

Abstract
The lung endothelium is exposed to mechanical stimuli through shear stress arising from blood flow and responds to altered shear by activation of NADPH (NOX2) to generate reactive oxygen species (ROS). This review describes the pathway for NOX2 activation and the downstream ROS-mediated signaling events on the basis of studies of isolated lungs and flow-adapted endothelial cells in vitro that are subjected to acute flow cessation (ischemia). Altered mechanical stress is detected by a cell-associated complex involving caveolae and other membrane proteins that results in endothelial cell membrane depolarization and then the activation of specific kinases that lead to the assembly of NOX2 components. ROS generated by this enzyme amplify the mechanosignal within the endothelial cell to regulate activation and/or synthesis of proteins that participate in cell growth, proliferation, differentiation, apoptosis, and vascular remodeling. These responses indicate an important role for NOX2-derived ROS associated with mechanotransduction in promoting vascular homeostasis.
Keywords: chemotransduction, mechanotransduction, KATP channel, NADPH oxidase, endothelial cell membrane potential, nitric oxide
INTRODUCTION
Reactive oxygen species (ROS) are well established as a cause of tissue injury via oxidative damage to lipids, proteins, DNA, and other biomolecules. The toxic effects of ROS constitute a major mechanism for inflammatory lung injury of diverse etiology and following exposure to various exogenous agents such as hyperoxia, paraquat, and bleomycin. ROS are generated normally in cells by the activity of several enzymes or through auto-oxidation of endogenous compounds. The endogenous generation of ROS was first recognized as a component in the bactericidal activity of phagocytic cells (1), and only several decades later did the scientific community realize that these molecules could also serve physiological signaling functions. This belated understanding of the physiological role of ROS contrasts with that of another free radical, ·NO, whose signaling function was recognized long before the appreciation of its potential toxicity. The lag in recognizing ROS as signaling molecules is related to their evanescence in tissues and the cumbersome and indirect methods available for their detection. Furthermore, the biological effects of ROS have generally been determined by the response to exogenous application of H2O2 or compounds that generate the superoxide anion ( ), almost always in concentrations that greatly exceed potential physiological levels. Finally, availability of and cell membrane permeability to detoxifying agents greatly influence the biological response to oxidants. Increasing evidence over the past two decades has indicated that ROS exert subtle effects when produced in a controlled and regulated manner in a specific subcellular location or compartment (2). In such a setting, ROS participate as second messengers to regulate cellular signaling pathways that control cellular and tissue homeostasis through the modulation of cell growth and proliferation, apoptosis, differentiation, and a myriad of other cellular processes.
This review focuses on the physiological role of ROS in the pulmonary vasculature and emphasizes our model of altered lung (endothelial) shear stress in which the endogenous generation of ROS and their physiological effects can be readily measured. Altered (acutely decreased) shear stress represents a physiological model for pulmonary ischemia.
REACTIVE OXYGEN SPECIES (ROS)
We now define ROS from a chemical standpoint and consider their sources in the intact cell. These sources include both endogenous production within the cell as well as exogenously administered drugs and poisons.
Definition of ROS
The term ROS describes a broad group of molecules that includes both free radical (e.g., , ·OH) and nonradical (e.g., H2O2) species derived from the reduction of O2. These species are of varying reactivity with tissue components. For example, the hydroxyl radical (·OH) is extremely reactive and oxidizes almost any biomolecule with which it comes in contact. and H2O2 are less reactive and interact with chemically defined targets. Several members of the ROS family have properties that enable them to function in signaling; i.e., they are generated at low levels in a precise location and show both specificity and reversibility in their reactivity with biomolecules. H2O2, which is present normally at ~10−8 Min the cytosol, can reversibly oxidize protein cysteines and is regarded as the major ROS-related signaling molecule. Less well understood is the role of in cell signaling; although this molecule is relatively short lived, it may participate in a limited range of signaling functions.
Cellular Sources of ROS
ROS generation in vivo can occur via various enzymatic pathways and via nonenzymatic processes. The former generate either or H2O2 by the transfer of one or two electrons, respectively, whereas the nonenzymatic pathway generates by auto-oxidation of a reduced compound. An important difference between these two general mechanisms is that enzymatic pathways, but not auto-oxidation, represent a potentially controlled reaction that can participate in cellular homeostasis.
Major enzymatic pathways for generation of ROS are as follows:
NADPH oxidase (NOX): This family of proteins (NOX2 is the prototype) utilizes NADPH to reduce molecular oxygen to (3). This is the only family of enzymes with the known primary function of generating ROS.
Xanthine oxidase: This enzyme catalyzes the oxidation of hypoxanthine to xanthine and then to uric acid. These reactions use molecular oxygen as an electron acceptor, resulting in the generation of or H2O2. Activity of the similar xanthine dehydrogenase enzyme does not generate ROS.
Cyclo-oxygenase/lipoxygenase (COX/LOX): These enzymes produce during the metabolism of arachidonate in the presence of NAD(P)H.
Nitric oxide synthase (NOS): With deficiency of the important cofactor tetrahydrobiopterin or in a low-pH medium, this enzyme can generate instead of ·NO.
Various amine oxidases: Monoamine oxidase, for example, generates H2O2 during the oxidation of dopamine.
Common nonenzymatic sources of ROS are as follows:
Mitochondrial electron transport chain (ETC): Under normal physiological conditions, approximately 1–3 % of electrons carried by the mitochondrial ETC leak out of the pathway and pass directly to oxygen, generating . This leakage represents an auto-oxidation reaction. There are several sites of possible auto-oxidation during the sequential transfer of electrons to ETC components in the process of oxidative phosphorylation. A major site is ubisemiquinone, the component at the interface between one- and two-electron transfer in the ETC.
Other cytochromes: A similar auto-oxidation reaction can occur with reduced members of the cytochrome P-450 family of enzymes.
Free iron (Fe2+): The presence of Fe2+ can catalyze the generation of ·OH from H2O2 and , thereby generating a more potent electrophile.
Quinones and other auto-oxidizable chemicals: A variety of endogenously produced compounds, especially those with a quinone moiety, can auto-oxidize on exposure to O2 with the generation of . An example is the metal-catalyzed oxidation of dopamine to a semiquinone; this compound can further auto-oxidize, thereby generating . Other endogenous auto-oxidizable chemicals include epinephrine, ascorbic acid (vitamin C), menadione (vitamin K), and certain thiols.
Exogenous agents: A variety of exogenously administered agents, including drugs and xenobiotics, demonstrate chemistry similar to that described for quinones. For example, the herbicide paraquat is reduced by NADPH-cytochrome P-450 reductase and auto-oxidizes to generate as the basis for its toxicity. The cancer therapeutic agents doxorubicin and bleomycin can also generate ROS.
ROS-MEDIATED SIGNALING IN THE PULMONARY CIRCULATION
ROS generated in endothelial cells exert their physiological effects by interaction with transcription factors, protein tyrosine phosphatases, protein tyrosine kinases, mitogen-activated protein kinases, and other components of various signaling cascades. Many of these modified proteins transmit their signal through phosphorylation and dephosphorylation of specific amino acid residues (serine, threonine, tyrosine, and histidine). ROS modify these signaling-related proteins through the oxidation of specific residues that result in reversible activation or inactivation of enzymatic activity. Excess ROS produced by these pathways can result in cell injury, and organisms have developed a variety of enzymatic and nonenzymatic defense mechanisms to protect against this possibility. The vast majority of studies of cell signaling have evaluated the response to various chemical agents (chemotransduction), many of which initiate the signaling process through cell membrane–associated receptors. Recent publications have reported that cells respond similarly to mechanical stresses (mechanotransduction), which may initiate the signaling process through forces acting on the cell membrane (4). The similarity between the chemo- and mechanotransduction processes is shown in Figure 1.
Figure 1.

Reactive oxygen species (ROS) generation upon chemotransduction or altered mechanotransduction. Chemical stimuli or stop of shear activate NADPH oxidase, type 2 (NOX2) and endothelial nitric oxide synthase (eNOS). The resultant generation of oxygen ( )- and nitrogen (·NO)-centered radicals results in signaling or injury, depending on the level of production and antioxidant defenses of the cell.
