

Abstract
Uveal (ocular) melanoma is an aggressive cancer that metastasizes in up to half of patients. Uveal melanoma spreads preferentially to the liver, and the metastatic disease is almost always fatal. There are no effective therapies for advanced metastatic disease, so the most promising strategy for improving survival is to detect metastasis at an earlier stage or to treat high-risk patients in an adjuvant setting. An accurate test for identifying high-risk patients would allow for such personalized management as well as for stratification of high-risk patients into clinical trials of adjuvant therapy.
We developed a gene expression profile (GEP) that distinguishes between primary uveal melanomas that have a low metastatic risk (class 1 tumors) and those with a high metastatic risk (class 2 tumors). We migrated the GEP from a high-density microarray platform to a 15-gene, qPCR-based assay that is now performed in a College of American Pathologists (CAP)-accredited Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory on a routine clinical basis on very small samples obtained by fine needle aspiration and on archival formalin-fixed specimens. We collaborated with several centers to show that our specimen collection protocol was easily learned and performed and that it allowed samples to be safely and reliably transported from distant locations with a very low failure rate. Finally, we showed in a multicenter, prospective study that our GEP assay is highly accurate for predicting which patients will develop metastatic disease, and it was significantly superior to the previous gold standard, chromosome 3 testing for monosomy 3. This is the only prognostic test in uveal melanoma ever to undergo such extensive validation, and it is currently being used in a commercial format under the trade name DecisionDx-UM in over 100 centers in the USA and Canada.
Keywords: Uveal melanoma, Metastasis, Prognosis, Gene expression profiling, Support vector machine, Machine learning algorithm
1 Introduction
Uveal melanoma is the most common primary malignancy of the eye, with an incidence of about 1,200–1,500 new cases per year in the USA, and it accounts for about 5 % of all melanomas [1–4]. Uveal melanomas can arise anywhere in the uveal tract of the eye, composed of the iris, ciliary body, and choroid. Uveal melanomas rarely exhibit regional lymphatic spread, but, rather, they metastasize hematogenously to the liver and, to a lesser extent, other sites such as lung and bone [5]. Clinical and histopathologic features associated with poor prognosis include larger tumor size, ciliary body involvement, advanced patient age, epithelioid cell type, extracellular matrix patterning, and extraocular tumor invasion [6–10]. The mortality rate at 15-year diagnosis of the primary tumor is about 50 % [11], and median survival after detection of metastatic disease is about 9 months [12].
1.1 Chromosomal Alterations as Prognostic Markers
Several recurring chromosomal abnormalities in uveal melanoma have been used for prognostication, including loss of 1p, 3, 6q, 8p, and 9p and gain of 1q, 6p, and 8q. Various techniques have been used to detect these changes, including standard karyotyping [13–19], fluorescence in situ hybridization (FISH) [20, 21], comparative genomic hybridization (CGH) [22–28], spectral karyotyping [29], microsatellite analysis (MSA) [30, 31], multiplex ligation-dependent probe amplification (MLPA) [32], and single-nucleotide polymorphisms (SNPs) [33]. Loss of one copy of chromosome 3 (monosomy 3) occurs in almost half of uveal melanomas and is the most prognostically significant of these chromosomal markers [31, 34]. The prognostic accuracy of chromosome 3 status can be improved by including other chromosomal information, including 6p and 8q gain, as well as clinical and histopathologic information, which results in multiple combinations of prognostic groups [32].
1.2 Transition from Chromosomal Markers to Gene Expression Profiling
Cytogenetic alterations provided an important step towards the development of accurate prognostic markers for uveal melanoma, but they have a number of significant drawbacks that limit their value for routine clinical use. These methods were developed from uveal melanomas that were treated by enucleation, which provides a large amount of tumor tissue. However, about 90 % of uveal melanomas are treated not by enucleation but by radiotherapy, in which case the only opportunity to obtain tumor tissue is by needle biopsy. Unfortunately, the amount of tumor material obtained by needle biopsy is often insufficient for chromosomal assay techniques.
Further problems with chromosomal prognostic testing include sampling error resulting from intratumoral heterogeneity [32, 35] and the complicated combination of chromosomal changes and clinicopathologic information that are needed to maximize prognostic accuracy [32]. Thus, several groups explored the use of gene expression profile (GEP) as a potentially more robust prognostic and accurate method. Analysis of uveal melanomas using high-density microarrays showed that tumors with disomy 3 exhibited a different GEP than those with monosomy 3 [36]. Our group went on to show that GEP could classify UMs into two prognostically significant groups using unsupervised clustering techniques without regard to chromosomal status [2]. Class 1 tumors had a low risk and class 2 tumors had a high risk of metastasis (see Fig. 1). Notably, the prognostic accuracy of this GEP classification outperformed clinical, pathological, and cytogenetic prognostic indicators [37], and this has been confirmed by several independent groups [38, 39]. A likely reason for the superiority of GEP over cytogenetic methods for prognostication is that cytogenetic markers are often distributed heterogeneously throughout the tumor and are thus prone to sampling error. In contrast, GEP represents a functional “snapshot” of the tumor’s microenvironment that is less variable across the tumor [40]. We migrated the GEP to an assay comprising 12 discriminating genes and 3 control genes performed on a microfluidics platform that could be used on a routine clinical basis on very small samples from fine needle biopsies [40]. The prognostic accuracy of this assay, and its superiority over chromosome 3 status for clinical prognostic testing, was recently validated in a prospective study involving ten centers across North America [41].
