

Abstract
A surface-enhanced Raman scattering (SERS) biosensor has been developed by incorporating a gold nanohole array with a SERS probe (a gold nanostar@Raman-reporter@silica sandwich structure) into a single detection platform via DNA hybridization, which circumvents the nanoparticle aggregation and the inefficient Raman scattering issues. Strong plasmonic coupling between the Au nanostar and the Au nanohole array results in a large enhancement of the electromagnetic field, leading to amplification of the SERS signal. The SERS sensor has been used to detect Ag(i) and Hg(ii) ions in human saliva because both the metal ions could be released from dental amalgam fillings. The developed SERS sensor can be adapted as a general detection platform for non-invasive measurements of a wide range of analytes such as metal ions, small molecules, DNA and proteins in body fluids.
Introduction
Heavy metals1–5 are routinely released into the ecosystem from both human and natural resources, ultimately accumulating in the human body via the food chain.6,7 Heavy metals such as mercury, lead, chromium and arsenic are acutely toxic to humans, but can only be detected in the body through physically invasive blood8,9 or privacy invading urine testing.10 Saliva is an alternative, non-invasive testing base from both the physical and privacy viewpoints,11 making it an excellent candidate for measurement of heavy metal uptake in the body. On the other hand, silver–mercury amalgams are widely used as dental fillings.12,13 The Ag(i) and Hg(ii) ions can release into saliva from the dental fillings.12 Therefore, it is important to monitor the Ag(i) and Hg(ii) ions in human saliva, especially for the patients with silver–mercury amalgams.
Currently metal ions are measured quantitatively by large-scale analytical techniques such as inductively coupled plasma mass spectrometry (ICP-MS) and atomic absorption spectroscopy14 which are performed in a centralized laboratory, making them unsuitable for rapid monitoring of metal ion exposure in the field or in point-of-care settings. Therefore, various portable devices such as electrochemical, surface plasmon resonance (SPR), surface-enhanced Raman scattering (SERS), colorimetric and fluorescent sensors have been developed for metal ion detection.3,15–18 Among these sensors, SERS stands out for its molecular distinguishing (or fingerprinting) and anti-interference capabilities,19–23 both of which allow detection in complex matrices such as body fluids.
Plasmonic Au and Ag colloidal particles have previously been used as SERS substrates to detect metal ions,24–27 but low sensitivity, instability and poor repeatability remain a concern, especially in body fluids where a high ionic strength and the presence of abundant bio-molecules (blood, serum, urine and saliva) aggravate these problems. These problems can be partially overcome, however, by the use of a sandwich-structured colloidal SERS substrate/probe in which the Raman reporter molecules are sandwiched between a plasmonic Au core and a thin silica shell.28 The sandwich-structured SERS substrates/probes are water-soluble and stable in aqueous solution even with a high ionic strength, which significantly improves the repeatability of measurements. Solid-state chip-based SERS substrates with periodic plasmonic patterns can further mitigate these issues, improving both sensitivity and repeatability.29–32 For example, two-dimensional (2D) periodic nanostructures support both localized surface plasmon resonance (LSPR) and surface plasmon polaritons (SPP),33–35 generating a high-density of “hot spots”, which enhance the SERS intensity, boosting signals beyond that of colloids.
In the present work, a metal ion biosensor is constructed by coupling a sandwich-structured SERS probe to a gold nanohole array pattern. This strategy combines the advantages of stability and increased sensitivity of the sandwich-structured probe with the large area signal enhancement of the nanohole array. For the SERS probe, malachite green isothiocyanate (MGITC) Raman reporter molecules are sandwiched between a gold nanostar and a thin silica shell. The silica shell prevents leaching of the Raman reporter molecules, renders the water solubility of particles and provides a platform for bioconjugation.28 The LSPR peak of the gold nanostar is overlapped with the excitation laser source (785 nm), providing optimal SERS enhancement. The gold nanohole array LSPR peak is tuned to 785 nm, creating a spatially distributed electromagnetic (EM) field with which the gold nanostar can hybridize only when the heavy metal ions are present. It is expected that the distributed plasmonic “hot spots” in the periodic pattern leads to significant amplification of the SERS signal. By taking advantage of the large SERS amplification capability of the Au nanostar-nanohole couple, a sensitive metal ion biosensor is developed for detection of Ag+ and Hg2+ ions in human saliva.