NADPH Oxidases
The predominant and most-studied source of ROS within the pulmonary vasculature is the NOX pathway. The NOX family has seven members, of which NOX2 and −4 are most highly expressed in the pulmonary endothelium. NOX2 was the first member of the NOX family to be described and is the best understood (5, 6). Active NOX2 is composed of two integral membrane proteins (gp91phox and p22phox) and four cytosolic proteins (Rac, p40phox, p47phox, and p67phox); the latter proteins translocate to the membrane upon activation and associate with the membrane subunits (Figure 2). Current usage identifies the gp91phox flavoprotein as NOX2. Phosphorylation of Rac (Rac1 in pulmonary endothelium) is the key step in initiating translocation of the cytosolic components. The activation step regulates production by the enzyme complex. In contrast, NOX4 appears to be constitutively active and does not require translocation of cytosolic subunits for ROS generation, although this point remains controversial (7).
Figure 2.
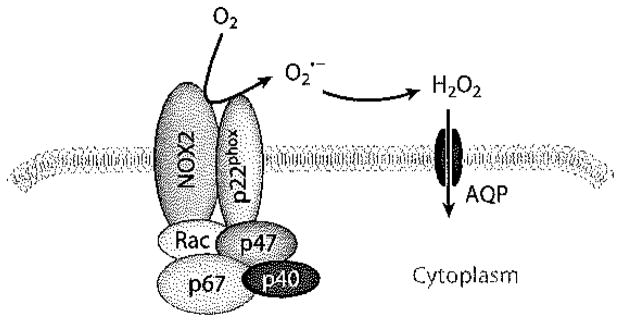
The assembled NADPH oxidase, type 2 (NOX2) complex. gp91phox (NOX2) and p22phox are integral plasma membrane components and together constitute cytochrome b558. The remaining components are normally cytosolic proteins that translocate to the plasma membrane following their phosphorylation. that is generated by NOX2 is converted to H2O2, which can diffuse across the plasma membrane, possibly through aquaporin (AQP) channels, to initiate cell signaling.
Chemotransduction
Cell signaling can be initiated by soluble factors that act through either receptor-mediated or receptor-independent mechanisms to promote ROS generation. This process, termed chemotransduction, is described in this section. Similarly, cells can respond to an altered physical environment to initiate a signaling cascade; this latter process is termed mechanotransduction and is considered subsequently.
Activation of NOX2
Cell signaling mediated by ROS can occur via the activation of chemoreceptors on the endothelial plasma membrane by various agonists. Representative agonists include angiotensin, thrombin, transforming growth factor-β, and platelet-derived growth factor. Each of these agonists initiates ROS generation through the activation of NOX2 activity. The interaction of angiotensin II (Ang II) with its receptor activates phospholipase C, which in turn hydrolyzes phosphatidylinositol-4,5-biphosphate to inositol 1,4,5-triphosphate (InsP3) and di-acylglycero1 (DAG) (8). DAG and Ca2+ released by InsP3 then activate protein kinase C (PKC) (8, 9), with subsequent phosphorylation of the cytosolic and possibly membrane components of NOX2 (10). NOX2 activation can also occur through nonreceptor mechanisms, such as stimulation by phorbol esters, that directly activate PKC (11, 12). Thus, PKC activation through either receptor-mediated or non-receptor-mediated mechanisms represents a key step in NOX2 activation, at least in response to some agonists.
Role of peroxiredoxin 6
Our recent studies using pulmonary microvascular endothelial cells have shown that peroxiredoxin 6 (Prdx6) serves as a link between PKC activity and NOX2 activation upon either Ang II or PMA (phorbol-12-myristate-13 acetate; also known as 12-O-tetradecanoylphorbol-13-acetate) treatment (13). The peroxiredoxins are a ubiquitously distributed family of peroxidases that function in cellular redox reactions (14, 15). Most of the peroxiredoxins utilize thioredoxin as the redox cofactor, although Prdx6 is GSH dependent (16). Prdx6 uniquely possesses phospholipase A2 (PLA2) in addition to its peroxidase activity (15, 16). Translocation of cytosolic NOX2 components and enzyme activation did not occur in endothelial cells that were Prdx6 null; transfection of Prdx6-null cells with constructs expressing the PLA2 activity of Prdx6 rescued the ROS response to agonist treatment (13). NOX2 activation was also suppressed by the treatment of lungs and cells with an agent [MJ33 (1-hecadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol)] that inhibits the PLA2 but not the peroxidase activity of Prdx6 (13). Thus, the PLA2 activity of Prdx6 is essential for NOX2 activation in pulmonary endothelial cells.
Prdx6 is a cytosolic enzyme that requires phosphorylation for binding to substrate lipids, such as those of the cell membrane, and its subsequent PLA2 activity (17). Prdx6 can be phosphorylated by mitogen-activated protein kinases (MAP kinases) in vitro (18), and MAP kinase inhibitors block Prdx6 phosphorylation in Ang II-treated endothelial cells (13). Erk and p38 MAP kinase are able to phosphorylate Prdx6 in vitro. Thus, we propose that agonist-mediated PKC activation in turn activates MAP kinases, resulting in Prdx6 phosphorylation and Prdx6 binding (translocation) to the plasma membrane (13). This paradigm constitutes the upstream events leading to translocation of the cytosolic components of NOX2 and activation of ROS generation. A Prdx6-dependent mechanism similar to that described for endothelium is also required for NOX activation in pulmonary alveolar macrophages (13).
Although the role of PLA2 activity hi NOX2 activation is clear, the product of the PLA2 reaction that is responsible for promoting NOX assembly and activation is less so, PLA2 activity liberates both a free fatty acid and a lysophospholipid, and there is evidence that either metabolite may be directly or indirectly responsible for activation of the NOX2 pathway (19–21). Prdx6 does not show a preference for arachidonate-containing phospholipids such that this eicosanoid precursor would not be preferentially liberated by Prdx6 activity (22).
Mechanotransduction
Mechanotransduction is the process by which altered physical forces are sensed by the cell and converted into biochemical signals. The concept is analogous to chemotransduction. The endothelium is normally exposed to dynamic fluid shear stress as a result of viscous drag of blood flow and to cyclic strain resulting from hydrostatic pressure. Shear stress is defined as the fluid frictional force per unit area and acts at the luminal surface (endothelial lining) of blood vessels; it can be calculated as 4μQ/πr3, where μ is the dynamic viscosity, Q is the volumetric flow rate, and r is the radius of the conducting vessel. Thus, the magnitude of a change of shear, in a perfused vessel of given radius, is directly proportional to the change in perfusate flow rate (ΔQ). The wall strain associated with altered hydrostatic (perfusion) pressure is the ratio of the change in matrix unit length or area to the original length (Δl/l0) or original area (ΔA/A0). Endothelial cells, located at the interface between blood and tissue, can sense changes in hemodynamic forces and respond appropriately to maintain homeostasis (23–26). These responses to pulsatile fluid shear stress and to cyclic circumferential strain contribute to the phenotype of endothelial cells.
Mechanotransduction in the lung
In the lung, the endothelium of major vessels is exposed to both shear stress as well as cyclic circumferential strain due to pulsations associated with the cardiac cycle. Both of these forces generated by the cardiac cycle can result in cell-mediated signaling responses. Although the effect of cyclic strain on lung cells has been studied relative to acute lung injury (27–29), less is known about strain-related signaling in the pulmonary endothelium. Expansion of the alveolus during ventilation is thought to be more of a change in conformation (unfolding) rather than one in surface area and thus does not significantly contribute to strain of the matrix and vascular structures. However, the relationship between respiration and circumferential strain at the capillary level is complex, depending on alteration of cardiac output with ventilation and the zones of the upright lung; these considerations are beyond the scope of this review.