Fig. 1.
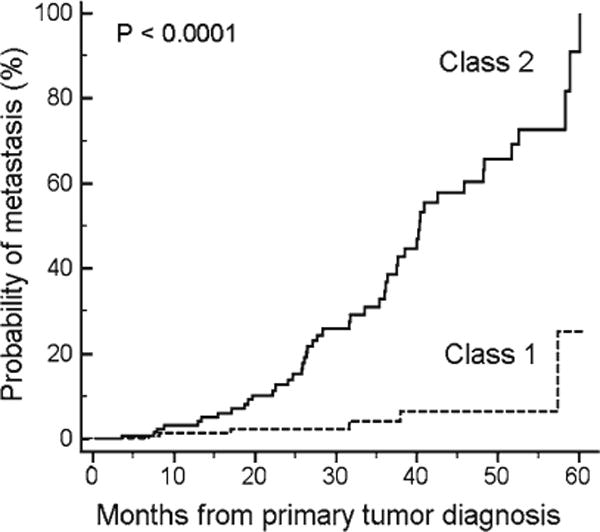
Prognostic performance of the 15-gene assay. Kaplan–Meier survival plot of 334 uveal melanoma patients with up to 5-year follow-up
1.3 Biological Insights from GEP
Aside from its clinical value, gene expression profiling has provided important insights into the pathobiology of UM. The GEP of class 1 tumors closely resembles that of normal uveal melanocytes and low-grade uveal melanocytic tumors, whereas the GEP of class 2 tumors shows reduced expression of melanocytic genes and instead resembles the transcriptome of primitive neural/ectodermal stem cells [42, 43].
The 12 discriminating genes in the GEP assay are indicated in Table 1. Many of these genes have been previously shown to be associated with cancer [40].
Table 1.
Genes included in the 15-gene expression profile
| Gene symbol | Gene name |
|---|---|
| Up-regulated in class 2 uveal melanomas | |
| CDH1 | E-cadherin |
| ECM1 | Extracellular matrix protein 1 |
| HTR2B | 5-Hydroxytryptamine (serotonin) receptor 2B |
| RAB31 | RAB31, member RAS oncogene family |
| Down-regulated in class 2 uveal melanomas | |
| EIF1B | Eukaryotic translation initiation factor 1B |
| FXR1 | Fragile X mental retardation, autosomal homolog 1 |
| ID2 | Inhibitor of DNA binding 2 |
| LMCD1 | LIM and cysteine-rich domains 1 |
| LTA4H | Leukotriene A4 hydrolase |
| MTUS1 | Microtubule-associated tumor suppressor 1 |
| ROBO1 | Roundabout, axon guidance receptor, 1 |
| SATB1 | SATB homeobox 1 |
| Control genes | |
| MRPS21 | Mitochondrial ribosomal protein S21 |
| RBM23 | RNA-binding motif protein 23 |
| SAP130 | Sin3A-associated protein, 130 kDa |
1.4 Class 2 Tumors and BAP1 Mutations
Our findings suggested that class 2 tumors have undergone mutations that lead to a loss of melanocyte cell identity and reversion to a stem-like phenotype. We used exome capture followed by next-generation sequencing to search for mutations that may be specifically associated with class 2 tumors [44]. We identified frequent inactivating mutations in the BRCA1-associated protein 1 (BAP1), located at chromosome 3p21.1, and loss of the other copy of chromosome 3, in the vast majority of class 2 tumors but in only one class 1 tumor which retained two copies of chromosome 3. BAP1 is a ubiquitin carboxy-terminal hydrolase that appears to play a major role in the developmental regulation of chromatin structure as a component of the Polycomb repressor complex PR-DUB [45]. We reported one uveal melanoma patient carrying a germline BAP1 mutation [44], and we identified another family with a germline BAP1 mutation in which uveal and cutaneous melanoma occurred in multiple family members (unpublished data). Subsequent to our report, there have been a growing number of cancers associated with somatic and germline BAP1 mutations, including uveal and cutaneous melanoma, mesothelioma, meningioma, lung cancer, breast cancer, and renal carcinoma [46–53]. Despite the strong correlation between BAP1 mutations and the class 2 signature, however, the latter continues to be much more accurate for clinical prognostic testing. As with chromosomal analysis, which suffers from intratumoral heterogeneity and consequent sampling error, BAP1 mutations can also be heterogeneously distributed within the tumor. Thus, we do not believe that either chromosomal analysis or BAP1 testing should replace the GEP assay for routine clinical use.