Experimental section
Chemicals and reagents
DNA sequences of 5′-NH2-(CH2)6-CTCCCCATA-3′ and 5′-NH2-(CH2)6-TATCCCCAG-3′ were designed for Ag+ detection. The 5′-NH2-(CH2)6-GTCTTTCTG-3′ and 5′-NH2-(CH2)6-CAGTTTGAC-3′ were designed for Hg2+ detection. All DNA molecules were purchased from Integrated DNA Technologies, Inc. (IDT, Coralville, IA). Malachite green isothiocyanate (MGITC) was purchased from Molecular Probes, Inc. (Carlsbad, CA). Chloroauric acid trihydrate (HAuCl4·3H2O), trisodium citrate dihydrate (Na3C6H5O7·2H2O, ACS, 90.0+%), sodium hydroxide, silver nitrate (AgNO3, Premion, 99.995%), Na2HPO4 (99.0%), NaH2PO4 (99.0%), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O, 99.0%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 98+%), copper(II) nitrate hemi(pentahydrate) (Cu(NO3)2·2.5H2O), yttrium(III) nitrate hexahydrate (Y(NO3)3· 6H2O, 99.9%), and indium(III) nitrate hydrate (In(NO3)3·xH2O) were purchased from Alfa Aesar (Ward Hill, MA). Sodium hydrobromide (NaBH4), poly(vinylpyrrolidone) (PVP, 10 000), N,N-dimethylformamide (DMF), 11-mercaptoundecanoic acid (MUA), 11-mercapto-1-undecanol (MU), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98+%), and chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O) were purchased from Sigma-Aldrich (St Louis, MO). Nickel(II) nitrate 6-hydrate (Ni(NO3)2·6H2O) was purchased from ScholAR Chemistry (Rochester, NY). Aluminium nitrate nonahydrate (Al(NO3)3·9H2O, 99+%) was from Acros Organics (Fair Lawn, NJ). Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 98%) was from Strem Chemicals (Newburyport, NA). 3-Triethoxysilylpropyl succinic anhydride (TEPSA) was purchased from Gelest Inc. (Morrisville, PA). Deionized (D.I.) water was produced by using a Milli-Q Millipore system (18.2 MΩ cm, Millipore Corp., Billerica, MA) and was used in the whole experiment for washing or reactions. All chemicals were directly obtained from commercial vendors and used without further purification. Quartz slides were purchased from AdValue Technology (Tucson, AZ). Human saliva samples were de-identified. Saliva samples were screened for mercury and silver using an Agilent 7500CE ICP-MS from Exova (Santa Fe Springs, CA). Mercury in the saliva sample was beyond the instrument detection capability while silver was determined to be 0.12 ppb.
Apparatus
A JEOL JSM-7600F scanning electron microscope (SEM) and a JEOL JEM-2100F transmission electron microscope (TEM) were used to observe the morphology of gold nanoparticles and nanohole arrays. UV-Visible spectra of gold nanoparticles were acquired by using a Shimadzu UV-2550 spectrometer. Transmission spectra of the Au nanohole arrays were recorded using an Ocean Optics USB 4000 spectrometer and DT-MINI-2-GS. Raman spectra were recorded by using a Renishaw InVia Raman Microscope with an excitation wavelength of 785 nm. An Oxygen Plasma Asher (March PX-250 Plasma Asher) was used to remove the organic groups adsorbed on the Au film substrates and to etch the polystyrene microspheres.
Preparation of Au nanostar@MGITC@SiO2 sandwich nanoparticles
Gold nanostars were prepared as reported previously.28 A HAuCl4·3H2O aqueous solution (1 mL, 1 wt%) was first diluted with water (90 mL), followed by the injection of sodium citrate (2 mL, 38.8 mM). A freshly prepared NaBH4 solution was then added. The mixed solution was stirred overnight to form a seed solution. Subsequently, PVP was dissolved in 50 mM of the above-prepared seed solution and stirring was continued for 24 h. Afterwards, 82 μL of 50 mM HAuCl4 aqueous solution was mixed with 15 mL of PVP in DMF, followed by rapid addition of 43 μL of PVP-coated gold seed solution. The reaction lasted about 13 h. The as-prepared gold nanostar solution was washed with absolute ethanol and D.I. water three times, respectively, and then re-dispersed in D.I. water for further use.
To make the sandwich nanoparticles, 15 μL of MGITC was first added to 1 mL of Au nanostar solution (the optical density was adjusted to be around 2.5) under stirring for 30 min. 200 μL of 1% TEOS was then injected to grow the silica shell. The mixture was stirred continuously for 30 min. Finally, the solution was left to stand for 2 days before being washed with ethanol. The final product was dispersed in ethanol for further use.
Fabrication of Au nanohole arrays
Au nanohole arrays were fabricated on the quartz slides by nanosphere lithography (NSL).36 The quartz slides were first cleaned with a Piranha solution at 90 °C for 2 h (Precaution: the Piranha solution is very dangerous and caution must be taken while handling it), and subsequently sonicated in acetone, methanol and D.I. water. A monolayer of polystyrene (PS) microspheres (600 nm in diameter) was dip-coated on the quartz slides. After drying under ambient conditions, the monolayer-coated quartz slides were etched with oxygen plasma, followed by deposition of a 5 nm thick Ti layer and then a 45 nm thick Au layer with an e-beam evaporator. Finally, the Au nanohole array was obtained by removing the PS template by sonication in methanol.