The effects of shear stress on the pulmonary endothelium have received greater attention. The endothelium of the major pulmonary vessels is exposed to cardiac-induced cyclic changes in shear, whereas blood flow in the capillaries approaches a more constant laminar flow. Under conditions of altered flow (altered shear stress), increased ROS production mediates signal transduction, leading to downstream physiological responses in pulmonary as well as systemic endothelial cells (30, 31). This review concentrates on the role of ROS in pulmonary vascular signaling, with a focus on the generation of ROS associated with a model of acutely altered shear stress. The review emphasizes changes in laminar flow rate and does not discuss the responses to oscillatory or turbulent flow that are more relevant to pathophysiology associated with vascular abnormalities in the systemic vasculature (32).
Shear stresses in the circulation
The shear stresses in human pulmonary arteries have been determined by magnetic resonance imaging. Time-averaged axial near-wall shear stress was estimated at approximately 5–7 dyn cm−2 in the main pulmonary arteries (33), although calculations based on physiological parameters of flow and vessel dimensions suggest a lesser value of ~2 dyn cm−2 (34). The measured near-wall shear in the main pulmonary artery is similar to that in the aorta (~8 dyn cm−2) (35). Thus, although the driving pressure in the aorta is significantly greater than in the pulmonary artery (consistent with the greater vascular resistance in the systemic versus pulmonary circulation), the flow velocity and shear in the major vessels appear to be not significantly different. The blood flow velocity in the pulmonary capillaries is significantly lower than in the large vessels, but because shear stress varies with the cube of the radius, the shear stress in the capillaries is estimated to be significantly greater than in the major vessels (34).
Response of pulmonary endothelium to altered shear stress
Altered endothelial mechanotransduction associated with altered shear stress or cyclic strain results in ROS-dependent signaling (31, 36–38). The model used in our studies of altered mechanotransduction in the lung is the abrupt reduction of shear stress, i.e., an acute decrease (lung ischemia) that can occur physiologically due to vascular obstruction or during lung transplantation (30). In the isolated rat lung, a decrease in perfusion of approximately 80% represents the threshold for eliciting a response (39). Unlike the systemic circulation, loss of blood flow (ischemia) in the lung is not equated with tissue hypoxia, provided that ventilation is unimpaired (40–42). This physiological property has enabled us to study the endothelial response to decreased perfusion (altered shear stress) without the confounding variables of diminished oxygen supply and the metabolic responses to hypoxia.
In an ex vivo intact lung model (rat and mouse), loss of flow induces pulmonary endothelial membrane depolarization followed by ROS generation due to activation of NOX2 (43,44). These studies have been carried out primarily by imaging of the lung by epifluorescence, confocal, or multiphoton techniques following cell labeling with various fluorophores (45). This response of the perfused lung to altered shear has been reproduced with an in vitro model by subjecting endothelial cells to a flow adaptation regimen (physiological shear stresses of 5–10 dyn cm−2 for 24–72 h) followed by abrupt flow cessation (46–48). Exposure of endothelial cells in culture to shear leads over time to a range of biochemical and structural alterations that constitute a flow-adapted state (26,49). Several systems that we have used for flow adaptation of cells in vitro along with some examples of data that were generated with the flow cessation model are described in Chatterjee & Fisher (45) and are shown schematically in Figure 3. Because endothelial cells in vivo are presumably flow adapted, we believe that flow-adapting cells in culture results in a more physiological in vitro model for the study of the effects of altered shear stress compared with the commonly used model of acutely increased shear with non-flow-adapted cells.
Figure 3.

Apparatus for flow-adapting endothelial cells in vitro to shear stress. Cells are seeded, are allowed to attach for 24 h, and are then subjected to flow (generally at shear stresses of 5–10 dyn cm−2) for 24–72 h. (a) Procedure for flow-adapting cells for fluorescence imaging. The upper-panel chamber (Warner Instruments, Hamden, CT) shows the parallel plate. The middle panel shows a schematic of the perfusion circuit. Ceils are seeded on cover slips inserted into the chamber. Flow-adapted wild-type, caveolin-1-null, and KIR6.2 (KATP channel)-null cells were evaluated under continuous-flow (control) or stopped-flow (ischemia) conditions in the presence of the membrane potential–sensitive fluorophore bisoxonol; images were acquired by confocal microscopy (lowerpanel). Depolarization indicated by increased fluorescence was observed in wild-type but not in caveolin-1-null and KATP channel-null cells (91). (b) Procedure for cellular and biochemical analysis of flow-adapted cells. Cells are seeded on fibronectin-coated polycarbonate capillaries encased in a housing for perfusion via luminal or abluminal ports (upper panel). For the initial cell attachment and the subsequent ischemia phases of the experiment, medium is perfused through the abluminal ports, which removes the shear stress but allows adequate oxygenation and provision of nutrients (black arrows). For flow adaptation, medium is perfused through the luminal ports (red arrows). The lower panel shows images of cells that have been trypsinized from the capillaries and labeled with PKH for analysis by fluorescence-activated cell sorting; cell generations are indicated by the differently colored peaks. Continuous laminar flow results in an antiproliferative state. Increased proliferation is shown by wild-type cells that were flow adapted and then subjected to flow cessation; increased proliferation is not seen in cells that do not generate ROS, either NOX2-null cells or cells treated with a KATP channel agonist (cromakalim) to prevent NOX2 activation (91).
ROS AS A SIGNAL TRANSDUCER WITH ALTERED SHEAR
Studies of chemotransduction involving Ang II and other signaling agonists have established the role of the NOX enzymes in signaling (11, 50–52). Studies using our mechanotransduction model of acute interruption of perfusion (acute loss of shear stress) to the isolated lung or to flow-adapted endothelial cells in culture reveal the rapid (within-seconds) appearance of ROS (43, 44). This reaction is inhibited by the presence of diphenyleneiodonium, a nonspecific flavoprotein inhibitor, and is abolished by knockout of gp91phox (NOX2) (44,46, 53). Studies described below document the important signaling role in the pulmonary endothelium of NOX2-derived ROS subsequent to loss of shear. ROS generation in these pathways represents a mechanism to transduce the signal generated in response to altered mechanical forces into a physiological response. ROS function as transducing agents by specifically and reversibly reacting with proteins to alter their structure, activity, and function.
Specificity of ROS in the Signaling Paradigm
NOX2 activation generates as its only product: . On the basis of its location as a plasma membrane–spanning protein, gp91phox (NOX2) transfers its electron across the cell membrane and generates at the extracellular face of the endothelial cell (54). The reaction generates a proton in the cytoplasm that requires neutralization to avoid cellular acidification (54); discussion of the potential mechanisms for proton removal is beyond the scope of this review. Superoxide is a short-lived species that dismutes to H2O2, either rapidly in a reaction catalyzed by superoxide dismutase (SOD) (rate constant >109 M−1s−1) or spontaneously at a somewhat slower (>105 M−1s−1) but still significant rate: . H2O2 can cross biological membranes, thereby enabling its interaction with intracellular signaling pathways. However, recent evidence suggests that H2O2 does not directly cross the lipid bilayer but rather passes through membranes via aquaporins, similarly to H2O (55). Because H2O2 is highly diffusible, it has a steep gradient from its sites of production. Furthermore, H2O2 is relatively specific in its chemical reactions, and its concentration is well regulated by various specific enzymes (catalase, glutathione peroxidase, peroxiredoxins). These characteristics make H2O2 an ideal signaling molecule (2,54). H2O2 functions in signaling through a reversible reaction with low-pKa cysteine residues on proteins to initially form a disulfide bond (–SS–) or sulfenic acid (–SOH) (56). Further oxidation of –SOH yields sulfinic (–SO2H) and sulfonic (–SO3H) acids, which are considered to be irreversible reactions (2,57). These oxidation products in some proteins (e.g., most peroxiredoxins, but not Prdx6) can be reduced by an energy (ATP)-dependent enzyme, sulfiredoxin (58, 59).
Although it is generally believed that H2O2 represents the primary ROS-related signal transducer, recent evidence suggests that may activate discrete signaling pathways independently of H2O2 (54, 60). Because it is short lived, must be produced in very close proximity to its target to be effective as a signaling molecule (61). Thus, if extracellular functions as a signal transducer, it most likely acts on receptor proteins on the extracellular face of the endothelial cell plasma membrane. Recent studies (see below) indicate that may penetrate the plasma membrane via Cl− channels, although the travel distances are likely quite short (62).