2 Materials
2.1 Tumor Tissue Preservation and RNA Isolation
Molecular Devices Picopure RNA Isolation Kit for fine needle aspiration biopsy (FNAB) (Molecular Devices, Sunnyvale, CA).
TRIzol RNA Isolation Reagent for Snap-Frozen Tumor Samples (Invitrogen, Carlsbad, CA).
RecoverAll Total Nucleic Acid Isolation Kit for Formalin-Fixed Paraffin-Embedded (FFPE) samples (Ambion, Austin, TX).
RNeasy Kit (Qiagen, Valencia, CA).
Microcentrifuge (Eppendorf 5415D or similar).
Nuclease-free pipette tips.
0.5 ml microcentrifuge tubes (Applied Biosystems).
2.2 Real-Time PCR
cDNA synthesis reagents: High Capacity cDNA Reverse Transcription Kit (Applied Biosystems Inc.).
- Pre-amplification reagents:
- TaqMan® Pre-Amp Master Mix Kit.
- 20× TaqMan® gene expression assays for the 12 discriminating genes and 3 control genes (Table 1).
Tris–EDTA buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0).
- RT-PCR reagents:
- TaqMan® Gene Expression Master Mix.
- TaqMan® Low Density Array Format 16 RT-PCR plate custom ordered to include the 12 discriminant genes and 3 control genes. The components of the TaqMan® Pre-Amp Mix Kit and Gene Expression Master Mix are the proprietary property of Applied Biosystems, Inc.
Centrifuge (Sorvall Legend T Plus with TTH-750 rotor).
RT-PCR instrument (7900HT Fast Real-Time PCR System).
2.3 Gene Expression Analysis and Class Assignment
Sequence Detection Systems (SDS) Software for 7900HT Fast Real-Time PCR System.
GIST Support Vector Machine learning algorithm software (http://www.chibi.ubc.ca/gist).
3 Methods
3.1 Preparation of RNA
3.1.1 Preparation of RNA from Needle Biopsy Samples
The preferred method of obtaining tumor tissue for the GEP assay is by FNAB (see Note 1). Once the sample is obtained in the needle hub, it is immediately handed off to an un-scrubbed assistant and expelled into an empty RNase-free tube in the operating room (see Fig. 2).
The needle is then placed in another RNase-free tube containing 200 μl of extraction buffer (XB) from the PicoPure® RNA isolation kit, which was drawn up into the needle hub to dislodge and collect additional tumor cells, and the XB is then transferred to the first (empty) tube.
The cap of the tube is closed, and the tube is flicked gently with the finger to suspend the tumor cells in the XB.
The collection tube is then snap frozen in liquid nitrogen prior to leaving the operating room.
For transport to the testing laboratory, tubes are placed on dry ice and mailed by overnight courier.
Once the specimen arrives in the laboratory, RNA is isolated using the PicoPure® kit (including the optional DNase step).
Pipet an equal volume of 70 % ethanol solution to the tube containing FNAB sample in extraction buffer by pipetting up and down ten times.
Pipet the mixture onto the membrane of the pre-cleansed purification column.
Spin at 100 × g for 2 min and immediately followed by a spin at 16,000 × g for 1 min.
Wash the column sequentially with wash buffer 1 and 2 and spin at 8,000 × g for 1 min. Follow with another wash with buffer 2, and spin at 16,000 × g to dry the column.
Elute RNA with 10–30 μl of DEPC-treated water or elution buffer (EB).
To remove genomic DNA from total RNA add 0.1 volume of 10× DNase I buffer and 0.5–1 μl of 2 U/μl DNase I to the RNA solution and incubate at 37 °C for 20–30 min.
Inactivate DNAse I with 0.1 volume of the DNAse inactivation reagent to the sample. Incubate in room temperature for 2 min, and spin at 10,000 × g for 1 min to pellet the DNase inactivation reagent. RNA can be further purified using RNeasy column (Qiagen) or used for subsequent steps.
Determine the concentration of RNA samples using a Nanodrop Fluorospectrometer. This procedure usually yields about 100 ng to 1.5 μg total RNA per FNAB.
Fig. 2.
