

Abstract
The alternative sigma factor E (σE) is critical for response to extracytoplasmic stress in Salmonella. Extensive studies have been conducted on σE-regulated gene expression, particularly at the transcriptional level. Increasing evidence suggests however that σE may indirectly participate in post-transcriptional regulation. In this study, we conducted sample-matched global proteomic and transcriptomic analyses to determine the level of regulation mediated by σE in Salmonella. Samples were analyzed from wild-type and isogenic rpoE mutant Salmonella cultivated in three different conditions: nutrient-rich and conditions that mimic early and late intracellular infection. We found that 30% of the observed proteome was regulated by σE combining all three conditions. In different growth conditions, σE affected the expression of a broad spectrum of Salmonella proteins required for miscellaneous functions. Those involved in transport and binding, protein synthesis, and stress response were particularly highlighted. By comparing transcriptomic and proteomic data, we identified genes post-transcriptionally regulated by σE and found that post-transcriptional regulation was responsible for a majority of changes observed in the σE-regulated proteome. Further, comparison of transcriptomic and proteomic data from hfq mutant of Salmonella demonstrated that σE-mediated post-transcriptional regulation was partially dependent on the RNA-binding protein Hfq.
Keywords: Salmonella, sigma factor E, proteomics, transcriptomics, post-transcriptional regulation, infection, virulence
Introduction
Salmonella enterica serovar Typhimurium (STM, referred to as Salmonella in the following) is a facultative intracellular bacterial pathogen capable of colonizing a wide range of hosts. In susceptible mice, STM causes systemic infection that resembles typhoid fever caused by the S. enterica serovar Typhi in human, which makes STM a paradigm for understanding intracellular pathogenesis.1,2 Within the host, Salmonella confronts a hostile environment while it proceeds through the digestive tract. It survives the low pH milieu of the stomach and out-competes natural gut flora. After invasion of the intestinal epithelium, Salmonella is consumed by underlying macrophages allowing for systemic dissemination in mice.3–5 It replicates inside different populations of immune cells and is most frequently found in monocytes and neutrophils where it reacts to a variety of stresses to maintain cellular integrality and evade innate host immunity.6,7 The high adaptability of Salmonella to various environments is largely dependent on its capability to integrate different environmental cues to achieve coordinated gene regulation under different stresses.
Salmonella utilizes multiple signal transduction systems that govern extracytoplasmic stress response.8 In the presence of environmental factors that lead to accumulation of misfolded proteins in the periplasm, the alternative sigma factor E (σE, encoded by rpoE) plays a major role in sustaining homeostasis. These stressors (e.g., heat shock, ethanol, osmotic stress, immune response, etc.) can initiate a proteolytic cascade that releases σE from sequestration by the antisigma factor RseA at the bacterial inner membrane.9 Free σE binds to core RNA polymerase and recognizes a specific σE-binding motif in DNA to initiate transcription.8,9 Functional σE is crucial for Salmonella intracellular survival, as null mutant of rpoE persist for less than 30 min inside primary macrophage.10
The regulon of σE in Salmonella and its close relative E. coli has been extensively studied.11–14 We recently showed that σE regulates approximately 58% of the entire Salmonella genome and that an almost equal number of genes are up- or down-regulated by this sigma factor under multiple growth conditions.15 The direct effect of σE on gene regulation via promoter recognition of the σE-binding motif is traditionally defined as an event that activates transcription. Therefore, we speculated that the down-regulation of gene expression may not be a direct effect of σE, but rather through the general regulators controlled by σE, or that small noncoding RNAs (sRNAs) recognized by σE as one of the major functions of sRNA are silencing trans-encoded target mRNAs. It has been shown that σE binds sRNAs RybB and MicA, both of which can function as global regulators.16,17 Thus, it is likely that σE is involved in post-transcriptional regulation through its effects on sRNA. In the context of post-transcriptional regulation, Hfq is a major mediator that binds to RNA and facilitates sRNA–mRNA interactions, which modulates the translation and decay of mRNA.18 In Salmonella, Hfq has been shown to post-transcriptionally regulate at least 20% of all possible proteins.19 Both RybB and MicA are regulated by Hfq in repressing outer membrane protein expression.16,20 We found that σE regulates hfq expression in both nutrient-rich and infection-like conditions, which brought up the question whether or not the regulation of σE on Hfq endows σE with the capacity for mediating post-transcriptional regulation.
To identify the level of regulation mediated by σE, we performed sample-matched global proteomic and transcriptomic analyses on wild type (WT) and rpoE-deletion mutant Salmonella cultured in nutrient-rich Luria–Bertani (LB) broth to log phase, in pH 5.8, low phosphate, low magnesium-containing medium (LPM) for 4 h (LPM 4h) or 20 h (LPM 20h). The microarray data of σE obtained in this experiment15 are used here to compare with proteomic data without further delving into transcriptional regulation. We also conducted proteomics and transcriptional analyses in parallel on the parent and isogenic hfq mutant to elucidate the role of Hfq in σE–dependent post-transcriptional regulation. We found that (1) σE affected 30% (344 proteins) of the observed proteome (1138 proteins) combining all three conditions, which involved a broad spectrum of Salmonella proteins needed for various biological processes; (2) post-transcriptional regulation accounts for the majority of σE-mediated protein-level regulation; and (3) up to 22%, 19%, and 29% of all σE–mediated post-transcriptional regulation in LB, LPM 4h, and LPM 20h, respectively, were likely to be dependent on Hfq.
Experimental Procedures
Bacterial Strains and Culture Conditions
STM ATCC14028s was used as the parent strain (WT) of all deletion and tagged strains in this study. The λ red recombination system was employed to delete or tag genes of interest as described before.21 Nonpolar in-frame gene deletion was carried out with modified pKD13 (pKD13-mod) plasmid (pKD13; GenBank accession no. AY048744), which replaces genes of interest with 135-nucleotide (nt) barcode sequences following homologous recombination.22 For HA tagging, pKD13–2HA plasmid was used as PCR template, which introduced a DNA fragment encoding 2HA prior to the stop codon sequence of target gene.19 The plasmid pHfq expressing Hfq was constructed by cloning a DNA fragment containing coding sequence of hfq on pWKS30 via EcoRI and XbaI. Bacterial strains and plasmids used in this study are listed in Table S6 of the Supporting Information. The primers used for tagging chromosomal genes of Salmonella are listed in Table S7 of the Supporting Information.
The WT and mutant strains were grown under three conditions: in LB medium to log phase (OD600, 0.5) and in LPM for 4 or 20 h. Briefly, bacteria were first cultured in LB medium for 16 h at 37 °C with shaking (200 rpm), then either diluted 100-fold into new LB medium and grown to log phase or washed with LPM, diluted 10-fold, and grown in LPM for 4 or 20 h. Cells were harvested by centrifugation, and for proteomic analysis, the pellets were directly frozen at −80 °C until needed; whereas for transcriptomic analysis, pellets were treated with RNAlater (Ambion) and then stored at −20 °C until they were processed. All the bacterial samples were prepared in triplicate.