Mitochondria-derived ROS and signal amplification
Mitochondria generate essentially by leakage of electrons from the ETC, as described above. ROS production by this mechanism is nonenzymatic and is unlikely to constitute a regulated signaling process, although this is an area of active investigation, and the disagreements may be largely semantic (63, 64). The amount of generated by mitochondria varies significantly with the mitochondrial metabolic state, and physiological changes in mitochondrial metabolism may result in increased generation (64). With respect to cell signaling, mitochondria can also be a secondary source of as a response to a primary ROS signal from the plasma membrane or from other intracellular sources (65). This process has been termed -induced generation. Our studies in endothelial cells have shown that the transmembrane flux of extracellularly generated (by the addition of Ang II, xanthine/xanthine oxidase, or KO2 to the medium) leads to a transient increase in cytosolic Ca2+ and intracellular ROS generation (60, 62). The proposed mechanism is the activation of plasma membrane InsP3 receptors resulting in Ca2+ release from intracellular stores, depolarization of the mitochondrial membrane, and activation of mitochondrial production (Figure 4). Another possible mechanism for secondary mitochondrial ROS production is H2O2-mediated oxidation of Fe3+-cytochrome c, with sequential transfer of the electron to generate NAD· and then (66). This amplification process represents another potential mechanism (in addition to H2O2 diffusion) for transmission of the initial extracellular ROS signal across the plasma membrane to serve an intracellular signaling function.
Figure 4.

Possible mechanism for -induced release by mitochondria. generated extracellularly by NOX2 can penetrate the pulmonary endothelial cell membrane through Cl−3 channels (Cl3 C) and can interact with the inositol 1,4,5-triphosphate (InsP3) receptor, resulting in Ca2+ release, depolarization of the mitochondrial inner membrane, and mitochondrial generation by auto-oxidation of components of the electron transport chain (62).
UPSTREAM EVENTS IN THE ACTIVATION OF ROS-MEDIATED SIGNALING
Sensors for Altered Shear
Although cells sense and respond to altered shear, the mechanism(s) by which endothelial cells sense mechanical forces is less clear. Several models have been proposed. As one possibility, proteins that are integral to the cell membrane may link the extracellular matrix to the cytoskeleton; altered external forces may lead to cytoskeletal distortion, which generates a biochemical response.
Alternatively, flow sensors situated on the cell surface may respond to mechanical forces (e.g., shear stress) at the cell membrane. These views of flow sensing are not mutually exclusive, and both mechanisms or an additional mechanism may cooperatively regulate the response to flow. The cell membrane proteins that have been considered responsible for mechanosensing by the endothelium include G protein–coupled receptors, ion channels, the glycocalyx, platelet–endothelial cell adhesion molecule-1 (PECAM-1), vascular endothelial cadherin (VE-cadherin), receptor tyrosine kinases, and tight junctions. Although several such proteins have been demonstrated to transduce strain, most require forces of supraphysiological magnitude that are unlikely to be relevant to normal signaling.
Adhesion receptor complex
Initial studies demonstrated that PECAM is involved in cell sensing of altered shear stress and requires phosphorylation for this effect (67). Subsequently, significant evidence indicates that shear sensing by endothelium involves a mechanosensory complex composed of vascular endothelial growth factor receptor 2 (VEGFR2) (Flk-1), VE-cadherin, and PECAM-1 (68–70). VEGFR2 is a transmembrane receptor tyrosine Kinase. VE-cadherin and PECAM-1 are transmembrane cell adhesion molecules that form homophilic dimers at cell-cell junctions but also complex with VEGFR2. The role of VE-cadherin does not require interaction with juxtaposed cadherins, and VE-cadherin is thus considered to be primarily an adaptor molecule in this complex (68). PECAM-1 can mediate Src-dependent transactivation of VEGFR2, which in turn can activate phosphatidylinositol-3 kinase (PI3K) and Akt, thereby initiating downstream signaling in response to shear stress (69, 70). The components of this complex are conserved in pulmonary endothelial cells, although its role in shear sensing has not yet been characterized in the pulmonary circulation.
Glycocalyx
The endothelial glycocalyx is an extracellular polymeric surface layer of variable thickness containing glycoproteins, proteoglycans, and their associated glycosaminoglycan side chains. Heparan sulfates are the predominant glycosaminoglycans found on the endothelial surface and are important in ·NO generation following mechanical stress (71), although no studies specifically deal with ROS generation and pulmonary circulation.
Caveolae
Caveolae, the flask-like invaginations within the plasma membrane of the endothelium, may also play an important role in sensing and/or coordinating the response to altered shear. These structures (or similar lipid-rich lipid rafts) are signaling hubs localizing several redox-sensitive and ROS-generating molecules in endothelial cells, including the membrane-associated subunits of the NOX2 complex (gp91phox, p22phox) (72). Ex vivo–perfused lungs and flow-adapted endothelial cells from caveolin-1-null mice that have caveolar disruption show a marked attenuation of the rate of ROS generation in response to loss of shear compared with the wild type (73). Similar attenuation of the response to loss of shear was observed following the treatment of wild-type cells with an agent (filipin) that disrupts caveolae (73); here, loss of caveolin may disrupt the NOX2 assembly mechanism (74). However, the ROS response (via NOX2) following thrombin treatment of caveolin-1-null cells was similar to that of wild type, indicating that the ROS-generating pathway was intact (73). Thus, caveolae appear to be involved in the sensing of altered shear rather than merely serving as a platform for enzymatic activity of the ROS-generating complex. VEGFR2 and possibly PECAM-1 also localize to caveolae so that the caveolar role in mechanosensing may be through PECAM-1-dependent, ligand-independent activation of VEGFR2, which then activates downstream signaling pathways (75). Thus, caveolae may function along with the adhesion receptor complex (VEGFR, VE-cadherin, PECAM) to constitute a mechanosensory receptor complex.
Role of Ion Channels
Studies using isolated cells suggest that an alteration of ion channel activity represents an early response to altered shear stress (26, 76). To determine possible changes in channel activity with altered shear under physiological conditions, we used intravital microscopy to monitor endothelial membrane potential in the isolated lung that was perfused with membrane potential–sensitive fluorescent dyes (45). Depolarization of the pulmonary microvascular endothelial cells in the intact lung was observed almost immediately following the cessation of flow (39, 43, 44). Perfusion of intact lungs with increasing K+ concentrations as calibrating signals indicated that endothelial cell membrane depolarization with flow cessation was equivalent to that observed by perfusion with ~12 mM KCI; this represents a depolarization of ~17 mV, assuming a resting membrane potential of −70 mV(77, 78). A similar membrane depolarization with stop of flow was observed using fluorescent dyes as well as whole-cell patch-clamp analysis of pulmonary endothelial cells that had been flow adapted in vitro (44,73,79). Thus, altered (decreased) shear results in membrane depolarization, suggesting a decreased open probability of channels that set the cell membrane potential.
The membrane potential of endothelial cells, as for most nonexcitable cells, is controlled predominantly by inwardly rectifying K+ (KIR) channels. On the basis of studies of isolated aortic endothelial cells, a KIR current was identified with increased shear, although the molecular identity of the responsible channel was not determined (80). Subsequently, an outwardly rectifying Cl− channel and various nonselective cation channels were also proposed to play a role in the response to altered shear (76, 81–83). Of these, a KIR channel has the most support for a role in the early response to altered shear, although other channels may be responsible for subsequent effects.