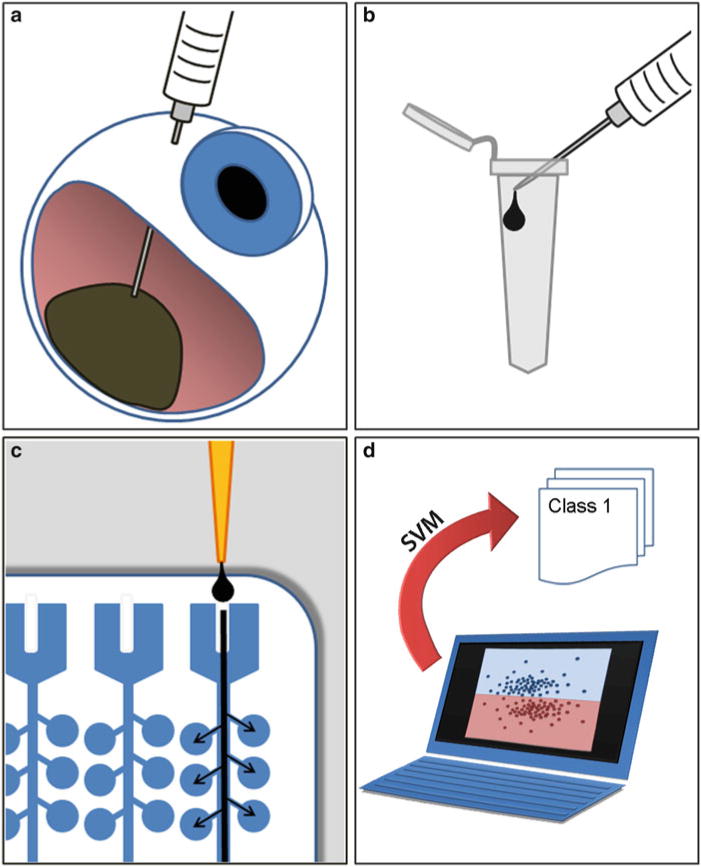
Work flow for 15-gene expression profile prognostic assay. (a) A needle biopsy of the uveal melanoma is performed prior to plaque brachytherapy or immediately after enucleation (eye removal). (b) The needle biopsy aspirate is immediately expelled into an empty tube, and then the same needle is used to draw up 200 μl of extraction buffer, which is then expelled into the first tube containing the tumor sample. (c) RNA is isolated, converted to cDNA, pre-amplified, loaded onto TaqMan® Expression Assays on microfluidics cards, and subjected to PCR using the 7900HT Real-Time PCR System. (d) Ct values are calculated and analyzed using support vector machine (SVM), which compares new test samples to a validated training set of samples. SVM assigns each new sample to class 1 or class 2
3.1.2 Preparation of RNA from Snap-Frozen Tumor Samples
For eyes that are undergoing enucleation, an alternative method for obtaining tumor samples is to open the globe immediately after the eye is removed and dissect a small piece of tumor tissue using a blade or a scissors.
The sample is wrapped in foil, immediately snap frozen in liquid nitrogen, and maintained in a frozen state (at least −80 °C).
When ready for analysis, part or all of the frozen tumor sample is thawed and immediately placed in TRIzol reagent.
RNA is isolated according to the TRIzol protocol, including the optional isolation step, and purified using RNeasy kits according to the manufacturer’s instructions.
Homogenize tissue samples in 1 ml of TRIzol reagent per 50–100 mg of tissues and incubate at room temperature for 5 min to permit the complete tissue dissociation. Centrifuge to remove cell debris.
Transfer supernatant to new tube, and add 0.2 ml of chloroform per 1 ml or TRIzol reagent. Vortex samples and incubate at room temperature for 2–3 min. Centrifuge the samples at 12,000 × g at 8 °C for 15 min.
Remove carefully upper aqueous phase containing RNA, and precipitate RNA by mixing with isopropyl alcohol. Use 0.5 ml of isopropyl alcohol per 1 ml of TRIzol reagent used for the initial homogenization. Incubate samples at 15–30 °C for 10 min and centrifuge at 12,000 × g at 8 °C for 10 min.
Remove the supernatant, and wash the RNA pellet twice with 75 % ethanol (1 ml of ethanol per 1 ml of TRIzol reagent) by vortexing and spinning at 7,500 × g at 8 °C for 5 min.
Air-dry RNA pellet for 5–10 min, and dissolve RNA in DEPC-treated water. Take OD at 260 and 280 nm to determine sample concentration and purity.
RNA samples are stored at −80 °C and handled as described for the biopsy method.
3.1.3 Preparation of RNA from Formalin-Fixed Paraffin-Embedded Samples
The GEP assay can be reliably performed on FFPE samples that are up to 3 years old. For this method, five 10 μm sections are obtained from tissue blocks, and tumor tissue is scraped away from surrounding normal material using a dissecting microscope (laser capture microdissection is not necessary) and collected in RecoverAll™.
Total RNA is isolated using the RecoverAll™ Total Nucleic Acid Isolation kit following the manufacturer’s protocol.
FFPE samples are deparaffinized using a series of xylene and ethanol washes. Tissue slices are placed in microcentrifuge tube, and 1 ml of xylene is added to the sample. Incubate the sample at 50 °C for 3 min to melt paraffin. Centrifuge at maximum speed at room temperature for 2 min.
