ps (Tuckerman et al., 1992). The temperature and pressure were controlled using a Nose-Hoover chain thermostat (Martyna et al., 1992) and Martyna-Tobias-Klein (Martyna et al., 1994) barostat, respectively. Three independent simulations of 100 ns each were performed for each system on a Nvidia GTX GPU card with coordinates saved every 1.2 ps for subsequent analysis. Molecular dynamics used for docking and modeling studies utilized similar system parameters but with a 1.2 ns simulation. Coordinates were saved every 1.2 ps and clustered by root mean square deviation of the residues within a 4 Å radius of the ligand. Representative structures of each cluster were tested for ability to dock endogenous ligands with minimal torsional strain.
Change in binding affinity towards the ligand ONO-9780307 caused by either deprotonation of the residue His40 or its mutation to alanine was calculated using the Residue Scanning functionality in BioLuminate (version 1.7, Schrödinger, LLC, New York, NY, 2014) which incorporates the Prime MM-GBSA approach (Beard et al. 2014). A cutoff of 1 kcal/mol was used as a threshold for defining potential effect from perturbation.
Quantum mechanical calculations for determining the energy penalty associated with the indane position observed in the crystal structures were carried out after geometry optimizations using Jaguar 8.6 (Schrödinger, LLC, New York, NY, 2014) at the B3LYP/6-31G** level of theory with and without torsional constraints on the indane ring region of the compound.
Functional analysis studies
HA-tagged full-length human receptor (LPA1) and disulfide stabilized full-length human receptor (dsLPA1), DNAs were constructed and cloned into pCXN2.1 (Niwa et al., 1991). Rat neuroblastoma B103 cells, maintained in complete media were transfected with each plasmid using GeneJET Plus (Thermo Scientific) (Fukushima et al., 1998). After 24 hr incubation, media was replaced with complete media containing 1 mg/ml G418 (Life Technologies) and the cells were cultured for an additional 2 weeks. Cells labeled with anti-HA antibody (Roche) followed by PE-conjugated anti rat IgG (eBioscience) as primary and secondary antibodies, respectively were sorted by FACS Aria II (BD Biosciences) using a PE-highly-positive gate, collected, and cultured in complete media containing Penicillin-Streptomycin (Life Technologies) to create stable lines.
For Ca2+ measurement, the stable cell lines were starved with FreeStyle™ 293 Expression Medium (Life Technologies) for at least 1 hr before loading dye according to the manufacture's instruction with minor modifications (FLIPR Calcium 4 Assay Kit; Molecular Device). Intracellular Ca2+ mobilization was monitored in response to increasing ligand concentration using a scanning fluorimeter (FLEXstation 3; Molecular Devices) at an excitation wavelength of 485 nm and an emission wavelength of 525 nm.
For cAMP assay, stable cell lines were starved with FreeStyle™ 293 Expression Medium (Life Technologies) overnight. The cells were incubated with ligands for 30 min at room temperature in Hank's Balanced Salt Solution containing 10 μM forskolin and 0.5 mM 3-isobutyl-1-methylxanthine. Intracellular cAMP levels were determined using a cAMP-Screen® System (Applied Biosystems) according to the manufacturer's instructions.
For neurite retraction and stress fiber formation assay, stable cell lines were starved with FreeStyle™ 293 Expression Medium (Life Technologies) overnight. The cells were incubated with ligands for 15 min at 37°C, washed and then fixed with 2% paraformaldehyde. Cells with round morphology lacking neurite extensions were counted and represented as a percentage of the observed cells. The f-actin stress fiber formation was studied using rhodamine phalloidin staining. Fixed cells were washed with 0.3% Triton-X100 in PBS, and stained with 1μg/mL rhodamine-phalloidin for 30 min at room temperature. After washing, the cells were mounted using Vectorshield (Vector Laboratory). Images were captured using a Nikon C2 laser-scanning confocal microscope.