Functionalization of Au nanostar@MGITC@SiO2 particles with DNA
The Au nanostar@MGITC@SiO2 sandwich particles were first diluted to 1.5 mL. 20 μL of TEPSA was then added. The mixed aqueous solution was incubated overnight to achieve COOH-terminated Au nanostar@MGITC@SiO2. After washing with ethanol, the precipitate was dissolved in 2 mL of the solution containing 50 mM NHS and 200 mM EDC. After incubation for 4 h, the COOH group was activated, and 50 μL of 20 μM ssDNA solution was then added. After overnight incubation, the solution was washed with a buffer solution, and added into the following buffer solution for further use. For Ag+ detection, the buffer solution used was a 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer solution containing 30 mM NaNO3, pH 7.0. For Hg2+ detection, the buffer solution employed was a 10 mM phosphate-buffered saline (PBS) solution, pH 7.0.
Functionalization of Au nanohole array chips
The Au nanohole array chips were cleaned by successive sonication in acetone, methanol and D.I. water for 10 min at each step. After being dried with nitrogen gas, the chips were cleaned by using an Oxygen Plasma Asher at 300 W for 2 min. The cleaned Au nanohole array chips were immersed in an aqueous solution containing 100 mM MUA and 100 mM MU overnight. The resulting MUA/MU functionalized substrates were washed with ethanol and D.I. water, and then incubated in 50 mM NHS and 200 mM EDC for activation. Afterwards, 50 μL of 20 μM ssDNA solution was added. After overnight incubation, the chips were successively washed with a buffer solution to remove the unattached ssDNA.
Finite-difference time-domain (FDTD) simulation
FDTD simulations were conducted with the commercially available Optiwave software. Simulation cells with 3 nm grid size were created to mimic the Au nanohole array as well as the Au nanostar. A monochromatic light source with 785 nm wavelength was used as the input plane wave to match the laser used in experiments. The refractive index was taken from the data of Palik.37 A refractive index of 1.53 was used for the quartz slides. Simulation was conducted using both x and y polarizations. Periodic boundary conditions were implemented to replicate the periodic nature of the Au nanohole arrays.
Results and discussion
Construction of SERS sensors
The bare gold nanostars were ∼80 nm in size (Fig. 1(a)). The silica shell in the Au nanostar@MGITC@SiO2 sandwich nanoparticles was ∼3 nm thick (Fig. 1(c) and (d)). The bare gold nanostars exhibited a LSPR peak centered at 785 nm (Fig. 1 (b)). Coating the Au nanostar surface with a thin silica layer led to a slight shift of the LSPR peak. The Au nanohole array had a hole diameter of 420 nm with a pitch (hole center-to-center distance) of 600 nm (Fig. 2(h)). The LSPR of the Au nanohole array can be found by reading the transmission valley or reflection peak. The transmission spectrum in Fig. 2(i) reveals that the LSPR was located at 785 nm, in agreement with the FDTD simulation result.
Fig. 1.

Au nanostar@MGITC@SiO2 sandwich nanoparticles. (a) SEM image of Au nanostars; (b) UV-Vis absorption spectra of the bare Au nanostar and the Au nanostar@MGITC@SiO2; (c) and (d) TEM images of the Au nanostar@MGITC@SiO2.
Fig. 2.
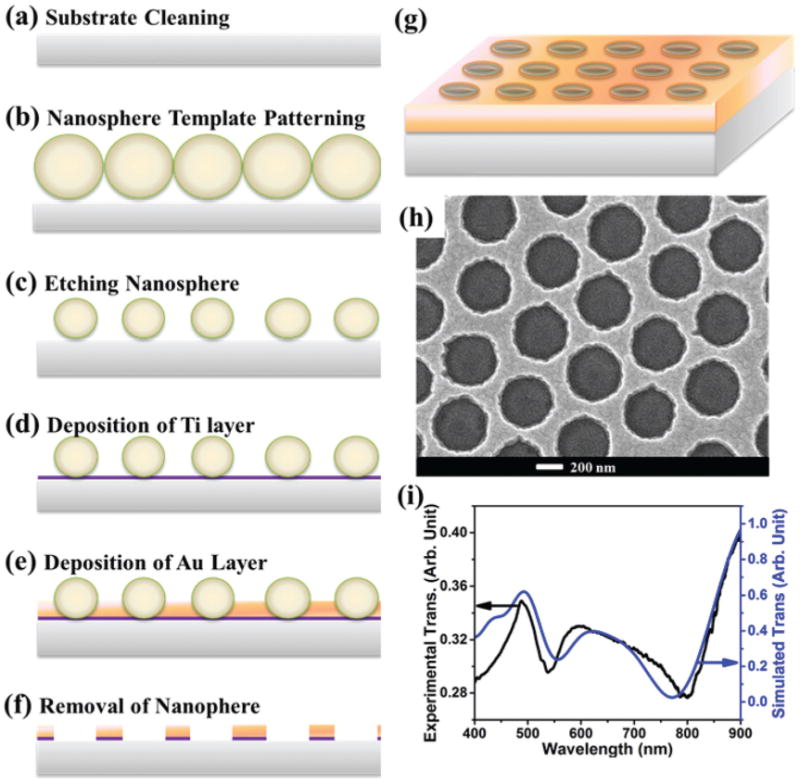
A gold nanohole array pattern. (a–g) Protocol for Au nanohole array fabrication; (h) SEM image of a gold nanohole array with a hole diameter of 420 nm at a pitch of 600 nm, and a film thickness of 50 nm; (i) experimentally measured (black curve) and FDTD simulated (blue curve) transmission of the Au nanohole array.