Remove the xylene, and wash the pellet with 1 ml of ethanol by vortexing. Centrifuge at maximum speed at room temperature for 2 min. Repeat the washing step with 100 % ethanol. Remove the ethanol, and air-dry the pellet.
Next, the samples are subjected to a rigorous protease digestion. Add digestion buffer (100–200 μl) to each sample and 4 μl of Protease K. Incubate the sample in heat blocks at 50 °C for 15 min and then at 80 °C for 15 min.
Add isolation additive/ethanol mixture according to the volume of digestion buffer (e.g., 790–200 μl, respectively) and mix.
RNA is purified using filter cartridge methodology. Pipet the sample/ethanol mixture on the cartridge and centrifuge at 10,000 × g for 30 s. Discard the flow through, and wash the filter cartridge with 700 μl of wash 1 buffer followed by 500 μl of wash 2/3. Centrifuge at 10,000 × g for 30 s each time.
Final step includes an on-filter nuclease treatment with DNase mix containing 6 μl of 10× DNase buffer, 4 μl DNase, and 50 μl nuclease-free water at 22–25 °C for 30 min.
Filter cartridge is washed with 700 μl of wash 1 and centrifuged at 10,000 × g for 30 s. Repeat this step with wash 2/3. RNA is eluted into either water or the low-salt elusion solution. RNA samples are stored at −80 °C and handled as described for the biopsy method.
3.2 Real-Time PCR
RNA samples quantified using the Nanodrop 1000 spectrophotometer are converted to cDNA using the High Capacity cDNA Reverse Transcription kit from Applied Biosystems. Add 50 ng to 1 μg of RNA to the cDNA transcription step in a final reaction volume of 20 μl.
The reverse transcription reaction is performed in a 96-well plate using the 7900HT Real-Time PCR System.
Combine cDNA reactions with 0.2× pooled TaqMan assay mix containing equal volumes of each of the 15 TaqMan assays used to amplify discriminant and control genes and TaqMan® Pre-Amp Master Mix.
Pre-amplification is carried out for 14 cycles in a 96-well plate using the 7900HT system and immediately placed on ice following completion of cycling.
Dilute pre-amplified samples 20-fold into sterile TE buffer and store at −20 °C until needed.
Perform PCR step using the 7900HT Real-Time PCR System with Applied Biosystems TaqMan® Gene Expression Assays and Gene Expression Master Mix following the manufacturer’s protocol.
Thaw, vortex, and centrifuge pre-amplified samples. Add an equal volume of 2× TaqMan® Gene Expression Master Mix to each reaction and mix thoroughly by vortexing. Centrifuge samples prior to loading to 96-well microfluidics plate.
TaqMan® Microfluidics Expression Arrays are custom ordered to include our 12 class discriminating genes, 3 endogenous control genes, and 18S rRNA as a manufacturer’s control, and each sample is analyzed in triplicate.
Add 100 μl of reaction mix to each fill port of the custom microfluidics plate.
Centrifuge the array to dispense approximately 2 μl of pre-amplified reaction mix per well. Verify that all wells have uniform volume following centrifugation step. The plate is ready to be run on the 7900HT instrument.
SDSv2.3 software is used to control the 7900HT system, and samples undergo 40 cycles of amplification during the procedure.
Ct values are calculated using the manufacturer’s software, and mean Ct values are calculated for all triplicate sets. Δ Ct values are calculated by subtracting the mean Ct of each discriminating gene from the geometric mean of the mean Ct values of the three endogenous control genes (see Note 2).
An “undetectable transcript” is defined as a transcript that exhibits no Ct value after 40 qPCR cycles. A “sample failure” is defined as a sample in which one or more endogenous control transcripts are undetectable after 40 qPCR cycles (see Note 3).
3.3 Analysis
We selected support vector machine (SVM) as the machine learning algorithm for this assay because it is a robust and widely accepted machine learning algorithm and because it outperformed other similar algorithms in our analyses.
SVM uses a training set of samples with known molecular class to assign new test samples. We have generated such a training set of samples from patients with very long follow-up.
SVM inputs the gene expression data of the training set as two sets of vectors (class 1 and class 2) in n-dimensional space and then constructs a hyperplane that maximizes the space between the two data sets [54]. SVM then classifies test samples by placing them on one or the other side of this hyperplane. The proximity of the sample to the hyperplane is inversely proportional to the discriminant score, which is a measure of confidence.
Initially, we were concerned that a low discriminant score may be associated with less accurate test results. However, this has not been the case. Nevertheless, we issue a reduced confidence warning if the score is below 0.1.
