

Summary
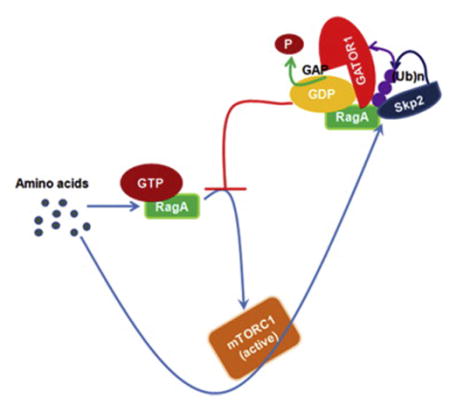
The regulation of RagAGTP is important for amino acid-induced mTORC1 activation. Although GATOR1 complex has been identified as a negative regulator for mTORC1 by hydrolyzing RagAGTP, how GATOR1 is recruited to RagA to attenuate mTORC1 signaling remains unclear. Moreover, how mTORC1 signaling is terminated upon amino acid stimulation is also unknown. We show that the recruitment of GATOR1 to RagA is induced by amino acids in an mTORC1-dependent manner. Skp2 E3 ligase drives K63-linked ubiquitination of RagA, which facilitates GATOR1 recruitment and RagAGTP hydrolysis, thereby providing a negative feedback loop to attenuate mTORC1 lysosomal recruitment and prevent mTORC1 hyperactivation. We further demonstrate that Skp2 promotes autophagy, but inhibits cell size and cilia growth through RagA ubiquitination and mTORC1 inhibition. We thereby propose a negative feedback that Skp2-mediated RagA ubiquitination recruits GATOR1 to restrict mTORC1 signaling upon sustained amino acid stimulation, which serves a critical mechanism to maintain proper cellular functions.
Graphical Abstract
Introduction
The mammalian target of rapamycin complex 1 (mTORC1) signaling regulates cellular functions in response to various stimuli, such as growth factors, stress and amino acids. Signal from growth factor is sensed by the TSC1/TSC2 tumor suppressor complex, which is a negative regulator of mTORC1 signaling through its role as a GTPase activation protein (GAP) of Rheb, a small guanosine triphosphate (GTP) binding protein. Growth factor stimulation inhibits the GAP activity of TSC1/TSC2 complex to promote Rheb GTP binding, which activates mTORC1 signaling(Sarbassov et al., 2005). In contrast, amino acids stimulate mTORC1 signaling independent of TSC1/TSC2 complex(Nobukuni et al., 2005; Roccio et al., 2006; Smith et al., 2005).
Deregulation of mTORC1 signaling leads to human disorders from cancer, metabolic diseases to aging(Guertin and Sabatini, 2007; Laplante and Sabatini, 2012; Zoncu et al., 2011). To maintain proper mTORC1 signaling, the level of mTORC1 signaling is precisely controlled by not only positive regulators but also negative regulators under physiological conditions where sufficient nutrients could otherwise over-activate mTORC1. For instance, negative feedback loops have been identified to restrict growth factor initiated Akt-mTORC1 signaling, partially through the phosphorylation and degradation of insulin receptor substrate 1 (IRS1)(Laplante and Sabatini, 2012). However, it remains unknown how the mTORC1 signaling upon sustained amino acid stimulation is regulated by negative feedback loops.
Recent findings showed that amino acids initiate mTORC1 signaling through translocating the mTORC1 complex to lysosome surface where it interacts with Rheb and is activated(Sancak et al., 2010; Sancak et al., 2008). Multiple protein complexes associated with lysosome surface are required for the mTORC1 lysosome localization and activation upon amino acid stimulation. Among them, the Rags complex directly interacts with mTORC1 and recruits mTORC1 to lysosome surface(Kim et al., 2008; Sancak et al., 2008). The Rags complex is a heterodimer of GTP binding proteins RagA or RagB with RagC or RagD. RagA and RagB, similar to RagC and RagD, are high homologs(Hirose et al., 1998; Sekiguchi et al., 2001). Amino acids induce the RagA/B bound to GTP, which is essential for mTORC1 recruitment and activation. RagA/B mutant constitutively bound to GTP activates mTORC1 signaling regardless of amino acid starvation. On the contrary, amino acid starvation increases RagA/B bound to guanosine diphosphate (GDP), leading to the inhibition of mTORC1 signaling(Efeyan et al., 2013; Kim et al., 2008; Sancak et al., 2008). Several regulators for RagA/B GTP/GDP nucleotide binding have recently been identified. The Ragulator complex with guanosine exchange factor (GEF) activity exchanges GDP to GTP of RagA/B upon amino acid stimulation. Sestrins was identified as the guanine nucleotide dissociation inhibitors (GDIs) of RagA/B, therefore inhibits amino acid induced exchanging of GDP to GTP(Peng et al., 2014). The GATOR1 complex displays GTPase activation protein (GAP) activity to switch RagA/B-bound GTP (RagA/BGTP) to GDP (RagA/BGDP)(Bar-Peled et al., 2013; Bar-Peled et al., 2012; Sancak et al., 2010). However, it is poorly understood how amino acids regulate the activity of these regulators for RagA/B nucleotide binding.