Fig. 3 shows the general experimental flow. To detect Ag+ ions, the Au nanostar@MGITC@SiO2 nanoparticles and the Au nanohole arrays were modified with single-stranded DNA (ssDNA) sequences of 5′-NH2-(CH2)6-CTCCCCATA-3′ and 5′-NH2-(CH2)6-TATCCCCAG-3′, respectively (Fig. 3). When the Ag+ ions were absent in the aqueous solution, the two ssDNAs cannot hybridize due to the presence of three mismatched C–C pairs. When the Ag+ ions are present, two ssDNAs hybridize to form a rigid double-stranded DNA (dsDNA) due to the strong affinity of C–Ag+–C (Fig. 3).38 To detect Hg2+ ions, the Au nanostar@MGITC@SiO2 nanoparticles and the Au nanohole array were functionalized with the ssDNA of 5′-NH2-(CH2)6-GTCTTTCTG-3′ and 5′-NH2-(CH2)6-CAGTTTGAC-3′, respectively. The Hg2+ ions were recognized by DNA via the formation of the T–Hg2+–T base pairs.39 The two different ions were tested to show that the sensor design can be easily converted to detect other types of analytes by simply changing the DNA.
Fig. 3.

The operating principle of the SERS sensor. The Au nanostar@MGITC@SiO2 sandwich nanoparticles and Au nanohole arrays were functionalized with ssDNA sequences. The two ssDNA sequences hybridize only when Hg2+ or Ag+ was present due to T–T or C–C mismatch.
During the operation of metal ion detection, first 35 μL of ssDNA-functionalized Au nanostar@MGITC@SiO2 nanoparticle solution was dropped on the ssDNA-functionalized Au nanohole array substrate, immediately followed by another drop of 35 μL of the sample containing the analyte. After incubation for 16 min, the Au nanohole array chip was rinsed with a buffer solution, and then dried with a nitrogen flow. Subsequently, the Raman spectrum was acquired. Since the number of dsDNA-bound SERS probes on the Au nanohole array pattern is proportional to the number of ions captured, the resulting SERS intensity correlates with the concentration of ions captured.
Detection of Ag+ ions
For detection of Ag(i), Ag(i) ions were dissolved in the 10 mM MOPS buffer solution. First the test conditions had to be optimized. The effects of the ionic strength and the pH value of the buffer solution on the sensing performance are shown in Fig. S1.† The sensing signal was found to be dependent on the incubation time. After incubation for 16 min, the sensing signal reached a constant. The final testing conditions were therefore in an optimized 10 mM MOPS buffer solution with 30 mM NaNO3 at pH 7.0 for 16 min of incubation time.
SERS spectra were acquired from the Au nanostar@-MGITC@SiO2 particles on the Au nanohole array pattern at different levels of Ag(i) ions in a 10 mM MOPS buffer solution containing 30 mM NaNO3 (Fig. 4(a)). The SERS peak at 1174 cm−1 was proportional to the logarithmic concentration of Ag+ (Fig. 4(b)). At a concentration of >10 nM, the SERS signal became saturated because the binding sites were fully occupied. The linear range was determined to be from 2 pM to 10 nM with a fitting equation (y = 1400x + 110, R2 = 99%), where y is the SERS intensity at 1174 cm−1, x is the logarithmic Ag(i) concentration (Fig. 4(b)). The performance was also compared with a SERS sensor constructed from a planar gold film chip instead of the nanohole array pattern (Fig. S2†). When the Au film was used, the slope of the calibration curve decreased (Fig. 4(b)), giving a fitting equation (y = 370x + 330, R2 = 98%) and indicating poorer sensitivity. This indicated that the Au nanohole array pattern improved the sensitivity of the SERS sensor due to its increased density of plasmonic “hot spots”. The limit of detection (LOD) was estimated with three times the standard deviation/slope (3σ/s) according to the International Union of Pure and Applied Chemistry (IUPAC) standard.40 The results showed a LOD of 1.7 pM for the Au nanohole array chip and 5 pM for the Au film chip.
Fig. 4.