KATP channels
The KIR family consists of seven subgroups (84, 85). A KIR of the KIR 2.1 family (cloned from bovine aortic endothelial cells) resulted in a shear-activated K+ current when expressed in Xenopus oocytes or mammalian HEK293 cells and was proposed as a mechanosensitive KIR channel (86). However, this result has not been confirmed under physiological conditions. Our studies have identified KATP channels as having a major role in the endothelial cell response to acutely decreased shear (87). A KATP channel is a heteromeric complex of a KIR protein of the 6 family, i.e., KIR 6.1 or KIR 6.2 with a sulfonylurea receptor (88). KIR 6.2 is the pore-forming component of KATP channels in endothelial cells of the pulmonary macro-and microvasculature (44, 87). With flow-adapted cells subjected to whole-cell patch-clamp studies, KATP channels deactivated upon the cessation of flow, providing a basis for the observed cell membrane depolarization (79). In the absence of KATP channels (KIR 6.2-null cells), the KIR currents were very low and were unaffected upon the removal of shear (79). Pretreatment of lungs with a KATP channel antagonist (glyburide) led to endothelial cell membrane depolarization, even with continued flow (36). Membrane depolarization with stop of flow was prevented in isolated lungs and flow-adapted cells by pretreatment with cromakalim, a classic agonist for KATP channels (43, 87, 89); this agonist is expected to maintain the KATP channels in an open configuration, even with the loss of shear. These results provide strong evidence that KATP channel closure (decreased open probability) is responsible for membrane depolarization in the stop-of-flow model.
Physiological role for KATP channels
An important question is whether the membrane depolarization observed With altered shear results in a downstream signaling response or is an epiphenomenon. Depolarization of the endothelial cell membrane by the addition of high K+ led to ROS generation during continued flow in both intact isolated lungs and endothelial cells in vitro, an effect that was abolished in NOX2-null cells (77, 78, 90). Treatment of lungs and cells with cromakalim to prevent depolarization associated with stop of flow also prevented ROS generation (36, 43, 91). These results indicate that a change in membrane potential triggers NOX2 activation and support the role of membrane depolarization in initiating the signaling cascade.
Although KATP channels can initiate the signaling cascade, what is their role in shear sensing? KATP channels are induced during in vitro flow adaption of pulmonary macro-and microvascular endothelial cells, an observation indicating that the KATP channels are not the primary sensor for altered shear but rather respond to an upstream signal (87). To support this observation, membrane depolarization with stop of flow was not observed in lungs or cells from caveolin-1-null mice (73). Thus, membrane depolarization due to KATP channel inactivation associated with loss of shear would represent a response to altered mechanical stress that is sensed by the mechanosensory complex.
Signal Transmission from the Membrane Potential to Activation of NOX2
The signaling pathway described thus far involves sensing of altered flow by a mechanosensory complex coupled to caveolae, resulting in endothelial cell membrane depolarization and then NOX2 assembly with ROS generation. But what are the signals that are activated by the change in endothelial cell membrane potential? On the basis of the above-described studies dealing with chemoreception, we evaluated PI3K and Akt as the signaling links. Our studies in this area are preliminary and have been published only in abstract form (92). Cessation of flow in our in vitro model of flow-adapted endothelial cells resulted in PI3K and Akt activation. PI3K and Akt activation was not seen in cells that were treated with cromakalim and that therefore did not depolarize with flow cessation. Similar observations were made in cells with knock out of the KATP channel. NOX2 activation with altered shear stress was greatly diminished by the presence of the PI3K inhibitor wortmannin. Lungs that were null for Akt-1, but not for Akt-2, failed to show ROS generation with stop of flow. Thus, Akt-1 is required for NOX2 activation in the mechanotransduction pathway. PI3K and Akt activation was also observed in cells that were null for NOX2, and thus such activation does not represent an effect of ROS. These results indicate that cell membrane depolarization with stop of flow precedes PI3K and Akt activation, which in turn is upstream of NOX2 generation. The mechanism for PI3K activation subsequent to membrane depolarization is not apparent, but an electrical potential–sensitive site in either this enzyme or a relevant upstream G protein is a possibility.
Activation of PI3K and Akt represents an initial (or early) phosphorylation event in the mechanotransduction signaling cascade. The effects of MAP kinase inhibitors indicate that these proteins also participate in the NOX2 activation process. Loss of shear resulted in Erk activation in flow-adapted bovine pulmonary artery endothelial cells (93). MAP kinases are responsible for phosphorylation of Prdx6 (18); the PLA2 activity of this protein is required for NOX2 activation with stop of flow (94), as described above for the response to Ang II (13). The major elements that lead from altered shear to NOX2 activation and ROS production are shown in Figure 5.
Figure 5.

Key elements in endothelial cell sensing and signal transduction following an abrupt decrease in flow in the pulmonary circulation. The signaling cascade leads to NOX2 activation and ROS generation. Abbreviations: MAP kinase, mitogen-activated protein kinase; PI3K, phosphatidylinositol-3 kinase; PLA2, phospholipase A2; Prdx6, peroxiredoxin 6.
CELLULAR AND PHYSIOLOGICAL RESPONSES TO THE ROS-DELIVERED SIGNAL
ROS can influence a broad spectrum of physiological processes through the modification of enzymes, transcription factors, and other metabolic intermediates. With respect to the endothelium, ROS modulates adhesion/migration, growth/proliferation, and survival/apoptosis. Some of these effects, e.g., modulation of apoptosis through the activation of caspase 3, may require supraphysiological concentrations of H2O2 and thus may represent a toxic response rather than a cell-signaling process (72). Studies with models of altered shear stress demonstrate that physiological levels of ROS can modulate several cell functions and may play an important role in vascular remodeling and angiogenesis.
Regulation of Gene Transcription
The modification of transcription factors by interaction with ROS can affect their translocation and binding to elements/promoters in the genome, representing an important redox-sensitive function (Figure 5). Nrf2 is a good example of a transcription factor that is ROS sensitive. Nrf2 binds to the ARE (antioxidant response element, also termed the electrophilic response element) in DNA, driving the transcription of several antioxidant enzymes, including Prdx6 (95). The role of Nrf2 in the response to altered shear stress has not yet been evaluated. Activator protein-1 (AP-1) and nuclear factor κB (NFκB), two other redox-sensitive transcription factors, are upregulated by ROS that are generated subsequent to acute flow cessation (46). These studies, carried out with bovine pulmonary artery endothelial cells, showed an increased expression in the p50 and p65 subunits of NFκB and in the c-jun and c-fos subunits of AP-1 (Figure 6). Additionally, the p50 subunit of NFκB requires cysteine oxidation to bind DNA as the basis for its redox sensitivity (96). Both NFκB and AP-1 have the potential to drive a proinflammatory phenotype in the endothelium through the upregulation of adhesion molecules such as vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin (97, 98).
Figure 6.

Downstream signaling by NOX2-generated ROS. ROS can activate several transcription factors that result in a diverse array of physiological effects. AP-1, activator protein-1; IκB, inhibitor of NFκB activation.
Cell Proliferation and Angiogenesis
ROS induce endothelial cell proliferation in various isolated cell models. An increase in the ROS-dependent proliferative profile was confirmed by fluorescence-activated cell sorting analysis with cells obtained using our model of altered shear stress in flow-adapted pulmonary microvascular endothelium (73, 91, 99). This proliferative phenotype was blocked by the administration of antioxidants or by knock out of NOX2, the source of ROS in this model (91, 99). Likewise, preventing cell membrane depolarization (KATP channel-null or cromakalim pretreatment) with altered shear in flow-adapted cells prevented the proliferative phenotype (91, 99). A critical question is whether the cell proliferation associated with altered mechanotransduction is random or represents an attempt at angiogenesis. Our studies have only recently begun to address this issue. Preliminary experiments with cells grown in Matrigel™ in vitro or placed subcutaneously in mice indicate new vessel formation compatible with angiogenesis (100). Ligation of the left pulmonary artery in mice resulted in proliferation of the systemic vasculature, arising mainly from intercostal vessels (101). This angiogenesis appears to be ROS dependent, although how the signal is transmitted from the pulmonary to the systemic circulation is not clear. From a teleological standpoint, angiogenesis would be a logical response to the acute loss of blood flow.