Standard lipids were obtained from commercial sources: 1-oleoyl-LPA and 1-arachidonoyl-LPA (Avanti Polar Lipids), AEA, 2-AG and pAEA (Cayman Chemical). Enzymatic synthesis was employed for 2-arachidonoyl-LPA that was enzymatically synthesized from 1,2-diarachidonoyl-sn-glycero-3-phosphate (diarachidonoyl-PA) (Avanti Polar lipids) using Rhizopus arrhizus lipase (Sigma) (see also Extended experimental procedures).
Supplementary Material
Acknowledgments
The authors thank Angela Walker for critical review of this manuscript and J. Smith, R. Fischetti, and N. Sanishvili for assistance in development and use of the minibeam and beamtime at GM/CA-CAT beamline 23-ID at the Advanced Photon Source, which is supported by National Cancer Institute grant Y1-CO-1020 and NIGMS grant Y1-GM-1104. This work was supported in part by the NIH grants MH051699, NS082092 and NS084398 to JC, and U54 GM094618 to RCS (target GPCR-235).
Footnotes
Accession Numbers : Coordinates and structure factors have been deposited in the PDB as LPA1-ONO-9780307 (4Z34); LPA1-ONO-9910539 (4Z35); and LPA1-ONO-3080573 (4Z36).
Supplemental Information: Supplemental information includes extended experimental procedures, two figures and two tables and can be found with this article online.
Author Contributions: JEC led the structure determination efforts. CBR led the construct development and expression efforts. DW performed the molecular dynamics simulations. MT and HK designed and synthesized antagonists. RO assisted in structural analysis for compound optimization efforts. SN evaluated biological effect of antagonists. GAR performed the ligand stability screening and assisted in crystallization efforts. MM and MTG assisted in purification and crystallization efforts. GWH finalized the structural refinement and quality control of the crystallization data. JV performed cloning for the functional analysis studies. CR developed large-scale expression protocols. YK and HM carried out the functional analyses, and with JC, conceived and wrote portions of the manuscript. RCS conceptualized the project and wrote portions of the manuscript. MAH led the project and wrote the main manuscript.
References
- Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Aoki J. Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol. 2004;15:477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Beard H, Cholleti A, Pearlman D, Sherman W, Loving KA. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PloS one. 2013;8:e82849. doi: 10.1371/journal.pone.0082849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito C, Romero JP, Tolon RM, Clemente D, Docagne F, Hillard CJ, Guaza C, Romero J. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci. 2007;27:2396–2402. doi: 10.1523/JNEUROSCI.4814-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond RA, Ijzerman AP. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci. 2006;27:92–96. doi: 10.1016/j.tips.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 micron size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Hla T, Spiegel S, Moolenaar WH. Lysophospholipid Receptors: Signaling and Biochemistry. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2013. pp. i–xviii. [Google Scholar]
- Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97:13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- Fukushima N, Kimura Y, Chun J. A single receptor encoded by vzg-1/lpA1/edg-2 couples to G proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc Natl Acad Sci U S A. 1998;95:6151–6156. doi: 10.1073/pnas.95.11.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, et al. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM, van Meeteren LA, Houben AJ, van Zeijl L, et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nature Struct Mol Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr KJ, Herr DR, Lee CW, Noguchi K, Chun J. Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc Natl Acad Sci U S A. 2011;108:15444–15449. doi: 10.1073/pnas.1106129108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa D, Hashidate T, Shimizu T, Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res. 2014;55:799–807. doi: 10.1194/jlr.R046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nature Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Justus CR, Dong L, Yang LV. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front Physiol. 2013;4:354. doi: 10.3389/fphys.2013.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoh H, Iwata T, Ono T, Suzuki T. Immunological characterization of sn-1,2-diacylglycerol and sn-2-monoacylglycerol kinase from pig brain. J Biol Chem. 1986;261:5597–5602. [PubMed] [Google Scholar]
- Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Sengupta S, Berk M, Kwak YG, Escobar PF, Belinson J, Mok SC, Xu Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006;66:7983–7990. doi: 10.1158/0008-5472.CAN-05-4381. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna GJ, Klein ML, Tuckerman M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J Chem Phys. 1992;97:2635–2643. [Google Scholar]
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- Matas-Rico E, Garcia-Diaz B, Llebrez-Zayas P, Lopez-Barroso D, Santin L, Pedraza C, Smith-Fernandez A, Fernandez-Llebrez P, Tellez T, Redondo M, et al. Deletion of lysophosphatidic acid receptor LPA(1) reduces neurogenesis in the mouse dentate gyrus. Mol Cell Neurosci. 2008;39:342–355. doi: 10.1016/j.mcn.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nature Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- Mirendil H, Lin ME, Chun J. Lysophospholipid Receptors: Signaling and Biochemistry. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2013. Lysophosphatidic Acid (LPA) Receptor Signaling; pp. 1–39. [Google Scholar]
- Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, Nureki O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nature Struct Mol Biol. 2011;18:205–212. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Moolenaar WH. Autotaxin: structure-function and signaling. J Lipid Res. 2014;55:1010–1018. doi: 10.1194/jlr.R046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma FR, Jalink K, Hengeveld T, Bot AG, Alblas J, de Jonge HR, Moolenaar WH. Serum-induced membrane depolarization in quiescent fibroblasts: activation of a chloride conductance through the G protein-coupled LPA receptor. EMBO J. 1996;15:63–72. [PMC free article] [PubMed] [Google Scholar]
- Salam NK, Adzhigirey M, Sherman W, Pearlman DA. Structure-based approach to the prediction of disulfide bonds in proteins. Protein Eng Des Sel. 2014;27:365–374. doi: 10.1093/protein/gzu017. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Shivakumar D, Harder E, Damm W, Friesner RA, Sherman W. Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J Chem Theory Comput. 2012;8:2553–2558. doi: 10.1021/ct300203w. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kodaka T, Kondo S, Nakane S, Kondo H, Waku K, Ishima Y, Watanabe K, Yamamoto I. Is the cannabinoid CB1 receptor a 2-arachidonoylglycerol receptor? Structural requirements for triggering a Ca2+ transient in NG108-15 cells. J Biochem. 1997;122:890–895. doi: 10.1093/oxfordjournals.jbchem.a021838. [DOI] [PubMed] [Google Scholar]
- Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nature Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97:1990–1998. [Google Scholar]
- Van Durme J, Horn F, Costagliola S, Vriend G, Vassart G. GRIS: glycoprotein-hormone receptor information system. Mol Endocrinol. 2006;20:2247–2255. doi: 10.1210/me.2006-0020. [DOI] [PubMed] [Google Scholar]
- van Loenen PB, de Graaf C, Verzijl D, Leurs R, Rognan D, Peters SLM, Alewijnse AE. Agonist-dependent effects of mutations in the sphingosine-1-phosphate type 1 receptor. European Journal of Pharmacology. 2011;667:105–112. doi: 10.1016/j.ejphar.2011.05.071. [DOI] [PubMed] [Google Scholar]
- Wang DA, Lorincz Z, Bautista DL, Liliom K, Tigyi G, Parrill AL. A single amino acid determines lysophospholipid specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J Biol Chem. 2001;276:49213–49220. doi: 10.1074/jbc.M107301200. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Hecht JH, Chun J. Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp Neurol. 1998;398:587–598. [PubMed] [Google Scholar]
- Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8:2032–2038. doi: 10.1021/mp200292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Hama K, Contos JJ, Anliker B, Inoue A, Skinner MK, Suzuki H, Amano T, Kennedy G, Arai H, et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435:104–108. doi: 10.1038/nature03505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung YC, Mutoh T, Lin ME, Noguchi K, Rivera RR, Choi JW, Kingsbury MA, Chun J. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Science Transl Med. 2011;3:99ra87. doi: 10.1126/scitranslmed.3002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.