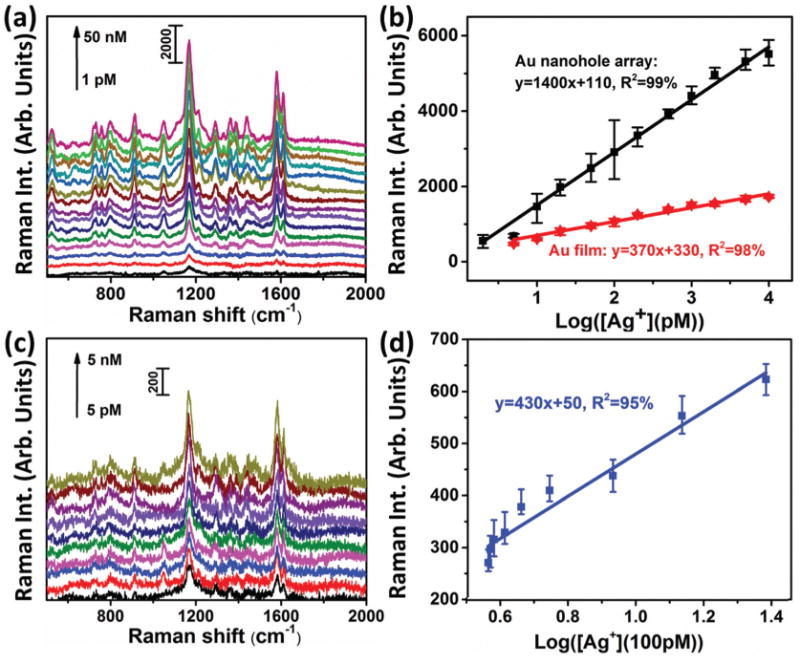
Detection of Ag(i) ions with the SERS sensor. (a) SERS spectra acquired from the Au nanostar@MGITC@SiO2 particles captured by the Au nanohole array pattern for different levels of Ag(i) ions in a 10 mM MOPS buffer solution containing 30 mM NaNO3; (b) fitting curves and equations of the SERS peak intensity at 1174 cm−1 vs. the logarithmic concentration of Ag(i) ions. The black curve is for the detection conducted on a Au nanohole array; the red curve is for the detection on a Au film. (c) SERS spectra obtained from the Au nanostar@MGITC@SiO2 nanoparticles captured by the Au nanohole array pattern for different levels of Ag(i) ions in a mixture of MOPS buffer (2/3 vol.) and human saliva (1/3 vol.); (d) the fitting curve and equation of the SERS peak intensity at 1174 cm−1 vs. the logarithmic concentration of Ag(i) ions.
A selectivity test was next conducted to check the anti-interference capability of the SERS sensor. Various metal ions such as Al3+, Ca2+, Cr3+, Cu2+, Fe3+, In3+, In3+, Ni2+, Y3+ and Zn2+ plus a mixture of the above ions with Ag+ were prepared with a concentration of 50 nM metal ions in the 10 mM MOPS buffer solution containing 30 mM NaNO3 at pH 7.0. The results showed that only the presence of Ag+ ions induced a high SERS intensity (Fig. S3†).
The Ag(i) ions were next spiked into a mixture of MOPS buffer (2/3 vol.) and human saliva (1/3 vol.). The SERS spectra obtained from these samples are shown in Fig. 4(c), and the SERS peak intensity at 1174 cm−1 is plotted as the logarithmic concentration of Ag(i) ions in Fig. 4(d). The fitting procedure gave an equation of y = 430x + 50, R2 = 95% in a linear range of 0.4-2.4 nM with a LOD of 0.17 nM.
Detection of Hg2+ in human saliva
To demonstrate the flexibility of the developed SERS sensor, the ssDNA sequences used were adapted to detect the Hg(ii) ions41 released alongside the Ag(i) ions in the Ag–Hg dental alloy saliva samples. The Hg2+ ions were first spiked into a mixture of PBS buffer (2/3 vol.) and human saliva (1/3 vol.) (Fig. 5(a)). The SERS peak at 1174 cm−1 again showed a linear dependence on the logarithmic concentration of Hg(ii) ions in the range of 5 pM (1 ppt) to 100 nM (20 ppb), yielding a calibration equation of y = 670x + 50, R2 = 98% (Fig. 5(b)). The LOD was estimated to be 2.3 pM based on three times the standard deviation/slope according to the IUPAC standard.40 Selectivity was also tested as shown in Fig. S4.† Studies have revealed that each additional amalgam filling increases the level of mercury in saliva by 7.5 nM (1.5 ppb) on average.41 For example, 9 amalgam fillings (mean number) in adults have yielded a median concentration of 58 nM (11.6 ppb) Hg(ii) in the pre-chewing saliva.42 Another study has investigated the Hg(ii) and Ag(i) levels in 53 patients and found that the medium values of Hg(ii) and Ag(i) levels in saliva were 47.5 nM (9.5 ppb) and 32 nM (3.2 ppb), respectively.43 Hence, the present SERS sensor can be used to measure the Hg(ii) level in human saliva of patients with the amalgam fillings.