Global Proteomic Analysis
Quantitative proteomic analysis was performed using the accurate mass and time (AMT) tag approach. Since the optimum LC-MS/MS running conditions for identifying and for quantifying peptides are different, in the AMT tag approach, peptide identification and quantification are performed in separated runs. Peptides are first identified by extensive 2D LC–MS/MS analysis to maximize the proteome coverage. Then the quantification is done by extracting the peak areas of peptides analyzed by 1D LC–MS/MS to diminish variations between runs due to prefractionation step.23
The WT, ΔrpoE, and Δhfq cells were mechanically ruptured by vortexing in the presence of zirconia/silica beads. Cell lysates were then subjected to ultracentrifugation, and resulting soluble and insoluble proteins were digested with trypsin, followed by solid phase extraction clean-up, as described previously.19 Peptides derived from digestion of soluble and insoluble proteins were pooled together and fractionated into 24 fractions by strong cation-exchange (SCX) chromatography.24 Each fraction or unfractionated sample (run in technical duplicates) was subjected to liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis. Peptides were loaded into capillary columns (75 μm × 65 cm, Polymicro) packed with C18 beads (3 μm particles, Phenomenex) connected to a custom-made four-column LC system.25 The elution was performed in an exponential gradient from 0–100% B solvent (solvent A, 0.1% FA; solvent B, 90% ACN/0.1% FA) for 100 min with a constant pressure of 10 000 psi and flow rate of approximately 400 nL/min. Eluting peptides were directly analyzed either on a linear ion-trap (LTQ XL, Thermo Scientific, San Jose, CA) (fractionated samples) or an orbitrap (LTQ Orbitrap XL, Thermo Scientific) (unfractionated samples) mass spectrometer using chemically etched nanospray emitters.26 Full scans were collected at 400–2000 m/z range (60K resolution at 400 m/z for Orbitrap scans), and the top ten most intense ions were subjected to low-resolution CID fragmentation once (35% normalized collision energy) before they were dynamically excluded for 60 s.
To identify peptides, all tandem mass spectra were converted into DTA files using default parameters and searched against the forward and reverse sequences of Salmonella Typhimurium 14028s (5634 sequences) using SEQUEST (v27.12). Database searches were performed considering (1) no enzymatic digestion specificity, (2) no post-translation modifications, (3) 0.5 Da fragment mass tolerance, and (4) 3.0 Da and 20 ppm mass tolerance for precursor ion for linear ion-trap and orbitrap data, respectively. Sequest results were filtered with Xcorr ≥ 1.9, 2.2, and 3.5 for singly-, doubly-, and triply-charged peptides, respectively, expectation value ≤ 0.01, and Peptide Prophet ≥ 0.5. Then identified peptides are used to build a database that contains the information on the peptide theoretical mass and normalized elution time (NET), named mass tag. The mass tags in the database were matched against the high-resolution LC–MS/MS runs using a mass accuracy ≤ 10 ppm and NET ≤ 0.025, and the peak areas were retrieved using VIPER. To ensure the quality of peptide matching, all peptides matched to the MT database were filtered with statistical tools for AMT tag confidence (STAC) using a score ≥ 0.7 and uniqueness probability ≥ 0.5.27 Additionally, peptides had to be present in more than half of the replicates of at least one sample, and proteins were required to have at least two peptides and at least one peptide with STAC ≥ 0.9. Peak areas were then normalized by linear regression and central tendency followed by fold change calculation and ANOVA test using DAnTE.28
Immunoblot Analysis
The HA-tagged WT and mutant strains were grown as described above. Cells were washed, and approximately 5 × 107 colony-forming units were pelleted and resuspended in Laemmli sample buffer, boiled for 5 min, and then loaded on SDS-PAGE. Proteins on the gel were then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). After blocking in Tris-buffered saline (TBS) plus 5% nonfat dry milk for 1 h, membranes were probed with anti-HA monoclonal antibody (Covance) and anti-DnaK monoclonal antibody (Stressgen). Membranes were washed and probed with peroxidase-conjugated antimouse IgG (Sigma). The immune complexes were detected via chemiluminescence using Western Lightning (PerkinElmer), and images were captured with ImageQuant LAS4000 (GE Healthcare Life Sciences).
Transcriptomic Analysis
For each of the three experimental conditions (LB, LPM 4h, and LPM 20h), we identified genes that were differentially expressed between Δhfq and WT strain of Salmonella. The samples were assayed to the Salmonella Typhimurium/Typhi microarray (version 8), a two-channel spotted array (70-mer probes) designed by the Pathogen Functional Genomics Resource Center at the J. Craig Venter Institute. The analysis consisted of quantifying spot intensities, background-correcting, normalizing the intensities, summarizing the intensities for replicate probes, removing low quality arrays, and finally finding differentially expressed genes.
For each of the arrays, we calculated a single, background-corrected intensity for the probes (spots). First, using the scanned array image, we quantified the probe intensities using the Spotfinder tool from the TM4Microarray Software Suite,29,30 which gave us an MEV file for each array. To load and manipulate the intensity data in the MEV files, we used Bioconductor's31 limma package.32 To get background-corrected intensities for the probes on each array, we used the maximum likelihood estimation for the normal-exponential convolution model,33 as implemented in Bioconductor's limma package.
After background correction, we summarized replicate probe intensities into a single, normalized expression value for each gene. First, we normalized all of the mutant and WT expression values using quantile normalization,34 as implemented in the normalize.quantiles function of the preprocessCore R package.35 Next, we summarized the replicate intensities (there were two identical probes per gene) by calculating their mean. Finally, for the WT arrays, we identified and removed any replicate samples that did not have at least a 0.7 correlation with other replicates, which resulted in 13, 12, and seven replicates that passed this array-level QC step in LB, LPM 4h, and LPM 20h conditions, respectively. Because of a smaller number of starting samples (two in LB, two in LPM 4h, and three in LPM 20h), no hfq-deletion strain arrays were removed; however, none of the replicate arrays had a correlation below 0.69. After the low-quality arrays were removed, we repeated the background-correction, normalization, and summarization steps.
By using the normalized expression values, we then identified differentially expressed genes between the Δhfq and WT strains. Since our sample size for the knockouts was small, we used the methodology described by Smyth et al., which involves using a moderated t-statistic that is more reliable for a small number of arrays.36 The differential expressions analysis was performed using functions available in the limma package.