MECHANOSIGNALING BEYOND ROS
Intracellular Ca2+ Flux
Intracellular Ca2+ concentrations as detected by fluorescent imaging rapidly increase upon the cessation of flow in flow-adapted endothelial cells in vitro and in lung endothelium in situ (43, 89, 102). This rise in Ca2+ was significantly inhibited by pretreatment with thapsigargin, an agent that depletes Ca2+ from intracellular stores. Thus, an increase in intracellular Ca2+ with stop of flow can occur by Ca2+ entry from the extracellular space, presumably via endothelial cell membrane Ca2+ channels. Using blockers or inhibitors, we identified this channel to be a T-type Ca2+ channel; Ca2+ influx in flow-adapted cells with loss of shear was blocked by mibefradil (which is semispecific as a T-type Ca2+ channel blocker), but not by nifedipine (an L-type Ca2+ channel blocker) (103). Furthermore, the estimated change in membrane potential with stop of flow (~20 mV) is compatible with the known voltage window for T-type channels. Ca2+ influx was blocked by a KATP channel agonist (cromakalim), as expected, because this treatment prevents cell membrane depolarization with flow cessation. Mibefradil had no effect on cell membrane depolarization with flow cessation, indicating that KATP channel closure preceded (and was responsible for) Ca2+ channel activation (103). Thus, activation of T-type Ca2+ channels as a result of endothelial cell membrane depolarization can allow Ca2+ influx with mechanotransduction (Figure 7).
Figure 7.

Mechanism for increased intracellular Ca2+ following an abrupt cessation of flow. The acute decrease in shear results in KATP channel closure, leading to endothelial cell membrane depolarization. Such depolarization in turn results in the opening of T-type voltage-gated Ca2+ channels (VGCC) in the plasma membrane, permitting Ca2+ entry (103). The increased intracellular Ca2+ can associate with calmodulin (CaM) to activate endothelial nitric oxide synthase (eNOS), resulting in ·NO generation (89, 93).
In the absence of thapsigargin treatment, the abrupt stop of flow can result in the release of Ca2+ from its intracellular stores. Although there may be more than one mechanism for this effect, we demonstrated that one possibility is associated with extracellular generated by NOX2. Extracellular can penetrate the endothelial cell through CI− channels and interact with membrane-associated proteins such as the IP3 receptor, an effect that results in intracellular Ca2+ release (60, 62). This effect has been described above as a stimulus for -mediated release by mitochondria, a Ca2+-dependent mechanism (Figure 3).
Nitric Oxide Synthase and ·NO Generation
In addition to ROS, flow-adapted endothelial cells and lung endothelium in situ produce ·NO with cessation of flow. In isolated rat lungs, ·NO production by the pulmonary endothelium was detected during the first minute after stop of flow and was preceded by a rise in intracellular Ca2+ (43, 89). Perfusion with a Ca2+-free medium inhibited ·NO production, implicating the activation of the Ca2+-dependent enzyme endothelial nitric oxide synthase (eNOS). Inhibitors of PI3K, Akt, and MAP kinases block ·NO generation with stop flow (89, 93), indicating that the upstream activating pathway is similar for eNOS andNOX2 (Figure 4). Thus, both and ·NO are generated intracellularly following flow cessation due to increased intracellular Ca2+, ·NO from eNOS activation (93), and from mitochondrial depolarization (62). ·NO and can react to form the highly reactive and potent nitrating agent ONOO− (the peroxynitrite anion), which effectively reduces the ·NO concentration. Extensive generation of nitrotyrosine moieties in lung proteins has been demonstrated as a result of lung ischemia (104). Whether such generation represents part of the signaling paradigm or is just an unfortunate (toxic) by-product is not known at this time. The generation of a vasodilator (NO) as part of the mechanosensory response can be considered an appropriate physiological response to the loss of blood flow.
SUMMARY AND CONCLUSIONS
ROS have now been recognized as important physiological mediators (in addition to their microbicidal activity) for the transmission of cellular signals. This review discusses results for ROS-mediated cell signaling by the pulmonary endothelium using the models of an intact lung and flow-adapted pulmonary microvascular endothelial cells in culture subjected to the physiological stimulus of acute reduction in perfusate flow. These models may correspond to the expected response to acute ischemia associated with a pulmonary artery embolism, lung transplantation, or other etiology for altered pulmonary blood flow. A key component of this response is endothelial NOX (NOX2). Although discovered in phagocytic cells, NOX2 is now well characterized in the endothelium. ROS generated by NOX2 participate in cell signaling, but overactivity of this enzyme can contribute to the oxidative stress that is evident in various vascular pathologies. Thus, information on NOX2 expression, activation, and assembly in response to various agonists and stimuli is critical for understanding the regulation of this enzyme in lung physiology and pathophysiology.
Our work on mechano- and chemotransduction of pulmonary endothelial cells sheds light on the seemingly diverse mechanisms that drive assembly of the NOX complex. Mechanotransduction activates NOX2 via depolarization of the endothelial cell plasma membrane, which in turn triggers the activation of kinases (PI3K and Akt) that drive NOX2 assembly. NOX2 agonists that activate PKC via either receptor-dependent or receptor-independent mechanisms work through pathways similar to mechanotransduction pathways, although the inciting signal is membrane perturbation of a different sort. An important new finding is that the PLA2 activity of Prdx6 serves as a link between the activation of intracellular kinases and the assembly of the oxidase in both chemotransduction as well as mechanotransduction pathways. The complex roles played by ROS in redox signaling and oxidative stress and the highly specific regulation of NOX2 may serve as the basis for the development of novel therapies that target redox pathways in a cell-specific manner at appropriate time points in the disease process.
Glossary
- ROS
reactive oxygen species
- NOX2
NADPH oxidase type 2
- NOS
nitric oxide synthase
- ETC
electron transport chain
- Ang II
angiotensin II
- InsP3
inositol 1,4,5-triphosphate
- PKC
protein kinase C
- Prdx6
peroxiredoxin 6
- PLA2
phospholipase A2
- MAP kinases
mitogen-activated protein kinases
- PECAM
platelet-endothelial cell adhesion molecule
- VE
cadherin, vascular endothelial cadherin
- VEGFR2
vascular endothelial growth factor receptor 2
- Akt
also termed protein kinase B
- PI3K
phosphatidyl inositol-3 Kinase
- KIR channels
inwardly rectifying K+ channels
- AP-1
activator protein-1
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Investig. 1973;52:741–44. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forman HJ, Torres M, Fukuto J. Redox signaling. Mol Cell Biochem. 2002;234/235:49–62. [PubMed] [Google Scholar]
- 3.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–89. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 4.Davies PF, Barbee KA, Volin MV, Robotewskyj A, Chen J, et al. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Annu Rev Physiol. 1997;59:527–49. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- 5.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–76. [PubMed] [Google Scholar]
- 6.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–61. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res. 2005;65:495–504. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 8.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 9.Touyz RM. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp Physiol. 2005;90:449–55. doi: 10.1113/expphysiol.2005.030080. [DOI] [PubMed] [Google Scholar]
