

Abstract
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome is a newly characterized autoinflammatory disorder, caused by mutations in PSMB8. It is characterized by early-onset fevers, accompanied by a widespread, violaceous and often annular, cutaneous eruption. While the exact pathogenesis of this syndrome is still obscure, it is postulated that the inflammatory disease manifestations stem from excess secretion of interferons. Based on preliminary blood cytokine and gene expression studies, the signature seems to come mostly from type I interferons, which are proposed to lead to the recruitment of immature myeloid cells into the dermis and subcutis. In this study, we systematically analyzed skin biopsies from 6 CANDLE syndrome patients by routine histopathology and immunohistochemistry methods. Skin lesions showed the presence of extensive mixed dermal and subcutaneous inflammatory infiltrate, composed of mononuclear cells, atypical myeloid cells, neutrophils, eosinophils and some mature lymphocytes. Positive LEDER and myeloperoxidase staining supported the presence of myeloid cells. Positive CD68/PMG1 and CD163 staining confirmed the existence of histiocytes and monocytic macrophages in the inflammatory infiltrate. CD123 staining was positive, demonstrating the presence of plasmacytoid dendritic cells. Uncovering the unique histopathologic and immunohistochemical features of CANDLE syndrome provides tools for rapid and specific diagnosis of this disorder as well as further insight into the pathogenesis of this severe, life-threatening condition.
Keywords: Immunohistochemistry, CANDLE syndrome, PSMB8, histopathology, autoinflammation, interferons
INTRODUCTION
Chronic, atypical, neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome1,2 is a newly characterized autosomal recessive autoinflammatory disease caused by mutations in PSMB8, which encodes the inducible β5i subunit of the immunoproteasome.3 Patients present with recurrent fevers and skin lesions beginning in early infancy. The skin lesions are characterized by recurrent and overlapping attacks of annular, erythematous or violaceous plaques that heal with residual purpura. With time, persistent violaceous edema develops around the eyelids and mouth. Recurrent episodes of multi-organ inflammation, as well as persistent, subclinical background inflammation, lead to delayed growth. Additionally, patients develop facial lipodystrophy and prominent abdomen, thus conferring the patients a very characteristic clinical phenotype.
The fact that the skin lesions of CANDLE syndrome appear during the first weeks or months of life allow for an early diagnosis of this progressive disorder. Furthermore, histopathology of the skin changes on light microscopy demonstrate characteristic features.1 There is a dense interstitial dermal infiltrate, extending into the subcutis, composed of mononuclear cells, many of them with atypical features exhibiting a myeloid appearance with bizarre nuclei, as well as neutrophils, which in some areas undergo karyorrhexis. However, the exact nature and origin of the atypical mononuclear cells in CANDLE syndrome have not been analyzed in detail. Thus, we performed an immunohistochemistry (IHC) study in skin biopsies from 6 patients with CANDLE syndrome to determine immunophenotypic characteristics of the inflammatory infiltrate. We demonstrate that the inflammatory infiltrate in skin biopsies is composed of a mixture of histiocytes and immature myeloid cells, but also contains significant numbers of plasmacytoid dendritic cells that most likely contribute to the pathogenesis of the syndrome by interferon (IFN) secretion.
MATERIALS AND METHODS
Skin biopsies were analyzed from 6 CANDLE syndrome patients, who have been reported previously. The syndrome was confirmed by detection of PSMB8 mutations in 5 of them;1-3 the 6th patient was deceased, but her affected sister had a homozygous PSMB8 mutation. For conventional light microscopy, skin biopsies were fixed in 4% formalin, embedded in paraffin, cut, and stained with hematoxylin-eosin (H&E). For immunohistochemical studies, representative sections of all cases were examined by the alkaline phosphatase anti-alkaline phosphatase (APAAP) technique using appropriate positive and negative controls. Automated immunostaining was performed on a BioTek Solutions Tech Mate (Tech-Mate 500; Biotech Solutions, Dako, Glostrup, Denmark). The antibodies used in this study targeted myeloperoxidase (MPO), CD117, CD163, CD68/KP1, CD68/PMG1, CD14, CD15, TdT, CD56, CD1a, CD33, CD123, and FoxP3. Their specificities and sources are given in Table 1. Chloracetate esterase (LEDER) stain, which stains hematopoietic cells of myeloid lineage (and mast cells), was performed in three cases using the Naphthol AS-D Chloroacetate (Specific Easterase) Kit from Sigma-Aldrich (91C-1KT) following standard laboratory protocols established by the histology section of the Laboratory of Pathology at the NIH.