Results
Effects of σE on Global Protein Abundances in Salmonella
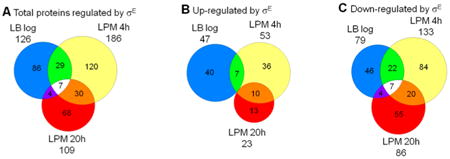
To determine the impact of σE on protein levels, we performed a comprehensive quantitative proteomic analysis of ΔrpoE mutant and WT Salmonella grown in nutrient-rich LB medium to log phase and in acidic minimal medium (LPM) that partially mimics the intracellular environment for 4 or 20 h. After harvesting, cells were lysed, and proteins were digested with trypsin. The resulting peptides were then analyzed by LC–MS/MS using the AMT approach for quantification. A total of 1138 Salmonella proteins were confidently identified and quantified, which corresponds to 25% coverage of the 4450 Salmonella annotated open reading frames.28 The proteomic data were expressed as log2 ratio of ΔrpoE mutant to WT strain, and the effects of σE on the Salmonella proteome were determined by an ANOVA analysis; changes were considered significant when meeting the threshold of p value ≤ 0.05 and fold change ≥ 1.5 (Table S1, Supporting Information). Out of the identified proteins, 126, 186, and 109 were altered by σE at LB log phase, LPM 4h, and LPM 20h conditions, respectively (Figure 1A). By combining all three conditions, our analysis revealed that 30% (344 proteins) of the observed proteome (1138 proteins) was regulated by σE. More proteins were down-regulated than up-regulated by σE in all three conditions. Differences were more pronounced in LPM 20h samples. In this condition representing sustained stress, the expression of 86 proteins was repressed by σE, whereas 23 proteins were activated (Figure 1B,C). There were seven proteins belonging to various categories that were commonly down-regulated by σE in all three conditions, while no protein was up-regulated by σE in both LB log and LPM 20h conditions, but both of these conditions have different overlaps with LPM 4h (Figure 1B,C).
Figure 1.
Overview of protein expression regulated by σE in Salmonella Typhimurium cultured under three growth conditions. Salmonella WT and rpoE-deletion strains were grown in LB medium to log phase or in acidic minimal medium (LPM) for 4 or 20 h in biological triplicates. Total protein was digested, and the peptides were analyzed by LC–MS/MS using AMT approach for quantification. The Venn diagrams show overlaps of (A) total proteins regulated by σE, (B) proteins up-regulated by σE, (C) and proteins down-regulated by σE in the three growth conditions.
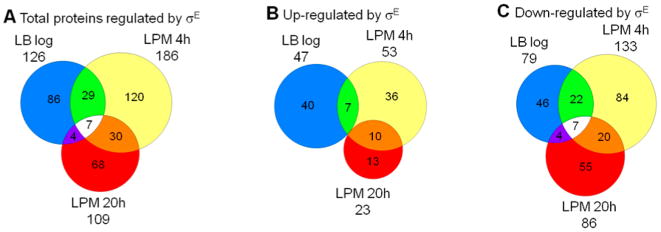
Proteomic results were verified by Western blot analysis of the relative protein levels of CspA and SrfN (STM0082) in the three growth conditions studied (Figure 2A,C). By using chromosomal HA-tagged fusion proteins, we found that CspA expression was significantly higher in the ΔrpoE strain compared to WT in LPM 4h condition, while the expression of SrfN in ΔrpoE was significantly lower than in WT in the same condition. In LB condition, the expression of both CspA and SrfN was comparable in WT and ΔrpoE strains. These findings were consistent with the proteomic analysis (Figure 2B,D). More validations were also found with SodC2 and PspA in LPM 4h condition (Figure 5D, first two lanes of each blot).
Figure 2.
Western blots and proteomics of CspA and SrfN levels from Salmonella WT and ΔrpoE strains in LB log, LPM 4h, and LPM 20h conditions. For Western blots (A, C), cspA or srfN gene was tagged with two HA at chromosomal level in WT and ΔrpoE strains. The same amount of cell lysates was loaded in each lane and probed for the indicated proteins and a control protein DnaK. For quantification, the ratio of CspA/DnaK or SrfN/DnaK was relativized to 100 in the WT background under LB condition. Proteomics data are represented by the log2 of peak areas ratios of (B) CspA and (D) SrfN in ΔrpoE divided by the WT strain.
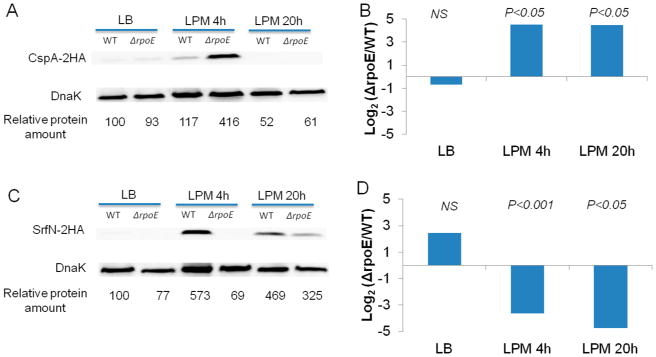
Figure 5.
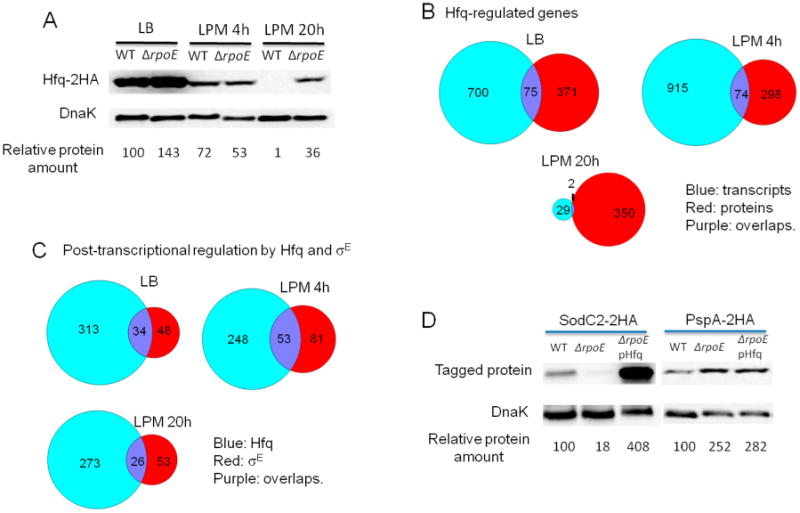
Hfq is involved in σE-mediated post-transcriptional regulation. (A) The effect of σE on Hfq expression in LB log, LPM 4h, and LPM 20h conditions. Western blots of Hfq-2HA in protein extracts from WT and ΔrpoE strains in the three conditions studied. DnaK was used as loading control. For quantification, the ratio of Hfq/DnaK was relative to the WT background in LB log condition (arbitrarily set to 100). (B) Venn diagrams showing transcripts and proteins regulated by Hfq in LB log, LPM 4h, and LPM 20h conditions. (C) Venn diagrams comparing post-transcriptional regulons of σE and Hfq. (D) Western blots of SodC2–2HA and PspA–2HA in protein lysates from WT, ΔrpoE strain, and ΔrpoE strain complemented with Hfq-expressing plasmid in LPM 4h condition. DnaK was used as loading control. For quantification, the ratio of SodC2/DnaK or PspA/DnaK was relative to the WT background.