- 10.Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, et al. Regulation of the phagocyte NADPH oxidase activity: Phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009;23:1011–22. doi: 10.1096/fj.08-114553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal. 2009;11:791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal. 2009;11:841–60. doi: 10.1089/ars.2008.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, et al. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem. 2011;286:11696–701. doi: 10.1074/jbc.M110.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–52. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 15.Fisher AB. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid Redox Signal. 2011;15(3):831–44. doi: 10.1089/ars.2010.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem. 2000;275:28421–27. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- 17.Manevich Y, Shuvaeva T, Dodia C, Kazi A, Feinstein SI, Fisher AB. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A2 activities. Arch Biochem Biophys. 2009;485:139–49. doi: 10.1016/j.abb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Feinstein SI, Manevich Y, Chowdhury I, Pak JH, et al. Mitogen activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipid A2 activity. Biochem J. 2009;419:669–79. doi: 10.1042/BJ20082061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bostan M, Galatiuc C, Hirt M, Constantin MC, Brasoveanu LI, Iordachescu D. Phospholipase A2 modulates respiratory burst developed by neutrophils in patients with rheumatoid arthritis. J Cell Mol Med. 2003;7:57–66. doi: 10.1111/j.1582-4934.2003.tb00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silliman CC, Elzi DJ, Ambruso DR, Musters RJ, Hamiel C, et al. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leukoc Biol. 2003;73:511–24. doi: 10.1189/jlb.0402179. [DOI] [PubMed] [Google Scholar]
- 21.Dana R, Leto TL, Malech HL, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J Biol Chem. 1998;273:441–45. doi: 10.1074/jbc.273.1.441. [DOI] [PubMed] [Google Scholar]
- 22.Akiba S, Dodia C, Chen X, Fisher AB. Characterization of acidic Ca2+-independent phospholipase A2 of bovine lung. Comp Biochem Physiol B. 1998;120:393–404. doi: 10.1016/s0305-0491(98)10046-9. [DOI] [PubMed] [Google Scholar]
- 23.Barbee KA, Mundel T, Lal R, Davies PF. Subcellular distribution of shear stress at the surface of flow-aligned and nonaligned endothelial monolayers. Am J Physiol Heart Circ Physiol. 1995;268:1765–72. doi: 10.1152/ajpheart.1995.268.4.H1765. [DOI] [PubMed] [Google Scholar]
- 24.Frangos J, McIntire L, Eskin S. Shear stress induced stimulation of mammalian cell metabolism. Biotechnol Bioeng. 1988;32:1053–60. doi: 10.1002/bit.260320812. [DOI] [PubMed] [Google Scholar]
- 25.Langille BL, Adamson SL. Relationship between blood flow direction and endothelial cell orientation at arterial branch sites in rabbits and mice. Circ Res. 1981;48:481–88. doi: 10.1161/01.res.48.4.481. [DOI] [PubMed] [Google Scholar]
- 26.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yerrapureddy A, Tobias J, Margulies SS. Cyclic stretch magnitude and duration affect rat alveolar epithelial gene expression. Cell Physiol Biochem. 2010;25:113–22. doi: 10.1159/000272056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaiahgari S, Yerrapureddy A, Hassoun PM, Garcia JG, Birukov KG, Reddy SP. EGFR-activated signaling and actin remodeling regulate cyclic stretch-induced NRF2-ARE activation. Am J Respir Cell Mol Biol. 2007;36:304–12. doi: 10.1165/rcmb.2006-0131OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168:1749–61. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S, Chapman KE, Fisher AB. Lung ischemia: a model for endothelial mechanotransduction. Cell Biochem Biophys. 2008;52:125–38. doi: 10.1007/s12013-008-9030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, Wang DL. Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:3570–77. doi: 10.1161/01.atv.17.12.3570. [DOI] [PubMed] [Google Scholar]
- 32.Jo H, Song H, Mowbray A. Role of NADPH oxidases in disturbed flow- and BMP4-induced inflammation and atherosclerosis. Antioxid Redox Signal. 2006;8:1609–19. doi: 10.1089/ars.2006.8.1609. [DOI] [PubMed] [Google Scholar]
- 33.Morgan VL, Graham TP, Jr, Roselli RJ, Lorenz CH. Alterations in pulmonary artery flow patterns and shear stress determined with three-dimensional phase-contrast magnetic resonance imaging in Fontan patients. J Thorac Cardiovasc Surg. 1998;116:294–304. doi: 10.1016/s0022-5223(98)70130-8. [DOI] [PubMed] [Google Scholar]
- 34.Ochoa CD, Wu S, Stevens T. New developments in lung endothelial heterogeneity: Von Willebrand factor, P-selectin, and the Weibel-Palade body. Semin Thromb Hemost. 2010;36:301–8. doi: 10.1055/s-0030-1253452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelfand BD, Epstein FH, Blackman BR. Spatial and spectral heterogeneity of time-varying shear stress profiles in the carotid bifurcation by phase-contrast MRI. J Magn Reson Imaging. 2006;24:1386–92. doi: 10.1002/jmri.20765. [DOI] [PubMed] [Google Scholar]
- 36.Al-Mehdi AB, Shuman H, Fisher AB. Intracellular generation of reactive oxygen species during nonhypoxic lung ischemia. Am J Physiol Lung Cell Mol Physiol. 1997;272:294–300. doi: 10.1152/ajplung.1997.272.2.L294. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzaki I, Chatterjee S, Debolt K, Manevich Y, Zhang Q, Fisher AB. Membrane depolarization and NADPH oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;288:336–43. doi: 10.1152/ajpheart.00025.2004. [DOI] [PubMed] [Google Scholar]
- 38.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:486–96. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 39.Al-Mehdi AB, Zhao G, Fisher AB. ATP-independent membrane depolarization with ischemia in the oxygen-ventilated isolated rat lung. Am J Respir Cell Mol Biol. 1998;18:653–61. doi: 10.1165/ajrcmb.18.5.2834. [DOI] [PubMed] [Google Scholar]
- 40.Fisher AB, Dodia C, Tan ZT, Ayene I, Eckenhoff RG. Oxygen-dependent lipid peroxidation during lung ischemia. J Clin Investig. 1991;88:674–79. doi: 10.1172/JCI115352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayene IS, Dodia C, Fisher AB. Role of oxygen in oxidation of lipid and protein during ischemia/reperfusion in isolated perfused rat lung. Arch Biochem Biophys. 1992;296:183–89. doi: 10.1016/0003-9861(92)90561-a. [DOI] [PubMed] [Google Scholar]
- 42.Al-Mehdi A, Shuman H, Fisher AB. Fluorescence microtopography of oxidative stress in lung ischemia-reperfusion. Lab Investig. 1994;70:579–87. [PubMed] [Google Scholar]
- 43.Song C, Al-Mehdi AB, Fisher AB. An immediate endothelial cell signaling response to lung ischemia. Am J Physiol Lung Cell Mol Physiol. 2001;281:993–1000. doi: 10.1152/ajplung.2001.281.4.L993. Erratum. 2002. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:167. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am J Physiol Lung Cell Mol Physiol. 2005;289:954–61. doi: 10.1152/ajplung.00210.2005. [DOI] [PubMed] [Google Scholar]
- 45.Chatterjee S, Fisher AB. Detection of ROS with altered shear stress in lung endothelium. In: Das DK, editor. Methods in Redox Signaling. New Rochelle, NY: Mary Ann Liebert; 2010. p. 213. [Google Scholar]
- 46.Wei Z, Costa K, Al-Mehdi AB, Dodia C, Muzykantov V, Fisher AB. Simulated ischemia in flow-adapted endothelial cells leads to generation of reactive oxygen species and cell signaling. Circ Res. 1999;85:682–89. doi: 10.1161/01.res.85.8.682. [DOI] [PubMed] [Google Scholar]
- 47.Manevich Y, Al-Mehdi A, Muzykantov V, Fisher AB. Oxidative burst and NO generation as initial response to ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol. 2001;280:2126–35. doi: 10.1152/ajpheart.2001.280.5.H2126. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, Chatterjee S, Wei Z, Liu WD, Fisher AB. Rac and PI3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxid Redox Signal. 2008;10:679–89. doi: 10.1089/ars.2007.1521. [DOI] [PubMed] [Google Scholar]
- 49.Boyd NL, Park H, Yi H, Boo YC, Sorescu GP, et al. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am J Physiol Heart Circ Physiol. 2003;285:1113–22. doi: 10.1152/ajpheart.00302.2003. [DOI] [PubMed] [Google Scholar]
- 50.Nakashima H, Suzuki H, Ohtsu H, Chao JY, Utsunomiya H, et al. Angiotensin II regulates vascular and endothelial dysfunction: recent topics of angiotensin II type-1 receptor signaling in the vasculature. Curr Vasc Pharmacol. 2006;4:67–78. doi: 10.2174/157016106775203126. [DOI] [PubMed] [Google Scholar]
- 51.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 52.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao G, Al-Mehdi AB, Fisher AB. Anoxia-reoxygenation versus ischemia in isolated rat lungs. Am J Physiol Lung Cell Mol Physiol. 1997;273:1112–17. doi: 10.1152/ajplung.1997.273.6.L1112. [DOI] [PubMed] [Google Scholar]