Footnotes
FNBA is typically performed in the operating room and may occur as an isolated procedure but more often is performed at the time of surgery for insertion of a radioactive plaque for brachytherapy or at the time of enucleation (eye removal). In the case of brachytherapy, the biopsy is performed immediately prior to attachment of the plaque to the surface of the eye. It is important to note that this assay has not been validated for tumors that were previously irradiated, which would be expected to alter global gene expression. In the case of enucleation, the biopsy is performed away from the operative field immediately after eye removal.
Since the amount of RNA in these samples is too low to evaluate for RNA quality using conventional electrophoretic methods, we have found it useful to use the geometric mean of the Ct values of the three endogenous controls as a measure of intact RNA template available for amplification in each sample. This is based on the assumption that the endogenous controls should be expressed at constant levels across all uveal melanomas, so a high Ct value should be a technical rather than biological aberration.
Sample failure in the prospective, multicenter study and on the current commercial platform is less than 5 % of samples, which is far superior to failure rates reported for all available chromosomal analytic platforms that have been subjected to peer review [41]. We found no relationship between sample failure and the concentration of RNA in the original sample as measured by NanoDrop, indicating that the GEP assay can detect RNA transcripts below the limits of the NanoDrop instrument. Rather, sample failure correlates with deviation from the SOPs for obtaining and processing samples. These deviations include failure to immediately snap freeze samples in the operating room and maintain them at −80 °C until analyzed and dilution of the 200 μl of extraction buffer with excess ocular fluid.
References
- 1.Harbour JW. Clinical overview of uveal melanoma: introduction to tumors of the eye. In: Albert DM, Polans A, editors. Ocular oncology. Marcel Dekker; New York: 2003. pp. 1–18. [Google Scholar]
- 2.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egan KM, Seddon JM, Glynn RJ, Gragoudas ES, Albert DM. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988;32:239–251. doi: 10.1016/0039-6257(88)90173-7. [DOI] [PubMed] [Google Scholar]
- 4.Ramaiya KJ, Harbour JW. Current management of uveal melanoma. Exp Rev Ophthalmol. 2007;2:939–946. [Google Scholar]
- 5.Diener-West M, Reynolds SM, Agugliaro DJ, Caldwell R, Cumming K, Earle JD, Hawkins BS, Hayman JA, Jaiyesimi I, Jampol LM, Kirkwood JM, Koh WJ, Robertson DM, Shaw JM, Straatsma BR, Thoma J. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123:1639–1643. doi: 10.1001/archopht.123.12.1639. [DOI] [PubMed] [Google Scholar]
- 6.Augsburger JJ, Gamel JW. Clinical prognostic factors in patients with posterior uveal malignant melanoma. Cancer. 1990;66:1596–1600. doi: 10.1002/1097-0142(19901001)66:7<1596::aid-cncr2820660726>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Gamel JW, McLean IW, Foster WD, Zimmerman LE. Uveal melanomas: correlation of cytologic features with prognosis. Cancer. 1978;41:1897–1901. doi: 10.1002/1097-0142(197805)41:5<1897::aid-cncr2820410534>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 8.de la Cruz PO, Jr, Spech CS, McLean IW. Lymphocytic infiltration in uveal malignant melanoma. Cancer. 1990;65:112–115. doi: 10.1002/1097-0142(19900101)65:1<112::aid-cncr2820650123>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 9.Folberg R, Pe’er J, Gruman LM, Woolson RF, Jeng G, Montague PR, Moninger TO, Yi H, Moore KC. The morphologic characteristics of tumor blood vessels as a marker of tumor progression in primary human uveal melanoma: a matched case-control study. Hum Pathol. 1992;23:1298–1305. doi: 10.1016/0046-8177(92)90299-i. [DOI] [PubMed] [Google Scholar]
- 10.Makitie T, Summanen P, Tarkkanen A, Kivela T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:1414–1421. [PubMed] [Google Scholar]
- 11.Kujala E, Makitie T, Kivela T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44:4651–4659. doi: 10.1167/iovs.03-0538. [DOI] [PubMed] [Google Scholar]
- 12.Kath R, Hayungs J, Bornfeld N, Sauerwein W, Hoffken K, Seeber S. Prognosis and treatment of disseminated uveal melanoma. Cancer. 1993;72:2219–2223. doi: 10.1002/1097-0142(19931001)72:7<2219::aid-cncr2820720725>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.Griffin CA, Long PP, Schachat AP. Trisomy 6p in an ocular melanoma. Cancer Genet Cytogenet. 1988;32:129–132. doi: 10.1016/0165-4608(88)90319-6. [DOI] [PubMed] [Google Scholar]