Table 1.
Immunhistochemical markers and special stain used for staining
| Monoclonal antibody | Source | Main cellular expression |
|---|---|---|
| CD163 | 10D6, Menarini, Berlin, Germany | Monocytes, macrophages |
| CD68/KP1 | KP1, DAKO, Hamburg, Germany | Monocytes, macrophages, myeloid precursors, neutrophils, mast cells |
| CD68/PMG1 | PG-M1, DAKO, Hamburg, Germany | Monocytes, macrophages |
| CD14 | 7, Menarini, Berlin, Germany | Monocytes, macrophages |
| Myeloperoxidase | Polyclonal rabbit, DAKO, Hamburg, Germany | Neutrophils, monocytes |
| CD33 | PWS44, Menarini, Berlin, Germany | Progenitor and other myeloid cells, monocytes |
| CD15 | C3D-1, DAKO, Hamburg, Germany | Granulocytes, myeloid leukemia |
| CD123 | 7G3, BD Biosciences, Heidelberg, Germany | Plasmacytoid dendritic cells |
| FoxP3 | 236A/E7, Abcam, Cambridge, UK | T regulatory cells |
| TdT | Polyclonal rabbit, DAKO, Hamburg, Germany | Immature pre-B and pre-T cells |
| CD56 | RCD56 (rabbit), Zytomed systems, Berlin, Germany | NK cells, some leukemias |
| CD1a | 010, DAKO, Hamburg, Germany | Langerhans cells |
| CD117 | Polyclonal rabbit, DAKO, Hamburg, Germany | Mast cells |
| LEDER (special stain) | 91C-1KT, Sigma-Aldrich, St. Louis, MO, USA | Hematopoietic cells of myeloid lineage and mast cells |
To score the positivity of IHC stains, these were considered negative (−) if no cells were stained with the marker; + if the marker was expressed by less than 25 % of the cells in the infiltrate; ++ if expressed by 25 % to 50 %; and +++ if it was expressed by 50 % or more of the cells in the infiltrate.
RESULTS
H&E-stained sections showed similar histopathologic features, consisting of perivascular and interstitial dermal infiltrates extending into the subcutis (Figure 1). The infiltrate was mainly composed of mononuclear cells, with many of them exhibiting large, vesicular, irregularly shaped nuclei, thus giving the impression of atypical myeloid cells. There were also scattered mature neutrophils, a variable number of eosinophils and some mature lymphocytes. Leukocytoclasis was often present, but true vasculitis with fibrinoid necrosis of the vessel walls was not found.
FIGURE 1.

Histopathologic features of CANDLE syndrome. A, Skin sections demonstrating a mixed interstitial and perivascular inflammatory infiltrate. B-D, Higher magnification of A, revealing abundant atypical myeloid cells combined with mature neutrophils, in addition to mature lymphocytes and some eosinophils. Leukocytoclasis is also present. H&E, Original magnifications, 10X (A), 20X (B), 40X (C), 100X (D).
In all samples, strong and diffuse staining with MPO was observed, revealing that the infiltrate was rich in myeloid cells (Figure 2, A,B). A positive LEDER stain performed in 3 cases further supported the presence of myeloid cells. However, CD15, which is usually expressed by mature neutrophils, monocytes and promyelocytes, showed negative results in all cases. Interestingly, all samples were also intensely positive for CD68/PMG1 (Figure 3, A,B), CD163 (Figure 3, C,D), and CD68/KP1 (not shown), indicating the presence of histiocytes and monocytic macrophages. Double-IHC with MPO and CD163 performed in 5 cases revealed a double population of MPO-positive myeloid cells and CD163-positive macrophages (Figure 4).