In this study, we identified that the interaction of GATOR1 and RagA is promoted by amino acids, therefore serving as a negative feedback regulator to terminate mTORC1 signaling and prevent its hyperactivation in response to sustained amino acid exposure. We found that amino acid stimulation induces the K63-linked ubiquitination of RagA, which recruits GATOR1 to hydrolyze RagAGTP and suppresses mTORC1 lysosomal recruitment and activation. We further identified that the Skp2 SCF E3 ligase is required for the K63-linked ubiquitination RagA, GATOR1 recruitment and mTORC1 inhibition under amino acid stimulation. Loss of Skp2 prolongs mTORC1 hyperactivation, enlarges cell size, enhances cilia growth, but reduces autophagy. We thereby reveal that Skp2-mediated RagA ubiquitination and GATOR1 recruitment serve as negative feedback regulators to terminate mTORC1 signaling under physiological conditions. Deregulation in this critical mechanism leads to disordered cellular functions.
Result
Amino acids promote the interaction of RagA with GATOR1 and RagA ubiquitination
Given that negative feedback loops have been demonstrated to attenuate growth factor mediated mTORC1 signaling, it is likely that amino acid-dependent mTORC1 signaling may be also regulated by negative feedback loops. To test this possibility, we examined the kinetic mTORC1 signaling upon sustained amino acid stimulation in primary mouse embryonic fibroblasts (MEFs). Amino acids triggered mTORC1 activation during short-term stimulation as determined by pS6K, pS6, and p4E-BP1. However, under long term amino acid stimulation up to 6 hours, the mTORC1 signaling significantly declined (Figure S1), revealing that negative feedback regulation mechanisms might be operated to restrict amino acid-dependent mTORC1 signaling.
The recent finding that GATOR1 complex removes GTP-bound status of RagA/B to inactivate mTORC1(Bar-Peled et al., 2013) drives us to examine whether the negative regulation of mTORC1 during amino acid treatment could be achieved by the recruitment of GATOR1 complex to RagA/B. RagA and RagB are highly homologous. Since RagA has been shown to be expressed more abundantly and widely than RagB(Efeyan et al., 2013), we therefore focused our study on the interaction of RagA with GATOR1. We found, to our surprise, that amino acid stimulation enhanced the interaction of endogenous DEPDC5 and Nprl3, the components of GATOR1 complex, with both ectopic expressed RagA (Figure 1A) and endogenous RagA (Figure 1B).
Figure 1. Amino acids promote the interaction of RagA with GATOR1 and RagA ubiquitination.
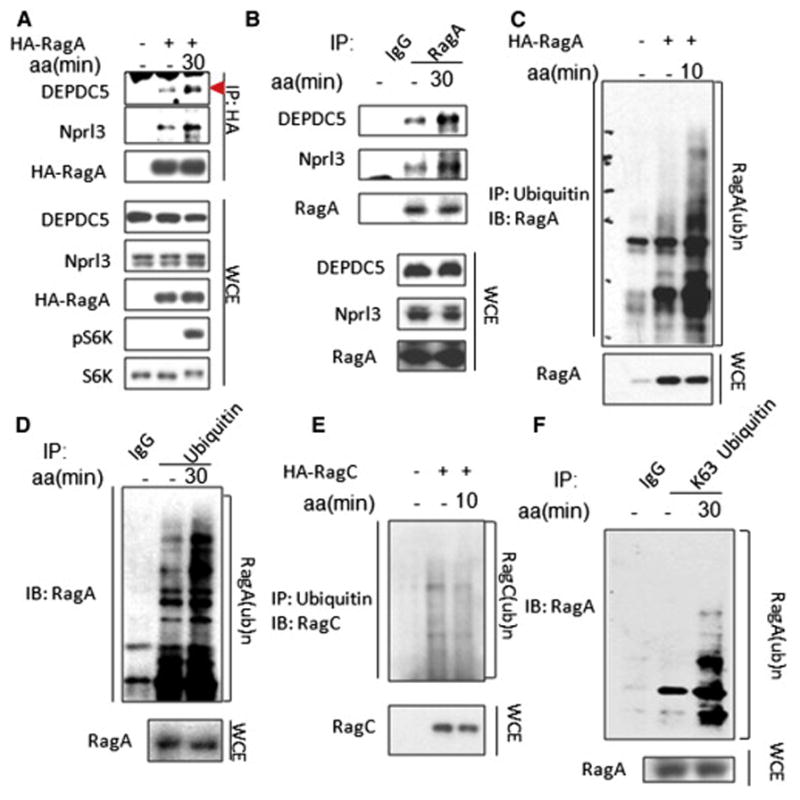
(A) Immunoprecipitation (IP) analysis of amino acid-dependent interaction between RagA and GATOR1 components (DEPDC5 and Nprl3) in HEK293T cells overexpressed with indicated proteins. Triangle indicates annotated protein. (B) IP analysis of amino acid-dependent interaction between endogenous RagA and GATOR1 components (DEPDC5 and Nprl3) in HEK293T cells. (C) IP analysis of amino acid-dependent RagA ubiquitination in HEK293T cells overexpressed with indicated proteins. (D) IP analysis of amino acid-induced ubiquitination of endogenous RagA in HEK293T cells. (E) IP analysis of RagC ubiquitination with or without amino acid stimulation in HEK293T cells overexpressed with indicated proteins. (F) IP analysis of amino acid-dependent K63-linked ubiquitination of endogenous RagA. Cells in (A–F) were starved for amino acids for 1 hour and restimulated with amino acids for the indicated time before lysis. See also Figure S1
To understand the mechanistic insight into how amino acids promote the interaction between GATOR1 with RagA, we examined the posttranslational modification of RagA. Interestingly, amino acids induced the ubiquitination of both ectopic expressed RagA (Figure 1C) and endogenous RagA (Figure 1D), but not RagC, another component of the Rags complex RagA-RagC (Figure 1E). We further demonstrated that amino acids induced lysine (K) 63-linked ubiquitination of endogenous RagA (Figure 1F).