- 14.Horsman DE, Sroka H, Rootman J, White VA. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990;45:249–253. doi: 10.1016/0165-4608(90)90090-w. [DOI] [PubMed] [Google Scholar]
- 15.Prescher G, Bornfeld N, Becher R. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990;82:1765–1769. doi: 10.1093/jnci/82.22.1765. [DOI] [PubMed] [Google Scholar]
- 16.Sisley K, Rennie IG, Cottam DW, Potter AM, Potter CW, Rees RC. Cytogenetic findings in six posterior uveal melanomas: involvement of chromosomes 3, 6, and 8. Genes Chromosomes Cancer. 1990;2:205–209. doi: 10.1002/gcc.2870020307. [DOI] [PubMed] [Google Scholar]
- 17.Wiltshire RN, Elner VM, Dennis T, Vine AK, Trent JM. Cytogenetic analysis of posterior uveal melanoma. Cancer Genet Cytogenet. 1993;66:47–53. doi: 10.1016/0165-4608(93)90148-f. [DOI] [PubMed] [Google Scholar]
- 18.Singh AD, Boghosian-Sell L, Wary KK, Shields CL, De Potter P, Donoso LA, Shields JA, Cannizzaro LA. Cytogenetic findings in primary uveal melanoma. Cancer Genet Cytogenet. 1994;72:109–115. doi: 10.1016/0165-4608(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 19.Prescher G, Bornfeld N, Friedrichs W, Seeber S, Becher R. Cytogenetics of twelve cases of uveal melanoma and patterns of nonrandom anomalies and isochromosome formation. Cancer Genet Cytogenet. 1995;80:40–46. doi: 10.1016/0165-4608(94)00165-8. [DOI] [PubMed] [Google Scholar]
- 20.McNamara M, Felix C, Davison EV, Fenton M, Kennedy SM. Assessment of chromosome 3 copy number in ocular melanoma using fluorescence in situ hybridization. Cancer Genet Cytogenet. 1997;98:4–8. doi: 10.1016/s0165-4608(96)00405-0. [DOI] [PubMed] [Google Scholar]
- 21.Patel KA, Edmondson ND, Talbot F, Parsons MA, Rennie IG, Sisley K. Prediction of prognosis in patients with uveal melanoma using fluorescence in situ hybridisation. Br J Ophthalmol. 2001;85:1440–1444. doi: 10.1136/bjo.85.12.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Speicher MR, Prescher G, du Manoir S, Jauch A, Horsthemke B, Bornfeld N, Becher R, Cremer T. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994;54:3817–3823. [PubMed] [Google Scholar]
- 23.Gordon KB, Thompson CT, Char DH, O’Brien JM, Kroll S, Ghazvini S, Gray JW. Comparative genomic hybridization in the detection of DNA copy number abnormalities in uveal melanoma. Cancer Res. 1994;54:4764–4768. [PubMed] [Google Scholar]
- 24.Ghazvini S, Char DH, Kroll S, Waldman FM, Pinkel D. Comparative genomic hybridization analysis of archival formalin-fixed paraffin-embedded uveal melanomas. Cancer Genet Cytogenet. 1996;90:95–101. doi: 10.1016/s0165-4608(96)00076-3. [DOI] [PubMed] [Google Scholar]
- 25.Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:313–317. [PubMed] [Google Scholar]
- 26.Hughes S, Damato BE, Giddings I, Hiscott PS, Humphreys J, Houlston RS. Microarray comparative genomic hybridisation analysis of intraocular uveal melanomas identifies distinctive imbalances associated with loss of chromosome 3. Br J Cancer. 2005;93:1191–1196. doi: 10.1038/sj.bjc.6602834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilic E, van Gils W, Lodder E, Beverloo HB, van Til ME, Mooy CM, Paridaens D, de Klein A, Luyten GP. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006;47:3703–3707. doi: 10.1167/iovs.06-0101. [DOI] [PubMed] [Google Scholar]
- 28.Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008;14:115–122. doi: 10.1158/1078-0432.CCR-07-1825. [DOI] [PubMed] [Google Scholar]
- 29.Naus NC, van Drunen E, de Klein A, Luyten GP, Paridaens DA, Alers JC, Ksander BR, Beverloo HB, Slater RM. Characterization of complex chromosomal abnormalities in uveal melanoma by fluorescence in situ hybridization, spectral karyotyping, and comparative genomic hybridization. Genes Chromosomes Cancer. 2001;30:267–273. [PubMed] [Google Scholar]
- 30.Tschentscher F, Prescher G, Zeschnigk M, Horsthemke B, Lohmann DR. Identification of chromosomes 3, 6, and 8 aberrations in uveal melanoma by microsatellite analysis in comparison to comparative genomic hybridization. Cancer Genet Cytogenet. 2000;122:13–17. doi: 10.1016/s0165-4608(00)00266-1. [DOI] [PubMed] [Google Scholar]
- 31.Scholes AG, Damato BE, Nunn J, Hiscott P, Grierson I, Field JK. Monosomy 3 in uveal melanoma: correlation with clinical and histologic predictors of survival. Invest Ophthalmol Vis Sci. 2003;44:1008–1011. doi: 10.1167/iovs.02-0159. [DOI] [PubMed] [Google Scholar]