FIGURE 2.
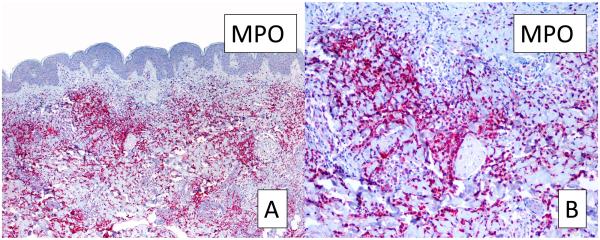
Myeloperoxidase stain for myeloid cells. A, Strong myeloperoxidase positivity reveals the presence of cells from a myeloid origin (original magnification, 10X). B, Higher magnification of A (40X). MPO: myeloperoxidase.
FIGURE 3.

Labeling of monocytes. A, CD68/PGM1 immunostain reveals the presence of monocytic cells (original magnification, 10X). B, Higher magnification of A (100X). C, positive CD163 staining (original magnification, 10X). D, Higher magnification of C (40X).
FIGURE 4.

Double immunostaining with MPO and CD163 reveals different cell populations co-existing in the same skin region. Original magnifications, 10X (A), 40X (B), 40X (C), 100X (D).
CD123, which identifies plasmacytoid dendritic cells, was positive in all cases, showing clustering of these cells in the infiltrate (Figure 5, A,B). Plasmacytoid dendritic cells are the most potent producers of Type I IFN.4 FoxP3 positivity was also noted (not shown), indicating the presence of significant numbers of T regulatory cells (Tregs) within the infiltrate.5
Figure 5.
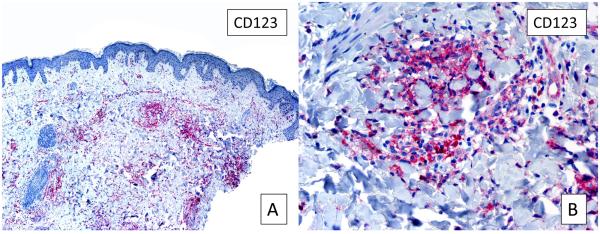
CD123 stain. A, Several foci of plasmacytoid dendritic cells are highlighted by CD123 (original magnification, 10X). B, Higher magnification of A (40X).
Numerous CD14 and CD33 were also seen (not shown), further demonstrating an important contribution of monocytes to the inflammatory infiltrate. CD117, CD15, TdT, CD56, and CD1a were negative (not shown), thus excluding the presence of mast cells, NK cells and Langerhans cells, as well as precursor hematological cells. A summary of the IHC results is shown in Table 2.
Table 2.