Functional Categories and Groups of Proteins Affected by σE
Salmonella proteins regulated by σE were classified according to the J. Craig Venter Institute (JCVI) functional categories (Table 1). Of the proteins with known functions, cellular processes and energy metabolism were the most representative categories within proteins up-regulated by σE in the LB log phase condition. Conversely, proteins down-regulated by σE in LB log phase condition were enriched in energy metabolism, cellular processes, and protein synthesis. When cells were grown in LPM for 4 h, transport and binding proteins was the most represented category among the up-regulated proteins. When compared to the LB log phase condition, the LPM 4h condition generally had more down-regulated proteins by σE in all categories, energy metabolism, cellular processes, and transport and binding proteins being the most represented ones. In the LPM 20h condition, fewer proteins were up-regulated by σE compared to LB and LPM 4h conditions, where cellular processes was most highly represented. However, the proteins down-regulated by σE in the same condition were enriched in energy metabolism, protein synthesis, and transport and binding proteins (Table 1). These results showed that σE alters proteins with a diversity of functions depending on the growth condition.
Table 1. Functional Categories of σE-Regulated Proteomea.
| Functional Categories | Total from | Total detected in proteome | Up-regulated by σE | Down-regulated by σE | ||||
|---|---|---|---|---|---|---|---|---|
| LB log | LPM 4h | LPM 20h | LB log | LPM 4h | LPM 20h | |||
| Amino acid biosynthesis | 130 | 48 | 2 | 2 | 1 | 2 | 1 | 2 |
| Biosynthesis of cofactors, prosthetic groups, and carriers | 167 | 60 | 1 | 4 | 4 | 6 | ||
| Cell envelope | 479 | 82 | 1 | 7 | 8 | 4 | ||
| Cellular processes | 289 | 85 | 8 | 6 | 6 | 11 | 13 | 7 |
| Central intermediary metabolism | 170 | 61 | 2 | 6 | 4 | 6 | 4 | |
| DNA metabolism | 166 | 32 | 1 | 4 | 3 | |||
| Energy metabolism | 610 | 216 | 8 | 4 | 4 | 15 | 31 | 16 |
| Fatty acid and phospholipid metabolism | 80 | 23 | 1 | 3 | 7 | |||
| Hypothetical proteins | 78 | 17 | 4 | 2 | 2 | 2 | ||
| Mobile and extrachromosomal element functions | 250 | 11 | 1 | 2 | 3 | |||
| Protein fate | 191 | 59 | 4 | 1 | 10 | 4 | ||
| Protein synthesis | 375 | 129 | 4 | 3 | 11 | 10 | 12 | |
| Purines, pyrimidines, nucleosides, and nucleotides | 81 | 50 | 1 | 1 | 4 | 4 | ||
| Regulatory functions | 305 | 46 | 1 | 2 | 2 | 3 | 7 | 8 |
| Signal transduction | 26 | 5 | 1 | 1 | ||||
| Transcription | 57 | 22 | 2 | 3 | ||||
| Transport and binding proteins | 628 | 78 | 2 | 14 | 4 | 9 | 13 | 11 |
| Unclassified | 332 | 44 | 3 | 1 | 2 | 3 | 4 | 2 |
| Unknown function | 670 | 145 | 11 | 12 | 5 | 7 | 8 | 11 |
Proteins regulated by σE are classified according to the JCVI (J. Craig Venter Institute), formerly TIGR (The Institute for Genomic Research), annotation system. Note that some proteins are annotated to multiple categories, accounting for the larger total gene count.
Further examination of protein abundances affected by σE across the three growth conditions according to functional categories suggested that three categories/groups of proteins exhibited distinct patterns in relation to growth conditions (Figure 3). The first category was transport and binding proteins (Figure 3A), in which the majority of proteins were not significantly regulated by σE in LB log phase. However, many proteins in this category were regulated by σE in the LPM 4h condition, where more proteins were up- rather than down-regulated by σE. In LPM 4h, the most significantly up-regulated protein was ModA (533 folds), a molybdate-specific periplasmic binding protein encoded by modABCD operon that functions to transport molybdate.37,38 In E. coli, molybdate is required for the production of molybdoenzymes such as nitrate reductase and formate dehydrogenase, which play an important role in anaerobic respiration.37 We speculate that up-regulating ModA expression might enhance Salmonella energy generation in infection-like conditions to meet the increased energy-consuming transport needed for intracellular survival. In the LPM 20h condition, most of the σE-regulated proteins in this category were down-regulated, where MalK (subunit of maltose transporter), CycA (d-alanine/d-serine/glycine transport protein), and GltJ (Glutamate/aspartate transporter) were reduced the greatest. The second category was protein synthesis (Figure 3B). Generally, σE repressed the expression of proteins involved in protein synthesis across all three conditions; however, in LB log phase and LPM 4h condition, some of the proteins in this category remained up-regulated by σE, unlike in the LPM 20h condition where each of these proteins were down-regulated. The third category was stress response proteins (Figure 3C), the majority of which belong to cellular processes or regulatory functions categories. This group of proteins was far less regulated by σE in LB log phase than in LPM 4h and 20h conditions because of the low stress in the nutrient-rich LB media compared to increased cellular stress in the infection-mimicking media conditions. Notably, some proteins involved in oxidative stress response were up-regulated by σE in LPM 4h (e.g., SodC1 and SodC2) and 20h (e.g., SodA and SodC2) conditions. Consistent with previous findings that phage shock protein and two-component regulatory system CpxR/CpxA play compensatory role to σE on extracytoplasmic stress response,39,40 we found that in the absence of σE the expression of PspA and CpxR increased by 42 and 19-fold, respectively, in the LPM 4h condition. Although PspE is encoded by pspABCDE operon, the regulation of PspE expression by σE was completely opposite to PspA, which may be related to the coexistence of the intrinsic pspE-specific promoter.41 In the LPM 20h condition, most of the stress response proteins were down-regulated by σE.
Figure 3.