Skp2 E3 ligase mediates amino acid-dependent RagA ubiquitination and GATOR1 recruitment
To understand which E3 ligase mediates RagA ubiquitination, we performed the E3 ligase screening using a panel of E3 ligases and identified Skp2 as a potential ubiquitin E3 ligase of RagA (Figure 2A). Skp2 is an F-Box protein, which forms one of the Skp1-Cul1-F-box (SCF) ubiquitin E3 ligase complexes to induce the ubiquitination of several protein substrates, such as p27 and Akt(Chan et al., 2012; Nakayama et al., 2000; Nakayama et al., 2004), and displays oncogenic activity(Lin et al., 2010; Lin et al., 2009). We found that ectopic expression of Skp2 promoted RagA ubiquitination, while Skp2 LRR mutant devoid of its E3 ligase activity compromised this ability (Figure S2A). We further demonstrated that purified Skp2 SCF complex could directly induce RagA ubiquitination in vitro (Figure S2B). Moreover, Skp2 triggers ubiquitination of RagA through lysine (K) 63 linkage, but not K48 linkage (Figure 2B). Importantly, Skp2 knockdown reduced amino acid-induced RagA ubiquitination (Figure 2C). Accordingly, our results suggest that Skp2 SCF complex is a direct E3 ligase for K63-linked ubiquitination of RagA in response to amino acid stimulation.
Figure 2. Skp2 E3 ligase mediates amino acid-dependent RagA ubiquitination and GATOR1 recruitment.

(A) Screening for E3 ligases of RagA. In vivo ubiquitination assay of RagA co-transfected with E3 ligases with flag or HA tags in HEK293T cells. Arrows indicate E3 ligases. (B) In vivo ubiquitination assay of K63-linked RagA ubiquitination by Skp2. His-ub, His-ubiquitin; WT, wild type; Ni–nitrilotriacetic acid (NTA), nickel bead precipitates. (C) IP analysis of amino acid-dependent RagA ubiquitination in control (shLuc) and Skp2 knockdown (shSkp2) HEK293T cells. (D) Immunofluorescence (IF) assay of partially co-localization between Skp2 and lysosome marker Lamp2 in MDA-MB-231 cells. Arrows indicate Skp2 and Lamp2 co-localization. Percentage of co-localization of Skp2 with Lamp2 is 11.4+/−3.7%, Quantitative data are represented as mean +/− S.D., p<0.001. (E) IP analysis of amino acid-dependent interaction between Skp2 and RagA in HEK293T cells. (F) IP analysis of the interaction between RagA and GATOR1 components (DEPDC5 and Nprl3) or Ragulator component p18 in Skp2 overexpression and empty vector control HEK293T cells. Triangle indicates annotated protein. (G) IP analysis of amino acid-dependent interaction between RagA and GATOR1 components (DEPDC5 and Nprl3) in Skp2 knockdown (shSkp2) and scramble control (shLuc) HEK293T cells. (H) IP analysis of amino acid-dependent interaction of RagA with Skp2 and GATOR1 components (DEPDC5, Nprl3 and Nprl2) in HEK293T cells treated with DMSO or rapamycin. Cells in (C, E,G, H) were starved for amino acids for 1 hour and restimulated with amino acids for the indicated time before lysis. See also Figure S2.
Since the Rags complex is localized in lysosome and mediates mTORC1 lysosomal localization and activation, we next determined whether Skp2 is also localized in lysosome. Immunofluorescence assay showed that Skp2 partially co-localized with Lamp2, a well-established lysosome marker (Figure 2D). The biochemical fraction assay further confirmed that Skp2 protein was localized in lysosome, although it was also in other compartments (Figure S2C). Skp2 could co-immunoprecipitate with RagA and RagC (Figure S2D), and amino acid stimulation further promoted the interaction between Skp2 and RagA (Figure 2E).
We next determined whether Skp2 is a critical regulator for the recruitment of GATOR1 to the RagA complex. We found that Skp2 overexpression promoted the interaction of GATOR1 components DEPDC5 and Nprl3 with RagA (Figure 2F), whereas Skp2 knockdown reduced this interaction during amino acid treatment (Figure 2G). Interestingly, mTORC1 inhibitor rapamycin abolished not only amino acid-driven interaction between RagA and GATOR1 components, but also amino acid-induced RagA and Skp2 interaction (Figure 2H). These data collectively suggest that Skp2 is required for the recruitment of GATOR1 to RagA under amino acid stimulation in a manner dependent on mTORC1 activation.
Skp2-mediated RagA-K15 ubiquitination recruits GATOR1 and inhibits RagA GTP binding
K63-linked ubiquitination is thought to serve as a scaffold to mediate protein-protein interaction, thereby regulating diverse signal transduction events(Chan et al., 2012; Chan et al., 2013; Yang et al., 2009). We speculated that Skp2-mediated K63-linked RagA ubiquitination may account for the recruitment of GATOR1 and negative regulation of mTORC1 signaling under amino acid treatment. To test this notion, we first determined the ubiquitination site(s) of RagA induced by Skp2. To this end, we made a series of RagA mutant constructs by mutating all conserved lysine (K) residues to arginine (R) and performed in vivo ubiquitination assay. Among these RagA mutants, only RagA-K15R mutant displayed substantial reduction in the basal ubiquitination compared to wild-type (WT) RagA (Figure 3A). We further showed that Skp2 promoted the ubiquitination of WT RagA, but not RagA-K15R mutant (Figure 3B), suggesting K15 is an ubiquitination site for Skp2.