Fig. 5.
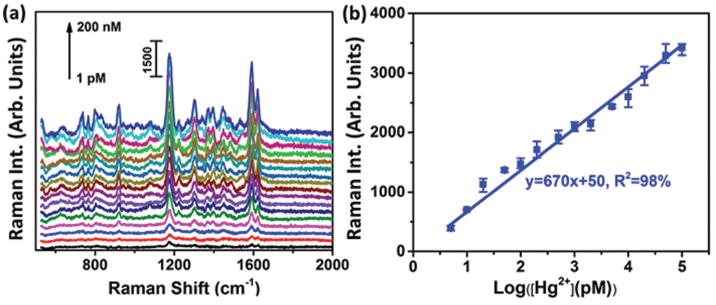
SERS sensor for Hg2+ detection. (a) SERS spectra of Au nanostar@MGITC@SiO2 on the Au nanohole array in the SERS sensor for the detection of Hg2+ in a mixed solution containing one portion of human saliva (1/3 vol.) and two portions of PBS buffer solution (2/3 vol.); (b) plots of the SERS peak intensity at 1174 cm−1 as a function of the logarithmic concentration of Hg2+.
FDTD simulations of the SERS sensor system
The success of the developed Raman sensor in detecting various analytes in a body fluid results from selectively coupling sandwich-structured SERS probes to a metal nanohole array. When the DNA and analyte change, the resulting field enhancement does not, making the stability against aggregation and large signal enhancement necessary for detection. FDTD simulations were performed to better understand the resulting EM field enhancement that occurs in the presence of an analyte.
The EM field was extracted for several nanostar–nanohole coupling arrangements in order to find the maximum enhancement which could be expected (Fig. 6). Four typical locations of the Au nanostar on the Au nanohole array pattern were investigated, as shown in Fig. 6. Point A is on the edge of the nanoholes. Point B is positioned in the middle of the gap between two nearest holes. Point C is located at the closest point between three nanoholes. Point D is at the center of a nanohole. The exact location of the nanostars cannot be imaged during sensing, so instead averaging over several key positions for coupling must be relied upon. Since LSPR has a decay length of only 10–30 nm,44 strong plasmonic coupling between the Au nanostar and the Au nanohole array should occur when the Au nanostar is close to the rim of nanoholes (Fig. 6(b)). This is reflected when comparing the nanostar alone (Fig. S5†) with the coupled position in Fig. 6(b).
Fig. 6.
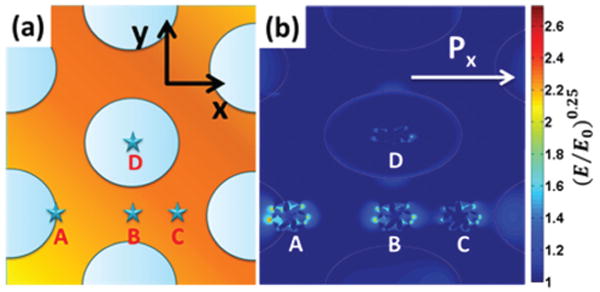
Simulated EM field distributions. (a) FDTD simulation cells with four Au nanostars located at point A (the rim of a nanohole), point B (the gap center between two nanoholes), point C (the gap center between three nanoholes), and point D (the nanohole center); (b) simulated EM field distribution of Au nanostars on Au nanohole arrays at points A, B, C, and D. The simulations were conducted under x polarization with a 785 nm laser source. (E/E0)0.25 was used to represent the EM field enhancement for easy visualization.
The idea that the maximum enhancement occurs around the edge is further supported by the fact that the peak SERS enhancement factor, which is defined as (E/E0)4, was calculated to be 4.5 × 106 for point A when the Au nanostar was at the right-side rim of a hole, yet it was 1.1 × 105 when the nanostar was located at the center of the nanohole. It is worth noting that the SERS enhancement factor (E/E0)4 was calculated to be 1.5 × 105 when a gold nanostar was placed on a planar Au film instead of a gold nanohole array (Fig. S6†). The one order of magnitude difference in enhancement correlates with the difference in experimental sensitivities, further proving the key role that LSPR–LSPR coupling played in enhancing the signal of the SERS sensor. Table S1† lists the maximum simulated SERS enhancement factor for all positions between x and y polarization. The EM field for y-polarization is shown in Fig. S7.†
It should be pointed out that the pitch of the gold nanohole array has a significant effect on the plasmonic properties, as shown in our previous study.36 The Au nanohole array with a periodicity of 600 nm displayed a LSPR band at around 785 nm, verified by both the experiments and simulation in Fig. 2(i). The overlap of the LSPR band with the excitation wavelength of 785 nm results in resonant enhancement of the electromagnetic field. In the meanwhile, the LSPR of the Au nanohole array can be effectively coupled with the LSPR field of the Au nanostar at 785 nm, which is important to further enhance the SERS signal, as confirmed in Fig. 6. Therefore, the Au nanohole array with a periodicity of 600 nm was fabricated.