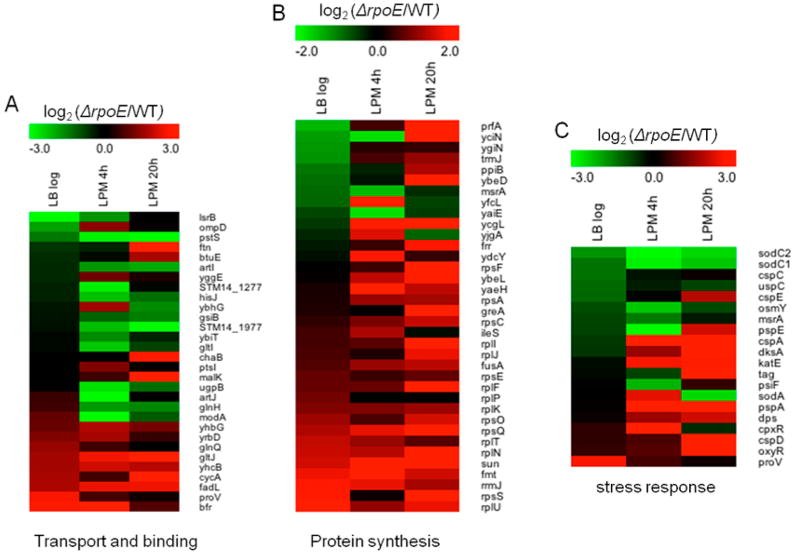
Heat maps of three groups of proteins that are differentially regulated by σE in Salmonella grown in LB to log phase, or in LPM for 4 or 20 h. Shown are proteins involved in (A) transport and binding, (B) protein synthesis, and (C) stress response. Green represents up-regulation of protein expression by σE, while red represents down-regulation.
Comparison of Proteomic and Transcriptomic Profiles of σE
To better understand the mechanisms of regulation, we compared the proteomic profile of σE-deficient cells with sample-matched transcriptomic data recently published.15 The expression of approximately 58% of Salmonella genes was affected by σE in at least one of the three conditions. When transcriptomic and proteomic data sets by Pearson Correlation were compared, a very low correlation (≤0.02) was observed between the abundances of mRNAs and proteins regulated by σE in all three growth conditions, which suggests that σE regulates the abundances of proteins in multiple levels both in transcriptional and translational processes.
Transcript and protein data were combined into scatter plots based on fold changes comparing ΔrpoE mutant to WT (expressed in log2 scale). The type of regulation was classified into four different mechanisms according to the significance of regulation on mRNA and protein levels: (1) regulated only at mRNA level, lacking significant change at protein level; (2) regulated only at protein level, lacking significant change at mRNA level; (3) regulated at both mRNA and protein levels, which is represented by negative correlation between mRNA and protein levels; and (4) regulated at mRNA level that has a direct impact on proteins level, which is represented by positive correlation between mRNA and protein levels (Figure 4; Table S2, Supporting Information). Of the 117 proteins differentially regulated by σE at LB log phase, 63 were solely regulated at protein level, and 54 were significantly regulated at both protein and mRNA levels. Of these 54 proteins, 22 showed the opposite trends at the mRNA and protein levels, which included ribosomal proteins (RplP, RplT, RpsE, RpsS, RplK, and RluC) and flagellar proteins (FliC, FlgN, FlgL, and FlgE). Thirty-two proteins showed the same trends in regulation at both levels, which included SPI-1 chaperones (SicA and InvB) and stress response proteins (CspC, CspE, and YecG). In the LPM 4h condition, there were 229 proteins regulated by σE at the mRNA level exclusively, which included a majority of ribosomal proteins. One hundred-sixteen proteins were regulated by σE exclusively at the protein level. This included select stress response proteins (CspA, DksA, KatE, OsmY, and SodC2). Eighteen proteins were regulated at both mRNA and protein level, and 43 proteins were regulated at protein level directly related to gene level regulation. In the LPM 20h condition, 210, 76, nine, and 19 proteins were regulated by σE by the above four classified mechanisms, correspondingly (Table 2). Some stress response proteins were regulated by σE at the protein level (CspA, Tag, SodA, SodC2, OmpA, OmpC, and OxyR), while others exhibited positive correlation between mRNA and protein levels (DksA, CspD, KatE, and PspA) (Figure 4C). We found that protein level regulation accounted for 25%, 43%, and 33% of total regulation by σE at LB log phase, LPM 4h, and LPM 20h condition, respectively. In this category, 73%, 76%, and 82% (for details of calculation, see Table 2) were regulated post-transcriptionally in the above conditions, respectively. These results suggest that post-transcriptional regulation accounts for the majority of σE-mediated protein-level regulation.
Figure 4.
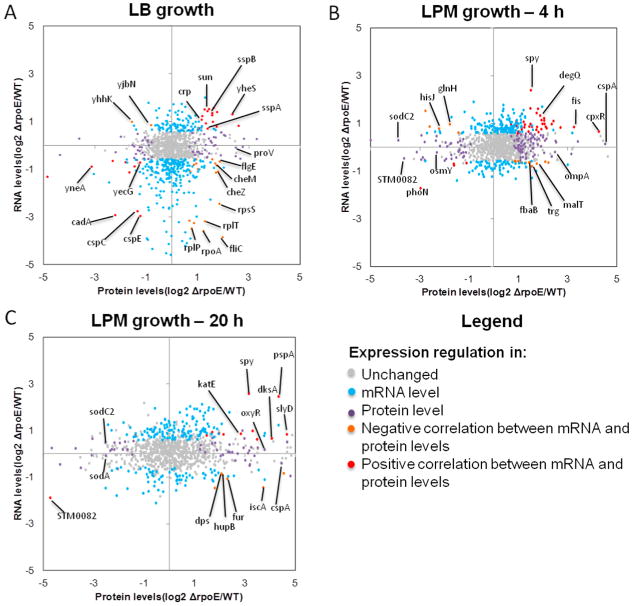
Scatterplot of fold changes of transcript versus protein expression regulated by σE in (A) LB log, (B) LPM 4h, and (C) LPM 20h conditions. The charts show log2-based fold changes of ΔrpoE compared to WT at mRNA level and protein level that were derived from transcriptomic and proteomic data, respectively. The legend describes the mechanism of regulations based on the changes on mRNA and protein levels. Each dot represents one gene/protein of Salmonella and was colored differently according to the way of regulation.
Table 2. Summary of Levels of Regulation Mediated by σE As Classified in Figure 4a.
| Growth conditions | Level of regulation | % of protein level regulation1 | % of post-transcriptional regulation2 | |||
| mRNA level only | Protein level only | Negative correlation | Positive correlation | |||
| LB log | 341 | 63 | 22 | 32 | 25 | 73 |
| LPM 4h | 229 | 116 | 18 | 43 | 43 | 76 |
| LPM 20h | 210 | 76 | 9 | 19 | 33 | 82 |
Superscripted 1 indicates the following: % of protein level regulation represents the percentage of protein level regulation accounting for total regulation and equals (protein level only + negative correlation + positive correlation)/(mRNA level only + protein level only + negative correlation + positive correlation). Superscripted 2 indicates the following: % of post-transcriptional regulation represents the percentage of post-transcriptional regulation accounting for protein level regulation and equals (protein level only + negative correlation)/(protein level only + negative correlation + positive correlation).