Figure 3. Skp2-mediated RagA-K15 ubiquitination recruits GATOR1 and inhibits RagA GTP binding.

(A) Screening for RagA ubiquitination sites. In vivo ubiquitination assay of RagA KR (lysine (K) mutated to arginine (R)) mutants. 3KR indicates the mutant with the lysine at residues 7, 8 and 9 mutated to arginine. (B) In vivo ubiquitination assay of WT RagA and RagA-K15R mutant by Skp2. (C) IP analysis of the interaction between WT RagA or RagA-K15R mutant and GAPTOR1 components (DEPDC5 and Nprl3) or Ragulator component p18 in HEK293T cells. Triangle indicates annotated protein. (D) GTP binding assay of RagA in empty vector and Skp2 overexpressed HEK293T cells. (E) GTP binding assay of RagA in control (shLuc) and Skp2 knockdown (shSkp2) HEK293T cells. (F) GTP binding assay of WT RagA and RagA-K15R mutant in HEK293T cells. (G) GTP binding assay of the WT RagA or RagA-K15R mutant in empty vector and Skp2 overexpressed HEK293T cells.
We next determined whether Skp2 facilitates the interaction between GATOR1 and RagA through promoting RagA ubiquitination. Compared with WT RagA, RagA-K15R mutant bound less to DEPDC5 and Nprl3 (Figure 3C). Our data therefore pinpointed the critical role of Skp2-driven RagA ubiquitination at K15 in recruiting GATOR1 to the RagA complex upon amino acid stimulation. Interestingly, we also found that RagA-K15R displayed less interaction with Ragulator component p18 compared to WT RagA (Figure 3C), consistent with the finding that Skp2 promoted the interaction between RagA and p18 (Figure 2F). This phenomenon might support a previously proposed model in which the loose interaction between RagA and Ragulator may facilitate the recruitment and activation of mTORC1(Bar-Peled et al., 2012).
GATOR1 acts as a GAP to hydrolyze RagAGTP to RagAGDP. Consistent with its role in promoting the GATOR1 and RagA interaction, Skp2 overexpression dramatically reduced the amount of RagAGTP (Figure 3D), whereas its knockdown significantly enhanced it (Figure 3E). Similar to Skp2 knockdown, the ubiquitination-deficient Rag-K15R mutant displayed much higher GTP binding compared to WT RagA (Figure 3F). We further showed that Skp2 inhibited the amount of RagAGTP by promoting RagA ubiquitination at K15, as RagA-K15R mutant abrogated the suppression effect of Skp2 on the GTP-bound status of RagA (Figure 3G). Our data collectively suggest that Skp2-driven RagA ubiquitination at K15 drives the interaction between GATOR1 and RagA leading to the reduction in GTP-bound RagA.
Skp2-mediated RagA-K15 ubiquitination negatively regulates amino acid-dependent mTORC1 lysosomal localization and activation
As the GTP-bound RagA critically regulates the interaction of Rags complex with mTORC1 and lysosomal localization of mTORC1, leading to mTORC1 activation in response to amino acids(Efeyan et al., 2013; Kim et al., 2008; Sancak et al., 2008), we hypothesized that Skp2-mediated RagA ubiquitination at K15 under amino acid stimulation would serve as a negative feedback loop to suppress the lysosomal localization and activation of mTORC1. As expected, we found that Skp2 overexpression reduced amino acid-dependent recruitment of mTOR to lysosome (Figure 4A), whereas Skp2 knockdown potentiated it (Figure 4B). Skp2 knockdown also enhanced the interaction of RagA-RagC complex with mTORC1 (Figure S3A). However, the interaction between mTORC1 complex components mTOR and Raptor was not affected by Skp2 (Figure S3B). Notably, we demonstrated that ectopic expression of RagA-K15R rescued the defect in mTOR lysosomal recruitment upon Skp2 overexpression (Figure 4C), revealing the critical role of RagA-K15 ubiquitination by Skp2 in the repression of mTORC1 lysosomal localization upon amino acid stimulation.
Figure 4. Skp2-mediated RagA-K15 ubiquitination negatively regulates amino acid-dependent mTORC1 lysosomal localization and activation.

(A) IF assay of amino acid-dependent co-localization of mTOR with lysosome marker Lamp2 in control (pBabe) and Skp2 overexpressed MDA-MB-231 cells. Quantitative data are represented as mean +/− S.D., (B) IF assay of amino acid-dependent co-localization of mTOR with lysosome marker Lamp2 in control (shLuc) and Skp2 knockdown (shSkp2) MDA-MB-231 cells. Quantitative data are represented as mean +/− S.D., ** indicates p<0.01. (C) IF assay of amino acid-dependent co-localization of mTOR with lysosome marker Lamp2 in MDA-MB-231 cells overexpressed with indicated proteins. Quantitative data are represented as mean +/− S.D., ** indicates p<0.01. (D) IB analysis of amino acid-dependent mTORC1 signaling in WT and Skp2−/− primary MEFs. (E) IB analysis of amino acid-dependent mTORC1 signaling in control (shLuc) and Skp2 knockdown (shSkp2) MDA-MB-231 cells. Triangle indicates annotated protein. (F) IB analysis of amino acid-dependent mTORC1 signaling in MDA-MB-231 cells transfected with indicated vectors. (G) IB analysis of amino acid-dependent mTORC1 signaling in the cells overexpressed with indicated proteins. Triangle indicates annotated protein. Cells in (A–G) were starved for amino acids for 1 hour and restimulated with amino acids for indicated time before lysis. See also Figure S3 and S4.