Conclusions
In summary, a facile SERS sensor system was developed for detection of analytes in body fluids by incorporating the Au nanostar and the Au nanohole array into a single sensing system. The high sensitivity of the sensor was due to the enhanced EM field that resulted from coupling between the LSPR of the nanohole and nanostar. The sensor was applied to Ag(i) and Hg(ii) detection in saliva, with the specific C–Ag+–C and T–Hg2+–T bindings, showing excellent selectivity. The SERS detection platform developed in this work can be adapted for analysis of a wide range of analytes if DNA sequences or proteins are designed as the molecular recognition probes for specific analysis purposes; this has provided the possibility of portable, non-invasive, and on-site detection of analytes in body fluids.
Supplementary Material
Acknowledgments
This work was supported by a NSF grant (CBET-1336205). The resources and facilities used were also partially supported by NSF (EPS 1003907). Cushing is supported by the NSF Research Graduate Fellowship (GRFP-1102689). The use of WVU shared facility is acknowledged.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr02142a
References
- 1.Aragay G, Pons J, Merkoci A. Chem Rev. 2011;111:3433–3458. doi: 10.1021/cr100383r. [DOI] [PubMed] [Google Scholar]
- 2.Duong TQ, Kim JS. Chem Rev. 2010;110:6280–6301. doi: 10.1021/cr100154p. [DOI] [PubMed] [Google Scholar]
- 3.Li M, Gou HL, Al-Ogaidi I, Wu NQ. ACS Sustainable Chem Eng. 2013;1:713–723. [Google Scholar]
- 4.Hoang CV, Oyama M, Saito O, Aono M, Nagao T. Sci Rep. 2013;3:1175. doi: 10.1038/srep01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long F, Zhu A, Shi HC, Wang HC, Liu JQ. Sci Rep. 2013;3:2308. doi: 10.1038/srep02308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mingorance MD, Valdes B, Oliva Rossini S. Environ Int. 2007;33:514–520. doi: 10.1016/j.envint.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 7.MacKenzie JM, Canil D. Earth Planet Sci Lett. 2008;269:487–495. [Google Scholar]
- 8.Li JQ, Cen DZ, Huang DL, Li XF, Xu JJ, Fu SL, Cai R, Wu XC, Tang M, Sun Y, Zhang JR, Zheng JF. Cell Biochem Biophys. 2014;70:1663–1669. doi: 10.1007/s12013-014-0110-6. [DOI] [PubMed] [Google Scholar]
- 9.Adam V, Hanustiak P, Krizkova S, Beklova M, Zehnalek J, Trnkova L, Horna A, Sures B, Kizek R. Electroanalysis. 2007;19:1909–1914. [Google Scholar]
- 10.Crinnion WJ. Altern Med Rev. 2009;14:3–8. [PubMed] [Google Scholar]
- 11.Schmalz G, Hiller KA, Garhammer P, Reitinger T. J Dent Res. 2001;80:1254–1254. [Google Scholar]
- 12.Kodaira H, Ohno K, Fukase N, Kuroda M, Adachi S, Kikuchi M, Asada Y. J Oral Sci. 2013;55:161–165. doi: 10.2334/josnusd.55.161. [DOI] [PubMed] [Google Scholar]
- 13.Yang HC, Pon LA. Drug Chem Toxicol. 2003;26:75–85. doi: 10.1081/dct-120020403. [DOI] [PubMed] [Google Scholar]
- 14.Lewen N, Mathew S, Schenkenberger M, Raglione T. J Pharm Biomed Anal. 2004;35:739–752. doi: 10.1016/j.jpba.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 15.Hanna CP, Tyson JF, Mcintosh S. Anal Chem. 1993;65:653–656. doi: 10.1021/ac00053a029. [DOI] [PubMed] [Google Scholar]
- 16.Yantasee W, Lin YH, Hongsirikarn K, Fryxell GE, Addleman R, Timchalk C. Environ Health Perspect. 2007;115:1683–1690. doi: 10.1289/ehp.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homola J. Chem Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 18.Xue XJ, Wang F, Liu XG. J Am Chem Soc. 2008;130:3244–3245. doi: 10.1021/ja076716c. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Cushing SK, Wu NQ. Analyst. 2015;140:386–406. doi: 10.1039/c4an01079e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haynes CL, Van Duyne RP. J Phys Chem B. 2003;107:7426–7433. [Google Scholar]