Involvement of Hfq in σE-Mediated Post-Transcriptional Regulation
Our microarray data showed that σE regulates hfq transcription in both LB log and LPM conditions. To find out if σE affects Hfq expression at protein level, we performed a Western blot comparing the expression of HA-tagged Hfq in WT and ΔrpoE strains (Figure 5A). In the LB log phase condition, the level of Hfq was higher in ΔrpoE mutant compared to that in WT. In the LPM 4h condition, the level of Hfq was lower in ΔrpoE than in WT. In the LPM 20h condition, a much higher level of Hfq was expressed in ΔrpoE than in WT. These data confirmed that σE regulates Hfq expression under the examined environmental conditions.
To investigate if Hfq plays a role in σE-mediated post-transcriptional regulation, we compared transcriptomic and proteomic data of WT and hfq-deletion Salmonella cultured under the same condition as the ΔrpoE strain (Table S3, Supporting Information). In the LB log phase condition, 775 RNAs and 446 proteins were regulated by Hfq, where 75 genes overlapped in both sets (Figure 5B). Within this overlap, 44 genes were positively correlated, and 31 were negatively correlated. In the LPM 4h condition, Hfq regulated 989 RNAs and 372 proteins with an overlap of 74 genes (Figure 5B) consisting of 38 and 36 positively- and negatively-correlated genes. There was a major reduction of RNAs regulated by Hfq in the LPM 20h condition. Thirty-one RNAs and 352 proteins were regulated by Hfq (Figure 5B). There were only two overlapping genes where one positively correlated and the other negatively correlated. Comparison across all growth conditions revealed a small overlap between proteins and RNAs regulated by Hfq (Figure 5B). Moreover, we found that Hfq regulated many proteins involved in general metabolism, stress response, virulence, and propanediol utilization, which is consistent with previous findings.19
When genes post-transcriptionally regulated by Hfq were compared to those post-transcriptionally regulated by σE, we found that under each condition studied, Hfq regulated a more extensive group of genes at this level than σE (Figure 5C; Table S4, Supporting Information). There were genes commonly regulated by both Hfq and σE, which accounted for a smaller proportion to Hfq than to σE. Although the overlapping genes comprised 41%, 39%, and 33% of all genes post-transcription-ally regulated by σE in LB, LPM 4h, and LPM 20h condition, respectively, further examination of the overlaps suggested that not all of these genes were regulated by Hfq and σE in the same direction (Table S5, Supporting Information). For instance, within the 34 overlapping genes found in the LB log condition, 16 genes were regulated by Hfq and σE oppositely (up-regulated vs down-regulated); in the LPM 4h condition, 27 of 53 overlapping genes were oppositely regulated by Hfq and σE; and in the LPM 20h condition, three of 26 genes were oppositely regulated. These desynchronized genes within the overlaps were likely regulated by σE and Hfq using distinct mechanisms. Therefore, the potential genes regulated by Hfq and σE dependent mechanism correspond to 22%, 19%, and 29% of all σE–mediated post-transcriptionally regulated genes in LB, LPM 4h, and LPM 20h, respectively.
To verify the genes that were regulated by σE through Hfq (listed in Table S5, Supporting Information), we selected SodC2, which was shown to be regulated by both σE and Hfq in a synchronized manner in LPM 4h and 20h conditions. The sodC2 gene fused with an HA tag was transformed into WT or ΔrpoE strains with or without a plasmid expressing Hfq controlled by the lac promoter. In this experiment, if σE regulated SodC2 expression through Hfq, the decreased expression of SodC2 in ΔrpoE strain should be compensated by the complementation of Hfq. The Western blot confirmed our hypothesis and showed that the SodC2 expression could be rescued by overexpressing Hfq (Figure 5D). As a negative control, we used PspA, which was down-regulated by σE (Figure 3) but not predicted to be post-transcriptionally regulated by Hfq. The results clearly show an increase of PspA abundance in ΔrpoE strain, which was not diminished by the overexpression of Hfq (Figure 5D). Hence, our results suggest that genes post-transcriptionally regulated by σE can be mediated through indirect regulation of Hfq.
Discussion
Gene expression regulated by alternative sigma factor σE has been extensively studied at the transcriptional level.11–13,42 Consensus sequences recognized by σE and genes directly regulated by σE were reported in multiple studies.13,14 In Salmonella, σE was found to repress gene expression through regulating two Hfq-dependent sRNAs, RybB and MicA,17 which suggests that σE is likely involved in post-transcriptional regulation indirectly. However, accurate characterization of σE on gene expression at the post-transcriptional level on a global scale had not yet been performed. Here, we performed sample-matched transcriptomic and proteomic analyses on WT and ΔrpoE strains grown under multiple conditions to understand the extent and the mechanism of σE-mediated post-transcriptional regulation. Global analyses revealed that a large portion of σE-mediated protein-level regulation actually occurred post-transcriptionally. Recently, we found that σE regulates a high percentage of all the annotated Salmonella genes (58%),15 including the transcription of hfq, a major post-transcriptional regulator. Therefore, the sample-matched method was also applied on WT and Δhfq strains to compare the post-transcriptional regulation mediated by Hfq and σE. We found that part of the post-transcriptional regulation mediated by σE was dependent on Hfq.
Gene regulation occurs at different levels. Post-transcriptionally regulated genes were often determined as genes regulated only at protein level but not at mRNA level, excluding genes that are regulated at both mRNA and protein levels.19 However, arbitrary exclusion of genes regulated at both levels from post-transcriptional regulation may generate false negatives. In this study, we looked more carefully at these genes and further divided them into positive and negative correlations. We included the negatively correlated genes as part of the post-transcriptionally regulated candidates since they are regulated after the transcription. Therefore, we classified genes that were regulated in “protein level only” and “negatively correlated” as candidates of post-transcriptional regulation. This new classification improved the accuracy of post-transcriptional regulation identification.
Although 20–30% of σE-mediated post-transcriptional regulation found in this study was a possible result of Hfq activity, the mechanism for the rest of the genes that are post-transcriptionally regulated by σE is still not clear. Post-transcriptional regulation of stress adaptation can be mediated by sRNA, riboswitches, RNA binding proteins, guanosine tetraphosphate (ppGpp), cold-shock proteins (Csp), transfer-mRNA (tmRNA), and others.18,43–47 We found that σE down-regulated CspA expression in infection-like conditions. Since CspA functions as an RNA chaperone to prevent formation of secondary structure in mRNA and thus facilitates translation initiation,48 it is possible that some genes are post-transcriptionally regulated by σE though its effects on CspA. Moreover, σE reduces RelA (ppGpp synthetase) production in LPM 4h condition (Table S1, Supporting Information), which may reduce the level of ppGpp and affect the feedback control of ribosomal protein synthesis via ppGpp.49 Hence, σE could presumably utilize other pathways for post-transcriptional regulation, which needs to be further elucidated.