We then investigated whether Skp2 negatively regulates amino acid-dependent mTORC1 signaling. We examined the kinetic mTORC1 signaling in WT and Skp2−/− primary MEFs. mTORC1 signaling gradually increased upon amino acid stimulation, but declined under long-term treatment in WT MEFs. However, Skp2 deficiency not only enhanced initial mTORC1 activation and but also caused sustained mTORC1 activation even under long-term amino acid treatment (Figure 4D). We further demonstrated that Skp2 knockdown in various cell lines enhanced mTORC1 activation (Figure 4E and Figure S4A), whereas Skp2 overexpression inhibited amino acid-dependent mTORC1 activation (Figure 4F and Figure S4B). In contrast to the wild type (WT) Skp2, E3 ligase-deficient Skp2-LRR mutant failed to inhibit mTORC1 signaling (Figure S4C). Notably, ectopic expression of Rag-K15R mutant, but not WT RagA, compromised Skp2’s suppressive effect on mTORC1 activity (Figure 4G). Accordingly, our data suggest that Skp2 serves as a negative feedback regulator for lysosomal localization and activation of mTORC1 by promoting RagA ubiquitination at K15.
Skp2 negatively regulates cell size through mTORC1 inhibition
The activation of mTORC1 promotes protein synthesis and increases cell size (Sarbassov et al., 2005; Zoncu et al., 2011). As Skp2 initiates a negative feedback to restrict amino acid-induced mTORC1 signaling, we therefore examined whether Skp2 may suppress cell size by inhibiting mTORC1 signaling. Indeed, Skp2 knockdown in various cell lines enhanced cell size (Figure S5A and S5B), and such effect was completely abolished by rapamycin (Figure S5C and S5D). We further found that there was no detectable pS6 signal in WT liver tissues by immunostaining, while liver tissues from Skp2−/− mice displayed significant high pS6 expression (Figure 5A) and enlarged cell size in hepatocytes, which was reversed by pretreatment of the mice with rapamycin (Figure 5B and 5C). Thus, Skp2 negatively regulates cell size in vitro and in vivo, at least in liver, by inhibiting mTORC1.
Figure 5. Skp2 negatively regulates cell size through mTORC1 inhibition.
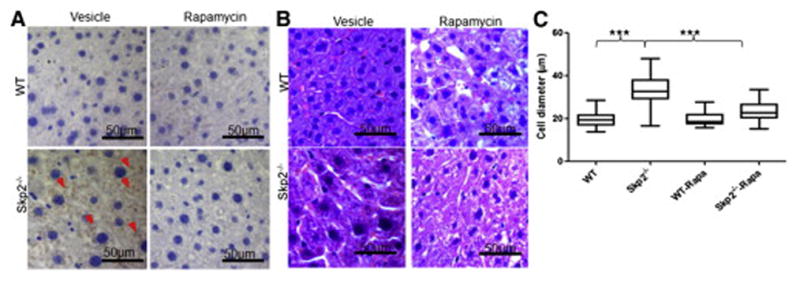
(A) Liver paraffin sections prepared from WT and Skp2−/− mice were stained with pS6. Arrows indicates the pS6 positive signals. The mice were pretreated with rapamycin or vesicle for 1 month before tissue collection. (B) Representative H&E staining of wild type (WT) and Skp2−/− liver paraffin sections. The mice were pretreated with rapamycin or vesicle for 1 month before tissue collection. (C) Statistic analysis of the long diameters of the liver cells in (B). Rapa indicates rapamycin. Quantitative data are represented as mean +/− S.D., *** indicates p<0.001. See also Figure S5.
Skp2-mediated RagA ubiquitination promotes autophagy through mTORC1 inhibition
While serving as a positive regulator of cell size, mTORC1 negatively regulates another pivotal cellular process termed autophagy. Autophagy is a critical event that maintains cell homeostasis, and deregulated autophagy is linked to various human disorders, such as aging and cancer(Choi et al., 2013; Hartleben et al., 2010). Consistent with mTORC1 activation, Skp2 knockdown suppressed autophagy, as determined by the decrease in the level of LC3-II, LC3-II/I ratio and reduction of GFP-LC3 punctate foci, and such suppressive effect was reversed by rapamycin (Figure 6A, B and Figure S6). As LC3-II level may be decreased through lysosomal degradation (Klionsky et al., 2008), we further showed that Skp2 knockdown decreased the LC3-II/I ratio both in the presence and absence of lysosomal inhibitor Bafilomycin A1 (Figure 6C), confirming that Skp2 knockdown reduced LC3-II/I ratio through suppressing autophagy. Conversely, Skp2 overexpression promoted autophagy, as demonstrated by the increase of LC3-II/I ratio (Figure 6D). Remarkably, ectopic expression of RagA-K15R mutant, but not WT RagA, abolished this promoting effect (Figure 6D).
Figure 6. Skp2-mediated RagA ubiquitination promotes autophagy through mTORC1 inhibition.

(A) IB analysis of LC3-II level using LC3B antibody and mTORC1 signaling in control (shLuc) and Skp2 knockdown (shSkp2) MDA-MB-231 cells treated with DMSO or rapamycin. Rapa indicates rapamycin. Triangle indicates annotated protein. (B) Imaging of GFP-LC3 foci in GFP-LC3 overexpressed control (shLuc) and Skp2 knockdown (shSkp2) MDA-MB-231 cells treated with DMSO or Rapamycin. (C) IB analysis of LC3-II/I ratio using LC3B antibody in control (shLuc) and Skp2 knockdown (shSkp2) HEK293T cells treated with 100nM Bafilomycin A1 (Bafi) and DMSO for 1 hour. (D) IB analysis of LC3-II/I ratio using LC3B antibody in the MDA-MB-231 cells overexpressed with indicated proteins. (E) IB analysis of LC3-II/I ratio using LC3B antibody and mTORC1 signaling in the lysates of isolated WT and Skp2−/− mouse tissues. See also Figure S6.