- 32.Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010;16:6083–6092. doi: 10.1158/1078-0432.CCR-10-2076. [DOI] [PubMed] [Google Scholar]
- 33.Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW. Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin Cancer Res. 2007;13:2923–2927. doi: 10.1158/1078-0432.CCR-06-2383. [DOI] [PubMed] [Google Scholar]
- 34.Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
- 35.Maat W, Jordanova ES, van Zelderen-Bhola SL, Barthen ER, Wessels HW, Schalij-Delfos NE, Jager MJ. The heterogeneous distribution of monosomy 3 in uveal melanomas: implications for prognostication based on fine-needle aspiration biopsies. Arch Pathol Lab Med. 2007;131:91–96. doi: 10.5858/2007-131-91-THDOMI. [DOI] [PubMed] [Google Scholar]
- 36.Tschentscher F, Husing J, Holter T, Kruse E, Dresen IG, Jockel KH, Anastassiou G, Schilling H, Bornfeld N, Horsthemke B, Lohmann DR, Zeschnigk M. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–2584. [PubMed] [Google Scholar]
- 37.Worley LA, Onken MD, Person E, Robirds D, Branson J, Char DH, Perry A, Harbour JW. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007;13:1466–1471. doi: 10.1158/1078-0432.CCR-06-2401. [DOI] [PubMed] [Google Scholar]
- 38.Petrausch U, Martus P, Tonnies H, Bechrakis NE, Lenze D, Wansel S, Hummel M, Bornfeld N, Thiel E, Foerster MH, Keilholz U. Significance of gene expression analysis in uveal melanoma in comparison to standard risk fac tors for risk assessment of subsequent metastases. Eye. 2007;22:997–1007. doi: 10.1038/sj.eye.6702779. [DOI] [PubMed] [Google Scholar]
- 39.van Gils W, Lodder EM, Mensink HW, Kilic E, Naus NC, Bruggenwirth HT, van Ijcken W, Paridaens D, Luyten GP, de Klein A. Gene expression profiling in uveal melanoma: two regions on 3p related to prognosis. Invest Ophthalmol Vis Sci. 2008;49:4254–4262. doi: 10.1167/iovs.08-2033. [DOI] [PubMed] [Google Scholar]
- 40.Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12:461–468. doi: 10.2353/jmoldx.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Onken M, Worley L, Char D, Augsburger J, Correa Z, Nudleman E, Aaberg TM, Jr, Altaweel M, Bardenstein D, Finger P, Gallie B, Harocopos G, Hovland P, McGowan H, Milman T, Mruthyunjaya P, Simpson E, Smith M, Wilson D, Wirostko W, Harbour J. Collaborative Ocular Oncology Group Report No. 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012;119(8):1596–1603. doi: 10.1016/j.ophtha.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006;66:4602–4609. doi: 10.1158/0008-5472.CAN-05-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang SH, Worley LA, Onken MD, Harbour JW. Prognostic biomarkers in uveal melanoma: evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008;18:191–200. doi: 10.1097/CMR.0b013e3283005270. [DOI] [PubMed] [Google Scholar]
- 44.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, Hovland P, Davidorf FH. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, Sander C, Delsite R, Powell S, Zhou Q, Shen R, Olshen A, Rusch V, Ladanyi M. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, Cox NJ, Dogan AU, Pass HI, Trusa S, Hesdorffer M, Nasu M, Powers A, Rivera Z, Comertpay S, Tanji M, Gaudino G, Yang H, Carbone M. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, Socci ND, Rutten A, Palmedo G, Abramson D, Offit K, Ott A, Becker JC, Cerroni L, Kutzner H, Bastian BC, Speicher MR. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Njauw CN, Kim I, Piris A, Gabree M, Taylor M, Lane AM, Deangelis MM, Gragoudas E, Duncan LM, Tsao H. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7(4):e35295. doi: 10.1371/journal.pone.0035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, Kinch L, Hambuch T, Jain S, Lotan Y, Margulis V, Sagalowsky AI, Summerour PB, Kabbani W, Wong SW, Grishin N, Laurent M, Xie XJ, Haudenschild CD, Ross MT, Bentley DR, Kapur P, Brugarolas J. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44(7):751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiesner T, Murali R, Fried I, Cerroni L, Busam K, Kutzner H, Bastian BC. A distinct subset of atypical spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol. 2012;36(6):818–830. doi: 10.1097/PAS.0b013e3182498be5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshikawa Y, Sato A, Tsujimura T, Emi M, Morinaga T, Fukuoka K, Yamada S, Murakami A, Kondo N, Matsumoto S, Okumura Y, Tanaka F, Hasegawa S, Nakano T, Hashimoto-Tamaoki T. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012;103:868–874. doi: 10.1111/j.1349-7006.2012.02223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Jr, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]