A total of 1138 proteins were observed in the proteome compared to the 4450 Salmonella annotated open reading frames; 75% of the ORFs were either not detected or the levels were too close to the background and they were excluded for further quantitative analysis. If the remaining ORFs were expressed, we expect that a larger range of functions would be affected by σE. Since the changes in the majority of σE-regulated proteome were caused by post-transcriptional regulation, which allows more rapid adaptation than for synthesis of proteins that must be transcribed first, this feature may contribute to the acute attenuation in virulence observed in rpoE null mutant.10
It is not surprising that σE regulates a considerable number of genes post-transcriptionally since increasing evidence has shown that post-transcriptional regulation is widespread in prokaryotes. In E. coli, large-scale measurement of protein expression suggested that only 47% of protein abundance is directly related to mRNA concentration.50 In L. interrogans, only 25% of the outer membrane proteins that were significantly regulated by temperature were indeed regulated at the transcriptional level.51 In P. fluorescens Pf-5, iron acquisition is regulated at both the transcriptional and post-transcriptional levels.52 Therefore, post-transcriptional regulation plays an important role in gene regulation for prokaryotes.
For survival and proliferation in various circumstances, Salmonella takes advantage of its highly complex regulatory network to rapidly adapt to the newly encountered environment.10,19,53 Our results suggest that extracytoplasmic stress regulator σE utilizes not only transcriptional, but also post-transcriptional mechanisms to enable Salmonella to rapidly adjust to changing conditions. Future systematic studies of multiple regulators combining multiple techniques will add new layers of information and lead to a deeper understanding of the global regulation process.
Supplementary Material
Acknowledgments
This research was supported by the NIH National Institute of General Medical Sciences (GM094623) and National Institute of Allergy and Infectious Diseases NIH/DHHS through Interagency agreement Y1-AI-8401. This work benefited from the investments in technology development from NIH NIGMS Grant No. 8 P41 GM103493 and the U.S. Genome Sciences Program under the Pan-omics project. Portions of this work were performed in the Environmental Molecular Science Laboratory, a U.S. Department of Energy (DOE) national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, WA. Battelle operates PNNL for the DOE under Contract No. DE-AC05-76RLO01830.
Abbreviations
- AMT
accurate mass and time tag
- LB
Luria–Bertani broth
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- LPM
acidic minimum medium low in phosphate and magnesium
- nt
nucleotide
- PVDF
polyvinylidene difluoride
- SCX
strong cation exchange
- sRNAs
small noncoding RNAs
- STM
Salmonella enterica serovar Typhimurium
- TBS
Tris-buffered saline solution
- WT
wild type
Footnotes
Supporting Information: Proteomic analysis of rpoE mutant Salmonella Typhimurium. Classification of genes regulated by σE into different mechanisms based on the changes on mRNA and protein levels. Transcriptomic and proteomic data of WT and hfq-deletion Salmonella cultured in LB to log phase and in LPM for 4 or 20 h. Genes post-transcriptionally regulated by Hfq or σE in LB log and LPM 4-h or LPM 20-h conditions. Genes post-transcriptionally regulated by both Hfq and σE. Strains and plasmids used in this study. List of primers used in this study. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ. Animal models of Salmonella infections: Enteritis versus typhoid fever. Microbes Infect. 2001;3:1335–1344. doi: 10.1016/s1286-4579(01)01495-2. [DOI] [PubMed] [Google Scholar]
- 2.Tsolis RM, Kingsley RA, Townsend SM, Ficht TA, Adams LG, Baumler AJ. Of mice, calves, and men. Comparison of the mouse typhoid model with other Salmonella infections. Adv Exp Med Biol. 1999;473:261–274. [PubMed] [Google Scholar]
- 3.Garcia-del Portillo F, Foster JW, Finlay BB. Role of acid tolerance response genes in Salmonella Typhimurium virulence. Infect Immun. 1993;61:4489–4492. doi: 10.1128/iai.61.10.4489-4492.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francis CL, Starnbach MN, Falkow S. Morphological and cytoskeletal changes in epithelial cells occur immediately upon interaction with Salmonella Typhimurium grown under low-oxygen conditions. Mol Microbiol. 1992;6:3077–3087. doi: 10.1111/j.1365-2958.1992.tb01765.x. [DOI] [PubMed] [Google Scholar]
- 5.Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, Le M, Berggren R, Parks WT, Fang FC. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- 6.Geddes K, Cruz F, Heffron F. Analysis of cells targeted by Salmonella type III secretion in vivo. PLoS Pathog. 2007;3:e196. doi: 10.1371/journal.ppat.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowley G, Spector M, Kormanec J, Roberts M. Pushing the envelope: Extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol. 2006;4:383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- 9.Osterberg S, del Peso-Santos T, Shingler V. Regulation of alternative sigma factor use. Annu Rev Microbiol. 2011;65:37–55. doi: 10.1146/annurev.micro.112408.134219. [DOI] [PubMed] [Google Scholar]
- 10.Yoon H, McDermott JE, Porwollik S, McClelland M, Heffron F. Coordinated regulation of virulence during systemic infection of Salmonella enterica serovar Typhimurium. PLoS Pathog. 2009;5:e1000306. doi: 10.1371/journal.ppat.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dartigalongue C, Missiakas D, Raina S. Characterization of the Escherichia coli sigma E regulon. J Biol Chem. 2001;276:20866–20875. doi: 10.1074/jbc.M100464200. [DOI] [PubMed] [Google Scholar]
- 12.Rezuchova B, Miticka H, Homerova D, Roberts M, Kormanec J. New members of the Escherichia coli sigmaE regulon identified by a two-plasmid system. FEMS Microbiol Lett. 2003;225:1–7. doi: 10.1016/S0378-1097(03)00480-4. [DOI] [PubMed] [Google Scholar]