To further determine whether Skp2 regulates mTORC1 signaling and autophagy in vivo, we isolated diverse tissues from WT and Skp2−/− mice for Western Blot analysis. Consistently, mTORC1 activity was much higher in various Skp2−/− tissues, such as in heart, liver and kidney, accompanied by the reduction in LC3-II/I ratio (Figure 6E). Altogether, these results suggest that Skp2-mediated RagA ubiquitination serves as a regulatory mechanism to induce autophagy through mTORC1 inhibition.
Skp2-mediated RagA ubiquitination suppresses cilia growth through mTORC1 inhibition
Cilia are microtubule cellular organelles, which are induced from the centrioles and play a role in chemical sensation, signaling transduction and cell growth control. Defects of cilia growth lead to a group of human disorders, termed ciliopathies(Nigg and Raff, 2009; Sang et al., 2011) and may be involved in cancer development(Nigg and Raff, 2009). Recently, two studies suggest there is a potential cross-talk between cilia and autophagy(Pampliega et al., 2013; Tang et al., 2013). As Skp2 regulates autophagy and cancer development, we examined whether Skp2 is a regulator of cilia formation. We found that Skp2 knockdown increased the average length of cilia, as determined by assessing the acetylated Tubulin (Ac-Tubulin), which was reversed by Rapamycin treatment (Figure 7A and 7B). In contrast, stable overexpression of Skp2 reduced cilia length. Notably, the inhibitory effect of Skp2 on cilia length was completely abrogated by the introduction of RagA-K15R mutant, but not by that of WT RagA (Figure 7C, 7D and Figure S7). These results suggest that Skp2-mediated RagA ubiquitination at K15 serves as a negative regulator of cilia growth through mTORC1 inhibition. Our data also indicate that mTORC1 has a positive role in cilia size control in mammalian cells, consistent with a recent finding that TORC1 signaling positively regulates cilia size in zebra fish(Yuan et al., 2012).
Figure 7. Skp2-mediated RagA ubiquitination suppresses cilia growth through mTORC1 inhibition.

(A) Representative of cilia growth in control (shLuc) and Skp2 knockdown (shSkp2) NIH3T3 cells treated with DMSO or Rapamycin. (B) Statistic analysis of cilia length in (A). Quantitative data are represented as mean +/− S.D., ** indicates p<0.01. (C) Representative of cilia growth in NIH3T3 cells overexpressed with indicated proteins. (D) Statistic analysis of cilia length in (C). Quantitative data are represented as mean +/− S.D., ** indicates p<0.01. (E) Schematic model for the molecular mechanism and biological functions of the negative feedback regulation for amino acid-dependent mTORC1 signaling. Skp2-mediated K63-linked ubiquitination of RagA upon amino acid stimulation recruits GATOR1 complex to RagA and hydrolyze RagAGTP, thereby serving as a negative feedback to terminate mTORC1 signaling and prevent its hyperactivation. Such regulatory mechanism critically regulates multiple cellular functions, including cell size, cilia growth and autophagy. See also Figure S7.
Discussion
It is well recognized that amino acid treatment promotes mTORC1 lysosomal localization and activation(Sancak et al., 2010; Sancak et al., 2008). However, the negative feedback regulators of preventing mTORC1 signaling hyperactivation driven by sustained amino acid stimulation have so far not been reported. We here identify a critical negative feedback regulatory mechanism by which mTORC1 signaling is shut off during continuous amino acid exposure (Fig. 7E). mTORC1 signaling is restricted by the negative feedback loop even in the early time point of amino acid stimulation. We show that amino acids induce mTORC1 activation and then drive an mTORC1 dependent negative feedback loop through recruiting the GATOR1 complex to RagA to terminate mTORC1 signaling and thus prevent mTORC1 hyperactivation. Amino acids elicit K63-linked ubiquitination of RagA, which is mediated by Skp2 E3 ligase and serves as a crucial event to recruit the GATOR1 complex to RagA and hydrolyze RagAGTP, thereby inhibiting the localization of mTORC1 to lysosome and mTORC1 activation. This finding provides the molecular insight into how mTORC1 signaling is restricted by a negative feedback loop upon amino acid exposure.
The GATOR1 complex is known to inhibit mTORC1 signaling by serving as a GAP for RagAGTP hydrolysis(Bar-Peled et al., 2013). However, how GATOR1 is recruited to the RagA remains unknown. We show that amino acids are physiological stimuli to drive the recruitment of GATOR1 and demonstrate that GATOR1 recruitment serves as a negative feedback regulator to terminate mTORC1 signaling under continuous amino acid stimulation. We further reveal that Skp2 SCF E3 ligase is an essential regulator for this process by inducing RagA ubiquitination, which facilitate the interaction of GATOR1 with RagA to hydrolyze RagAGTP. Our finding therefore provides the molecular clue to how GATOR1 is recruited to RagA to inhibit mTORC1 signaling during amino acid stimulation.