A summary of all immunohistochemistry stains in the 6 samples studied
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| CD163 | ++ | ++ | ++ | ++ | ++ | ++ |
| CD68/KP1 | +++ | +++ | ND | ND | ND | ND |
| CD68/PMG1 | ++ | ++ | ++ | ++ | ++ | ++ |
| CD14 | +++ | +++ | ++ | ++ | ++ | ++ |
| CD33 | ++ | ++ | +++ | ++ | ++ | +++ |
| MPO | +++ | +++ | +++ | +/- | ++ | +++ |
| LEDER stain | + | + | ++ | ND | ND | ND |
| CD123 | + | + | + | +/− | + | + |
| FoxP3 | + | + | ++ | − | +/− | ++ |
| CD15 | − | − | − | − | − | − |
| TdT | − | − | − | − | − | − |
| CD56 | − | − | ND | ND | ND | ND |
| CD1a | − | − | ND | ND | ND | ND |
| CD117 | − | − | ND | ND | ND | ND |
−: negative; +: <25 % of the cells in the infiltrate; ++: 25% − 50 % of the cells in the infiltrate; +++: >50 % of the cells in the infiltrate. ND: Not Done
DISCUSSION
CANDLE patients have a primary defect in the ubiquitin proteasome system that degrades intracellular proteins derived from self or foreign structures and that has a major role in the removal of misfolded proteins.6-8 Proteins are marked for destruction by ubiquitination to allow their recognition by the proteasome or the immunoproteasome. Certain triggers, such as infection or cellular stress, lead to the secretion of type I (IFN-α, −β) and type II (IFN-γ) IFNs, that activate the JAK kinase pathway. IFN stimulation produces increased amounts of waste proteins.9 PSMB8 missense mutations lead to decreased catalytic activity of the immunoproteasome, whereas a PSMB8 nonsense mutation causing premature termination codon leads to the inability to assemble the immunoproteasome.3 While the exact pathophysiology of CANDLE syndrome is still obscure, it is postulated that hematopoietic and likely also non-hematopoietic cells are unable to dispose of the waste proteins and that the excess accumulation of ubiquitinated proteins causes further cellular stress. This leads to a vicious cycle of cellular stress and further IFN release.
Our results indicate that the inflammatory cutaneous infiltrate in CANDLE syndrome is polymorphous and includes a mixture of immature myeloid cells, macrophages, plasmacytoid dendritic cells, and Tregs. Therefore, it mirrors the suggested pathophysiology of the syndrome. CD123-positive plasmacytoid dendritic cells are triggered to rapidly release Type I IFNs,4 and may be an important local population. Cells lacking intact proteasome function induce IFN-α (unpublished observations) and can contribute to the activation of the JAK kinase pathway and production of proinflammatory mediators. Interestingly, most macrophages in the lesional skin biopsies are CD68 and CD163 positive, the latter a marker that is more associated with M2-like macrophages, which are involved in immunosuppression and constructive tissue remodeling. They inhibit release of proinflammatory cytokines, scavenge debris, and promote tissue repair10,11 and may represent the “tissue’s attempt” to remodel and resolve the inflammatory lesion, as phagocytosis of apoptotic cells is an important role of M2-like macrophages for maintenance of cellular homeostasis.12 Alternatively, they may resemble macrophages seen in subacute necrotizing lymphadenitis.13 The IFN signatures in the blood of CANDLE patients do not allow us to definitively distinguish type I and type II IFN stimulation but are in general thought to be more associated with type I IFN.
The other innate immune cell subset found in the tissue, are granulocytes with an immature appearance including a non-segmented nuclei that stain with myeloid markers (MPO, LEDER stain). It remains unresolved why the granulocytes in the tissue remain immature, however an immature granulocyte signature is found in the peripheral blood of systemic lupus erythematosus patients together with an IFN signature,14 suggesting that the histopathology findings may be consistent with an IFN-mediated process in the skin. However, a better understanding of the tissue cytokines and the role of IFNs on the tissue migration of granulocytes and monocytes will likely shed light on that phenomenon in the future. During disease flares patients develop leukopenia and thrombocytopenia which is consistent with the known bone marrow suppressive effects of IFN in line with observations that high doses of therapeutically administered IFN-α to patients with hepatitis cause bone marrow suppression.15 The recruitment of mononuclear cells into the tissue may be triggered by skin resident cells including fibroblasts and other cells that are stressed in the context of IFN. Patients with CANDLE syndrome are unable to upregulate their proteasome production in stress situations to keep up with the degradation of “waste proteins” or immature proteins that are over-produced in the presence of particularly IFN mediated inflammation16 and thus amplify a vicious cycle of more IFN production leading to more cell stress and the further recruitment of inflammatory cells into the tissue.
The presence of FoxP3 Tregs in the infiltrate is of unknown pathophysiological significance, but can be explained as an attempt to downregulate the cycle of proinflammatory reactions and establish tissue homeostasis.