- 54.Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–56. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–92. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 56.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–28. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 57.Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol. 2004;14:679–86. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 58.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–26. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 59.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–84. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 60.Madesh M, Hawkins BJ, Milovanova T, Bhanumathy CD, Joseph SK, et al. Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J Cell Biol. 2005;170:1079–90. doi: 10.1083/jcb.200505022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–54. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 62.Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell. 2007;18:2002–12. doi: 10.1091/mbc.E06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuzaki S, Szweda PA, Szweda LI, Humphries KM. Regulated production of free radicals by the mitochondrial electron transport chain: cardiac ischemic preconditioning. Adv Drug Deliv Rev. 2009;61:1324–31. doi: 10.1016/j.addr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011;15:1517–30. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Velayutham M, Hermann C, Zweier JL. Removal of H2O2 and generation of superoxide radical: role of cytochrome c and NADH. Free Radic Biol Med. 2011;51:160–70. doi: 10.1016/j.freeradbiomed.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osawa M, Masuda M, Kusano K, Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: Is it a mechanoresponsive molecule? J Cell Biol. 2002;158:773–85. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–31. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 69.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, et al. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA. 2002;99:9462–67. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93:354–63. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 71.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136–42. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 72.Yang B, Rizzo V. TNF-α potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:954–62. doi: 10.1152/ajpheart.00758.2006. [DOI] [PubMed] [Google Scholar]
- 73.Milovanova T, Chatterjee S, Hawkins BJ, Hong N, Sorokina EM, et al. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta. 2008;1783:1866–75. doi: 10.1016/j.bbamcr.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. 2004;22:985–92. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- 75.Oshikawa J, Urao N, Kim HW, Kaplan N, Razvi M, et al. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS ONE. 2010;5:e10189. doi: 10.1371/journal.pone.0010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fisher AB, Chien S, Barakat AI, Nerem RM. Endothelial cellular response to altered shear stress. Am J Physiol Lung Cell Mol Physiol. 2001;281:529–33. doi: 10.1152/ajplung.2001.281.3.L529. [DOI] [PubMed] [Google Scholar]
- 77.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, et al. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+ Circ Res. 1998;83:730–37. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- 78.Al-Mehdi AB, Shuman H, Fisher AB. Oxidant generation with K+-induced depolarization in the isolated perfused lung. Free Radic Biol Med. 1997;23:47–56. doi: 10.1016/s0891-5849(96)00574-6. [DOI] [PubMed] [Google Scholar]
- 79.Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation. 2006;13:633–44. doi: 10.1080/10739680600930255. [DOI] [PubMed] [Google Scholar]
- 80.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–70. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 81.Barakat AI, Leaver EV, Pappone PA, Davies PF. A now-activated chloride-selective membrane current in vascular endothelial cells. Circ Res. 1999;85:820–28. doi: 10.1161/01.res.85.9.820. [DOI] [PubMed] [Google Scholar]
- 82.Ohno M, Cooke JP, Dzau VJ, Gibbons GH. Fluid shear stress induces endothelial transforming growth factor β-1 transcription and production. Modulation by potassium channel blockade. J Clin Investig. 1995;95:1363–69. doi: 10.1172/JCI117787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suvatne J, Barakat AI, O’Donnell ME. Flow-induced expression of endothelial Na-K-Cl cotransport: dependence on K+ and Cl− channels. Am J Physiol Cell Physiol. 2001;280:216–27. doi: 10.1152/ajpcell.2001.280.1.C216. [DOI] [PubMed] [Google Scholar]
- 84.Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol. 1995;5:268–77. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 85.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–91. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 86.Hoger JH, Ilyin VI, Forsyth S, Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA. 2002;99:7780–85. doi: 10.1073/pnas.102184999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol. 2003;285:959–67. doi: 10.1152/ajpcell.00511.2002. [DOI] [PubMed] [Google Scholar]
- 88.Yokoshiki H, Sunagawa M, Seki T, Sperelakis N. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am J Physiol Cell Physiol. 1998;274:25–37. doi: 10.1152/ajpcell.1998.274.1.C25. [DOI] [PubMed] [Google Scholar]
- 89.Al-Mehdi AB, Song C, Tozawa K, Fisher AB. Ca2+- and phosphatidylinositol 3-kinase-dependent nitric oxide generation in lung endothelial cells in situ with ischemia. J Biol Chem. 2000;275:39807–10. doi: 10.1074/jbc.C000702200. [DOI] [PubMed] [Google Scholar]
- 90.Al-Mehdi AB, Ischiropoulos H, Fisher AB. Endothelial cell oxidant generation during K+-induced membrane depolarization. J Cell Physiol. 1996;166:274–80. doi: 10.1002/(SICI)1097-4652(199602)166:2<274::AID-JCP4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 91.Milovanova T, Chatterjee S, Manevich Y, Kotelnikova I, Debolt K, et al. Lung endothelial cell proliferation with decreased shear stress is mediated by reactive oxygen species. Am J Physiol Cell Physiol. 2006;290:66–76. doi: 10.1152/ajpcell.00094.2005. [DOI] [PubMed] [Google Scholar]
- 92.Chatterjee S, Browning E, Hong NK, DeBolt K, Sorokina E, et al. PI3 kinase/Akt activation trigger ROS production in a model of pulmonary ischemia. FASEB J. 2010;24:796–6. [Google Scholar]
- 93.Wei Z, Al-Mehdi AB, Fisher AB. Signaling pathway for nitric oxide generation with simulated ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:2226–32. doi: 10.1152/ajpheart.2001.281.5.H2226. [DOI] [PubMed] [Google Scholar]
- 94.Chatterjee S, Feinstein S, Hong NK, Debolt K, Fisher AB. Paradoxical response of ROS production in peroxiredoxin 6 null mice to ischemia. FASEB J. 2007;21:894–5. [Google Scholar]
- 95.Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, Feinstein SI. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med. 2009;46:146–53. doi: 10.1016/reeradbiomed.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–20. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 97.Closa D, Folch-Puy E. Oxygen free radicals and the systemic inflammatory response. IUBMB Life. 2004;56:185–91. doi: 10.1080/15216540410001701642. [DOI] [PubMed] [Google Scholar]
- 98.Wiggins JE, Patel SR, Shedden KA, Goyal M, Wharram BL, et al. NFkappa;B promotes inflammation, coagulation, and fibrosis in the aging glomerulus. J Am Soc Nephrol. 2010;21:587–97. doi: 10.1681/ASN.2009060663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Milovanova T, Manevich Y, Haddad A, Chatterjee S, Moore JS, Fisher AB. Endothelial cell proliferation associated with abrupt reduction in shear stress is dependent on reactive oxygen species. Antioxid Redox Signal. 2004;6:245–58. doi: 10.1089/152308604322899314. [DOI] [PubMed] [Google Scholar]
- 100.Chatterjee S, Hong NK, Yu K, Fisher AB. Endothelial mechanotransduction with loss of shear is a signal for angiogenesis. FASEB J. 2010;24:602–3. [Google Scholar]
- 101.Nijmeh J, Moldobaeva A, Wagner EM. Role of ROS in ischemia-induced lung angiogenesis. Am J Physiol Lung Cell Mol Physiol. 2010;299:535–41. doi: 10.1152/ajplung.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tozawa K, Al-Mehdi AB, Muzykantov V, Fisher AB. In situ imaging of intracellular calcium with ischemia in lung subpleural microvascular endothelial cells. Antioxid Redox Signal. 1999;1:145–54. doi: 10.1089/ars.1999.1.2-145. [DOI] [PubMed] [Google Scholar]
- 103.Wei Z, Manevich Y, Al-Mehdi AB, Chatterjee S, Fisher AB. Ca2+ flux through voltage-gated channels with flow cessation in pulmonary microvascular endothelial cells. Microcirculation. 2004;11:517–26. doi: 10.1080/10739680490476367. [DOI] [PubMed] [Google Scholar]
- 104.Ischiropoulos H, Al-Mehdi AB, Fisher AB. Reactive species in ischemic rat lung injury: contribution of peroxynitrite. Am J Physiol Lung Cell Mol Physiol. 1995;269:158–64. doi: 10.1152/ajplung.1995.269.2.L158. [DOI] [PubMed] [Google Scholar]