RagA GTP binding is essential for mTORC1 lysosomal localization and activation. We reveal that Skp2 is a negative regulator of RagA GTP binding by recruiting GATOR1 to hydrolyze RagAGTP. Interesting, Sestrins have also been identified as RagA GTP binding negative regulators. Sestrins act as the GDIs of RagA/B and inhibit the exchange of GDP to GTP upon amino acid stimulation(Peng et al., 2014). Although both Skp2 and Sestrins inhibit RagA GTP binding and mTORC1 signaling, Skp2 functions through a negative feedback loop to remove GTP on RagA and terminate mTORC1 signaling, while Sestrins inhibit the initial mTORC1 induction through inhibiting the dissociation of GDP. These two mechanisms, theoretically non-overlapping, may work together to prevent the hyperactivation of amino acid-dependent mTORC1 signaling.
We identify Skp2 E3 ligase as a critical negative regulator of amino acid-dependent mTORC1 signaling. It remains to be determined how mTORC1 promotes Skp2-RagA interaction and subsequent RagA ubiquitination, thereby eliciting the negative feedback loop for mTORC1. Previous studies demonstrate that both mTORC1 and its downstream kinase S6K1 induce negative feedback regulation of growth factor-dependent mTORC1 signaling through directly phosphorylating IRS1(Hartman et al., 2009; Tzatsos and Kandror, 2006; Um et al., 2004). It is likely that Skp2 may be phosphorylated through an mTORC1-dependent manner, as Skp2 activity has been shown to be regulated by protein phophorylation(Lin et al., 2009). We therefore speculate that Skp2 may represent a substrate of an mTORC1-dependent kinase, such as mTOR itself, S6K1 or kinase regulated by mTORC1, and such phosphorylation may serve a molecular switich to initiate amino acid-dependent negative feedback of mTORC1.
mTORC1 deregulation leads to cellular abnormalities including defects in autophagy, cilia growth, and cell size. The fine-tune control of mTORC1 activity is of significance in maintaining cellular homeostasis. Our study suggests that Skp2-mediated K63-linked ubiquitination of RagA may serve as such a controller. Indeed, we show here that Skp2 deficiency cells also display enlarged cell size, enhanced cilia growth and autophagy inhibition, and such phenotypes can be rescued by rapamycin treatment. Our study therefore establishes that Skp2 is a negative regulator for cell size and cilia formation, but serves as a positive regulator for autophagy through mTORC1 inhibition. While our study identifies mTORC1 signaling inhibition as a critical mechanism for Skp2-mediated cell size, clila growth and autophagy control, we can not rule out the possibility that other pathways regulated by Skp2 may be involved in these cellular processes. It was shown that Skp2-mediated p27 degradation is also involved in cell size regulation (Kossatz et al., 2004). Therefore, it is interesting to understand how these two pathways may coordinate to regulate Skp2-mediated cell size control.
In summary, our study reveals that Skp2-mediated K63-linked ubiquitination of RagA upon amino acid stimulation serves as a negative feedback loop to prevent mTORC1 hyperactivation and to regulate autophagy, cell size, and cilia growth by promoting the recruitment of GATOR1 to the RagA complex. This regulatory mode may be globally deregulated in human disorders, such as age-related syndromes and cancers.
Experimental procedures
Mouse models
Genetic Skp2 knockout mice have been described previously(Lin et al., 2010). For the pretreatment of mice with rapamycin, rapamycin was dissolved in the solution containing 5% Tween80 plus 5% PEG400 and injected peritoneally into mice (4mg/kg) 3 times per week for 1 month. For histology analysis, the mouse tissues were fixed in 10% formalin and then embedded in paraffin before processed to hematoxylin and eosin (H&E) staining, immunohistochemistry staining by standard procedure.
Cell culture and materials
HEK293T, MDA-MB-231, PC3, NIH3T3 cells were cultured in DMEM medium supplied with 10%FBS. Primary mouse embryonic fibroblast (MEF) cells were isolated from day 13.5 mouse embryo and cultured in DMEM medium supplied with 10%FBS. HA-RagA, HA-RagC plasmids were modified from previous described constructs1. Flag-Skp2, pBabe-Skp2, Skp2 LRR mutant, and His-ubiquitin WT, K48R mutant and K63 mutant were described previously10,14. RagA-K15R mutant was generated by site mutagenesis. Antibody to Skp2 is from Invitrogen; antibodies to RagA, RagC, mTOR, Raptor, phospho-S6K T389 (pS6K), S6K, phospho-S6 S235/236 (pS6), S6, phospho-4E-BP1 T37/T46 (p4E-BP1), 4E-BP1, LC3B, p18, acetylated Tubulin from Cell Signaling; antibody to γ-Tubulin from Abcam; antibody to Nprl3 from Atlas Antibodies; antibodies to DEPDC5, Nprl2, ubiquitin, and HRP-conjugated anti-mouse and anti-rabbit secondary antibodies from Santa Cruz; antibody to lys63 specific ubiquitin from Millipore; antibody to flag from Sigma; antibody to HA from Convance. Fluorescence conjugated (Alexa 488 or Alexa 555) anti-mouse and anti-rabbit secondary antibodies are from Invitrogen. γ-aminohexyl-GTP-sepharose bead suspension is from Jena bioscience; Ni–nitrilotriacetic acid (NTA), nickel bead suspension from Invitrogen; protein agarose A/G bead suspension from Santa Cruz.
Amino acid starvation and stimulation
For amino acid starvation, the cells were washed with PBS and replaced with amino acid free RPMI 1640 for one hour. For amino acid stimulation, the starvation medium was replaced with normal RPMI 1640 without FBS for indicated times.