- 21.Mayer KM, Hafner JH. Chem Rev. 2011;111:3828–3857. doi: 10.1021/cr100313v. [DOI] [PubMed] [Google Scholar]
- 22.Banholzer MJ, Millstone JE, Qin LD, Mirkin CA. Chem Soc Rev. 2008;37:885–897. doi: 10.1039/b710915f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao J, Dieringer JA, Zhang XY, Schatz GC, Van Duyne RP. J Phys Chem C. 2008;112:19302–19310. [Google Scholar]
- 24.Tan EZ, Yin PG, Lang XF, Zhang HY, Guo L. Spectrochim Acta, Part A. 2012;97:1007–1012. doi: 10.1016/j.saa.2012.07.114. [DOI] [PubMed] [Google Scholar]
- 25.Ma W, Sun MZ, Xu LG, Wang LB, Kuang H, Xu CL. Chem Commun. 2013;49:4989–4991. doi: 10.1039/c3cc39087j. [DOI] [PubMed] [Google Scholar]
- 26.Eshkeiti A, Reddy ASG, Narakathu BB, Joyce MK, Bazuin BJ, Atashbar MZ. IEEE Sens. 2012:434–437. [Google Scholar]
- 27.Du YX, Liu RY, Liu BH, Wang SH, Han MY, Zhang ZP. Anal Chem. 2013;85:3160–3165. doi: 10.1021/ac303358w. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Cushing SK, Zhang JM, Lankford J, Aguilar ZP, Ma DL, Wu NQ. Nanotechnology. 2012;23:115501. doi: 10.1088/0957-4484/23/11/115501. [DOI] [PubMed] [Google Scholar]
- 29.Gopalakrishnan A, Chirumamilla M, De Angelis F, Toma A, Zaccaria RP, Krahne R. ACS Nano. 2014;8:7986–7994. doi: 10.1021/nn5020038. [DOI] [PubMed] [Google Scholar]
- 30.Tan JMR, Ruan JJ, Lee HK, Phang IY, Ling XY. Phys Chem Chem Phys. 2014;16:26983–26990. doi: 10.1039/c4cp03679d. [DOI] [PubMed] [Google Scholar]
- 31.Yan B, Thubagere A, Premasiri WR, Ziegler LD, Dal Negro L, Reinhard BM. ACS Nano. 2009;3:1190–1202. doi: 10.1021/nn800836f. [DOI] [PubMed] [Google Scholar]
- 32.Das G, Chirumamilla M, Toma A, Gopalakrishnan A, Zaccaria RP, Alabastri A, Leoncini M, Di Fabrizio E. Sci Rep. 2013;3:1792. doi: 10.1038/srep01792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Correia-Ledo D, Gibson KF, Dhawan A, Couture M, Vo-Dinh T, Graham D, Masson JF. J Phys Chem C. 2012;116:6884–6892. doi: 10.1021/jp3009018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahon JM, Henzie J, Odom TW, Schatz GC, Gray SK. Opt Express. 2007;15:18119–18129. doi: 10.1364/oe.15.018119. [DOI] [PubMed] [Google Scholar]
- 35.Couture M, Liang YZ, Richard HPP, Faid R, Peng W, Masson JF. Nanoscale. 2013;5:12399–12408. doi: 10.1039/c3nr04002j. [DOI] [PubMed] [Google Scholar]
- 36.Zheng P, Cushing SK, Suri S, Wu N. Phys Chem Chem Phys. 2015 doi: 10.1039/C4CP05291A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palik ED. Handbook of Optical Constants of Solids. Academic Press; Orlando: 1985. [Google Scholar]
- 38.Ono A, Cao S, Togashi H, Tashiro M, Fujimoto T, Machinami T, Oda S, Miyake Y, Okamoto I, Tanaka Y. Chem Commun. 2008:4825–4827. doi: 10.1039/b808686a. [DOI] [PubMed] [Google Scholar]
- 39.Miyake Y, Togashi H, Tashiro M, Yamaguchi H, Oda S, Kudo M, Tanaka Y, Kondo Y, Sawa R, Fujimoto T, Machinami T, Ono A. J Am Chem Soc. 2006;128:2172–2173. doi: 10.1021/ja056354d. [DOI] [PubMed] [Google Scholar]
- 40.Gilfrich JV, Birks LS. Anal Chem. 1984;56:77–79. [Google Scholar]
- 41.Li M, Wang QY, Shi XD, Hornak LA, Wu NQ. Anal Chem. 2011;83:7061–7065. doi: 10.1021/ac2019014. [DOI] [PubMed] [Google Scholar]
- 42.Krausß P, Deyhle M, Maier KH, Roller E, Weiß HD, Cledon P. Toxicol Environ Chem. 1997;63:29–46. [Google Scholar]
- 43.Lygre GB, Høl PJ, Eide R, Isrenn R, Gjerdet NR. Clin Oral Investig. 1999;3:216–218. doi: 10.1007/s007840050104. [DOI] [PubMed] [Google Scholar]
- 44.Willets KA, Van Duyne RP. Annu Rev Phys Chem. 2007;58:267–297. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.