- 13.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol. 2006;4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skovierova H, Rowley G, Rezuchova B, Homerova D, Lewis C, Roberts M, Kormanec J. Identification of the sigmaE regulon of Salmonella enterica serovar Typhimurium. Microbiology. 2006;152:1347–1359. doi: 10.1099/mic.0.28744-0. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Overall C, Nakayasu ES, Kidwai A, Jones M, Johnson R, Nguyen N, McDermott J, Ansong C, Heffron F, Cambronne ED, Adkins JN. Analysis of the Salmonella regulatory network suggests involvement of SsrB and H-NS in σE-regulated SPI-2 gene expression. Front Microbiol. 2015;6:27. doi: 10.3389/fmicb.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gogol EB, Rhodius VA, Papenfort K, Vogel J, Gross CA. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci U S A. 2011;108:12875–12880. doi: 10.1073/pnas.1109379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansong C, Yoon H, Porwollik S, Mottaz-Brewer H, Petritis BO, Jaitly N, Adkins JN, McClelland M, Heffron F, Smith RD. Global systems-level analysis of Hfq and SmpB deletion mutants in Salmonella: Implications for virulence and global protein translation. PLoS One. 2009;4:e4809. doi: 10.1371/journal.pone.0004809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Udekwu KI, Darfeuille F, Vogel J, Reimegard J, Holmqvist E, Wagner EG. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 2005;19:2355–2366. doi: 10.1101/gad.354405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon H, Gros P, Heffron F. Quantitative PCR-based competitive index for high-throughput screening of Salmonella virulence factors. Infect Immun. 2011;79:360–368. doi: 10.1128/IAI.00873-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmer JS, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev. 2006;25:450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adkins JN, Mottaz HM, Norbeck AD, Gustin JK, Rue J, Clauss TR, Purvine SO, Rodland KD, Heffron F, Smith RD. Analysis of the Salmonella Typhimurium proteome through environmental response toward infectious conditions. Mol Cell Proteomics. 2006;5:1450–1461. doi: 10.1074/mcp.M600139-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Livesay EA, Tang K, Taylor BK, Buschbach MA, Hopkins DF, LaMarche BL, Zhao R, Shen Y, Orton DJ, Moore RJ, Kelly RT, Udseth HR, Smith RD. Fully automated four-column capillary LC–MS system for maximizing throughput in proteomic analyses. Anal Chem. 2008;80:294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly RT, Page JS, Tang K, Smith RD. Array of chemically etched fused-silica emitters for improving the sensitivity and quantitation of electrospray ionization mass spectrometry. Anal Chem. 2007;79:4192–4198. doi: 10.1021/ac062417e. [DOI] [PubMed] [Google Scholar]
- 27.Stanley JR, Adkins JN, Slysz GW, Monroe ME, Purvine SO, Karpievitch YV, Anderson GA, Smith RD, Dabney AR. A statistical method for assessing peptide identification confidence in accurate mass and time tag proteomics. Anal Chem. 2011;83:6135–6140. doi: 10.1021/ac2009806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polpitiya AD, Qian WJ, Jaitly N, Petyuk VA, Adkins JN, Camp DG, Anderson GA, Smith RD. DAnTE: A statistical tool for quantitative analysis of omics data. Bioinformatics. 2008;24:1556–1558. doi: 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 30.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 31.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth GK. In: Limma: Linear models for microarray data. Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Springer; New York; 2005. [Google Scholar]
- 33.Silver JD, Ritchie ME, Smyth GK. Microarray background correction: Maximum likelihood estimation for the normal-exponential convolution. Biostatistics. 2009;10:352–363. doi: 10.1093/biostatistics/kxn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 35.Bolstad BM. preprocessCore: A collection of pre-processing functions. R package [Google Scholar]
- 36.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 37.Grunden AM, Shanmugam KT. Molybdate transport and regulation in bacteria. Arch Microbiol. 1997;168:345–354. doi: 10.1007/s002030050508. [DOI] [PubMed] [Google Scholar]
- 38.Rech S, Wolin C, Gunsalus RP. Properties of the periplasmic ModA molybdate-binding protein of Escherichia coli. J Biol Chem. 1996;271:2557–2562. doi: 10.1074/jbc.271.5.2557. [DOI] [PubMed] [Google Scholar]
- 39.Connolly L, De Las Penas A, Alba BM, Gross CA. The response to extracytoplasmic stress in Escherichia coli is controlled by partially overlapping pathways. Genes Dev. 1997;11:2012–2021. doi: 10.1101/gad.11.15.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker LA, Bang IS, Crouch ML, Fang FC. Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol Microbiol. 2005;56:1004–1016. doi: 10.1111/j.1365-2958.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- 41.Huvet M, Toni T, Sheng X, Thorne T, Jovanovic G, Engl C, Buck M, Pinney JW, Stumpf MP. The evolution of the phage shock protein response system: Interplay between protein function,genomic organization, and system function. Mol Biol Evol. 2011;28:1141–1155. doi: 10.1093/molbev/msq301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabir MS, Yamashita D, Koyama S, Oshima T, Kurokawa K, Maeda M, Tsunedomi R, Murata M, Wada C, Mori H, Yamada M. Cell lysis directed by sigmaE in early stationary phase and effect of induction of the rpoE gene on global gene expression in Escherichia coli. Microbiology. 2005;151:2721–2735. doi: 10.1099/mic.0.28004-0. [DOI] [PubMed] [Google Scholar]
- 43.Vogel J. A rough guide to the noncoding RNA world of Salmonella. Mol Microbiol. 2009;71:1–11. doi: 10.1111/j.1365-2958.2008.06505.x. [DOI] [PubMed] [Google Scholar]
- 44.Park SY, Cromie MJ, Lee EJ, Groisman EA. A bacterial mRNA leader that employs different mechanisms to sense disparate intracellular signals. Cell. 2010;142:737–748. doi: 10.1016/j.cell.2010.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. ppGpp conjures bacterial virulence. Microbiol Mol Biol Rev. 2010;74:171–199. doi: 10.1128/MMBR.00046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horn G, Hofweber R, Kremer W, Kalbitzer HR. Structure and function of bacterial cold shock proteins. Cell Mol Life Sci. 2007;64:1457–1470. doi: 10.1007/s00018-007-6388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dulebohn D, Choy J, Sundermeier T, Okan N, Karzai AW. Trans-translation: The tmRNA-mediated surveillance mechanism for ribosome rescue, directed protein degradation, and nonstop mRNA decay. Biochemistry. 2007;46:4681–4693. doi: 10.1021/bi6026055. [DOI] [PubMed] [Google Scholar]
- 48.Phadtare S. Recent developments in bacterial cold shock response. Curr Issues Mol Biol. 2004;6:125–136. [PubMed] [Google Scholar]
- 49.Dennis PP, Ehrenberg M, Bremer H. Control of rRNA synthesis in Escherichia coli: A systems biology approach. Microbiol Mol Biol Rev. 2004;68:639–668. doi: 10.1128/MMBR.68.4.639-668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 51.Lo M, Cordwell SJ, Bulach DM, Adler B. Comparative transcriptional and translational analysis of leptospiral outer membrane protein expression in response to temperature. PLoS Negl Trop Dis. 2009;3:e560. doi: 10.1371/journal.pntd.0000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim CK, Hassan KA, Tetu SG, Loper JE, Paulsen IT. The effect of iron limitation on the transcriptome and proteome of Pseudomonas fluorescens Pf-5. PLoS One. 2012;7:e39139. doi: 10.1371/journal.pone.0039139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDermott JE, Yoon H, Nakayasu ES, Metz TO, Hyduke DR, Kidwai AS, Palsson BO, Adkins JN, Heffron F. Technologies and approaches to elucidate and model the virulence program of Salmonella. Front Microbiol. 2011;2:121. doi: 10.3389/fmicb.2011.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.