Immunoblotting (IB), immuoprecipitation (IP) and immuofluorescence (IF)
Cells or mouse tissues were lysed in RIPA buffer in the presence of proteinase inhibitor cocktail and phosphatase inhibitors (10 mM Na-pyrophosphate, 10 mM Na-glycerophosphate, 50mM Na-fluoride) and subjected to immunoblotting by indicated antibodies. For immunoprecipitation, the lysates were incubated with primary antibodies overnight, followed by incubation with protein agarose A/G beads for 3 hours rotating at 4°C. The beads were washed 4 times with lysis buffer and analyzed by immunoblotting. For lys63 specific immunoprecipitation, cells were lysed and sonicated in 6M urea buffer containing 50mM Tris, 300mM NaCl, 0.5%NP40 in the presence of proteinase inhibitor cocktail. The lysate was then diluted to 4M urea before performing immuoprecipitation. For the analysis of interactions between RagA, GATOR1, Ragulator and Skp2, the cells were treated with crosslinker DSP before lysis as previously described2,3. For immunofluorescence, cells were fixed for 10mins with 4%PFA, permeabilized for 10min with 0.3%Triton, blocked for 1 hour with 2% BSA and incubated with primary antibodies overnight at 4°C, followed by incubation with fluorescence conjugated secondary antibodies for 1 hour at room temperature. The cells were then co-mounted with DAPI and all pictures were taken with confocal microscope. The co-localization percentages of Skp2, mTOR with Lamp2 were calculated using imageJ plugin JACoP.
In vivo and in vitro ubiquitination assay
In vivo and in vitro ubiquitination assay were described previously10,14. For the in vivo ubiquitination assay, HEK293T cells were transfected with the indicated vectors. After 48 hours, cells were lysed in the denatured buffer (6M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole) and incubated with Ni–nitrilotriacetic acid (NTA), nickel beads for 3 hours rotating at room temperature, followed by beads washing and immunoblotting analysis. For the in vitro RagA ubiquitination assay, flag-Skp2 SCF complex and HA-RagA were purified by immunoprecipitation followed by elution with flag or HA peptides from HEK293T cells transfected with flag-Skp2 and HA-RagA, respectively. Purified flag-Skp2 SCF and HA-RagA were incubated at 37°C for 3 hours along with E1 (UBE1) and E2 (UbcH5c, UbcH7 and Ubc13/Uev1) in the 20ul reaction buffer (20 mM Hepes (pH 7.4), 10 mM MgCl2, 1 mM DTT, 59 μM ubiquitin, 1 mM ATP, 30 μM creatine phosphate, and 1 U of creatine kinase) and subjected to immunoblotting analysis.
GTP binding assay
The GTP binding assay was performed as previously described15–17. Briefly, cells were lysed in the lysis buffer (1xPBS, 1%Triton) supplied with protein inhibitor cocktail. The lysates were incubated with γ-aminohexyl-GTP-sepharose beads for 2 hours rotating at 4°C, followed by washing 4 times with lysis buffer and subjected to analysis by immunoblotting.
Cilia length measurement
2×105 NIH3T3 cells were seeded in a well of 2 wells chamber slide (NUNC). From the following day, cilia were induced by serum starvation for 24 hours. Cells were treated with or without 100nM rapamycin during the serum starvation. After serum starvation, cells were fixed for 5 minutes with cold methanol and blocked for 1 hour with 3% BSA. The cells were then incubated with mouse anti-acetylated Tubulin and rabbit anti-γ-Tubulin overnight at 4°C, followed by incubation with anti-mouse and anti-rabbit secondary antibodies conjugated with Alexa 488 or Alexa 555. Cilia pictures were taken by confocal microscope and cilia length were measured by ImageJ software.
Statistic analysis
Statistical significance was identified by Student’s t-test. p values of less than 0.05 were considered statistically significant. ** indicates p<0.01
Supplementary Material
Highlights.
Skp2 mediates amino acid-induced RagA ubiquitination and GATOR1 recruitment
Skp2 is a negative feedback regulator of amino acid-dependent mTORC1 signaling
Skp2 promotes autophagy but inhibits cell size, cilia growth by mTORC1 repression
Acknowledgments
We thank Drs. David Sabatini for original plasmids, and Dr. B. Zhen and S. Zhang for technical assistance. We also thank all the members of the Lin’s lab for their valuable comments and suggestions. This work was supported by the MD Anderson Prostate SPORE Development Award, R. Lee Clark Scholar Award, MD Anderson prostate moonshot grant, NIH grants, and CPRIT grant to H.K.L, and a grant supported by the Health and welfare surcharge of tobacco products from Taiwan (MOHW103-TD-B-111-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–1111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493:679–683. doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, Henske EP. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose E, Nakashima N, Sekiguchi T, Nishimoto T. RagA is a functional homologue of S. cerevisiae Gtr1p involved in the Ran/Gsp1-GTPase pathway. J Cell Sci. 1998;111(Pt 1):11–21. doi: 10.1242/jcs.111.1.11. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004;18:2602–2607. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Orhon I, Patel B, Sridhar S, Diaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Li MO. Sestrins Function as Guanine Nucleotide Dissociation Inhibitors for Rag GTPases to Control mTORC1 Signaling. Cell. 2014;159:122–133. doi: 10.1016/j.cell.2014.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccio M, Bos JL, Zwartkruis FJ. Regulation of the small GTPase Rheb by amino acids. Oncogene. 2006;25:657–664. doi: 10.1038/sj.onc.1209106. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem. 2001;276:7246–7257. doi: 10.1074/jbc.M004389200. [DOI] [PubMed] [Google Scholar]
- Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–18727. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Li J, Diener DR, Choma MA, Rosenbaum JL, Sun Z. Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc Natl Acad Sci U S A. 2012;109:2021–2026. doi: 10.1073/pnas.1112834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.