Laboratory studies in CANDLE syndrome support a proposed pathomechanism of IFN-mediated pathology. In fact, microarray analyses in patients with CANDLE syndrome have shown an intense expression of IFN signature genes, and high levels of proteins induced by IFN have been reported in the serum of patients with CANDLE syndrome, such as IFN-derived protein 10.3
To our knowledge, this study is the most comprehensive IHC characterization of the cutaneous infiltrate in CANDLE syndrome. It sheds further light on the pathophysiological mechanism of the syndrome, and emphasizes its diverse immunological and inflammatory responses, which are mostly IFN-derived. The unique combination of histopathologic and IHC features presented in this study is virtually pathognomonic of CANDLE syndrome, and should permit early diagnosis of the disease, even in the absence of clinical suspicion or clinical information. Given that many of the characteristic features of the syndrome, such as facial lipodystrophy or prominent abdomen, appear later in the disease course, early histopathological recognition allows intervention, such as with the JAK1/2 inhibitor baricitinib (and clinical trials are underway, NCT01724580),17 which could prevent severe morbidity and mortality.
ACKNOWLEDGMENTS
This work was supported in part by the NIAMS Intramural Research Program (IRP) at the National Institutes of Health (NIH); The Authority for Research and Development, Hebrew University of Jerusalem (to A.Z.), and the Young clinician’s grant, Hadassah – Hebrew University Medical Center (to Y.R.).
Source of Funding:
This work was supported in part by the NIAMS Intramural Research Program (IRP) at the National Institutes of Health (NIH) (to Dr. Raphaela Goldbach-Mansky); The Authority for Research and Development, Hebrew University of Jerusalem (to Dr. Abraham Zlotogorski); and the Young clinician’s grant, Hadassah – Hebrew University Medical Center (to Dr. Yuval Ramot).
Footnotes
Conflicts of Interest
For the remaining authors no conflicts of interest or funding are declared.
REFERENCES
- 1.Torrelo A, Patel S, Colmenero I, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. 2010;62:489–495. doi: 10.1016/j.jaad.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 2.Ramot Y, Czarnowicki T, Maly A, Navon-Elkan P, Zlotogorski A. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a case report. Pediatr Dermatol. 2011;28:538–541. doi: 10.1111/j.1525-1470.2010.01163.x. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64:895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jegalian AG, Facchetti F, Jaffe ES. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol. 2009;16:392–404. doi: 10.1097/PAP.0b013e3181bb6bc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 6.Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Neurodegener Dis. 2012;10:7–22. doi: 10.1159/000334283. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–7. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 9.Ebstein F, Kloetzel PM, Krüger E, Seifert U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci. 2012;69:2543–2558. doi: 10.1007/s00018-012-0938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 11.Shabo I, Stål O, Olsson H, Doré S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. 2008;123:780–786. doi: 10.1002/ijc.23527. [DOI] [PubMed] [Google Scholar]
- 12.Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brüne B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood. 2006;108:1635–1642. doi: 10.1182/blood-2006-04-014852. [DOI] [PubMed] [Google Scholar]
- 13.Jang SJ, Jeon HM, Kim D, Yang W-I. Myeloperoxidase positive histiocytes in subacute necrotizing lymphadenitis express both CD11c and CD163. Basic and Applied Pathology. 2011;4:110–115. [Google Scholar]
- 14.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peck-Radosavljevic M, Wichlas M, Homoncik-Kraml M, Kreil A, Hofer H, Jessner W, Gangl A, Ferenci P. Rapid suppression of hematopoiesis by standard or pegylated interferon-alpha. Gastroenterology. 2002;123:141–151. doi: 10.1053/gast.2002.34175. [DOI] [PubMed] [Google Scholar]
- 16.Krüger E, Kloetzel PM. Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Curr Opin Immunol. 2012;24:77–83. doi: 10.1016/j.coi.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. 2013;147:155–174. doi: 10.1016/j.clim.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]