

Abstract
Mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene account for >70% of X-linked retinitis pigmentosa (XLRP) and 15–20% of all inherited retinal degeneration. Gene replacement therapy for RPGR-XLRP was hampered by the relatively slow disease progression in mouse models and by difficulties in cloning the full-length RPGR-ORF15 cDNA that includes a purine-rich 3′-coding region; however, its effectiveness has recently been demonstrated in four dogs with RPGR mutations. To advance the therapy to clinical stage, we generated new stable vectors in AAV8 or AAV9 carrying mouse and human full-length RPGR-ORF15-coding sequence and conducted a comprehensive long-term dose-efficacy study in Rpgr-knockout mice. After validating their ability to produce full-length proteins that localize to photoreceptor connecting cilia, we evaluated various vector doses in mice during a 2-year study. We demonstrate that eyes treated with a single injection of mouse or human RPGR-ORF15 vector at an optimal dose maintained the expression of RPGR-ORF15 throughout the study duration and exhibited higher electroretinogram amplitude, thicker photoreceptor layer and better targeting of opsins to outer segments compared with sham-treated eyes. Furthermore, mice that received treatment at an advanced age also showed remarkable preservation of retinal structure and function. Retinal toxicity was observed at high vector doses, highlighting the importance of careful dose optimization in future clinical experiments. Our long-term dose-efficacy study should facilitate the design of human trials with human RPGR-ORF15 vector as a clinical candidate.
Introduction
Dysfunction or death of photoreceptors is responsible for irreversible loss of vision in inherited retinal degenerative diseases that are genetically heterogeneous with wide variations in clinical phenotype. Of these, retinitis pigmentosa (RP) refers to a diverse group of dystrophies that often exhibit progressive impairment of rod photoreceptor function, leading eventually to both rod and cone death (1). As a result, an RP patient initially experiences night blindness, which is followed by progressively diminishing peripheral vision and eventually loss of central vision. RP afflicts 1 in 4000 births, with more than one million affected individuals worldwide (1). Almost 50% of all RP cases are referred as ‘simplex’ with no family history. X-linked forms of RP (XLRP) account for 10–20% of RP and show clinically more severe findings (2,3). Though two major X-linked genes have been discovered, retinitis pigmentosa GTPase regulator (RPGR) mutations account for >70% of XLRP (2–4). In some pedigrees, genetic defects in RPGR cause cone dystrophy, cone-rod dystrophy and/or atrophic macular degeneration (5). Furthermore, RPGR mutations have been identified in roughly 15% of males in simplex cases (6) and even in presumably ‘dominant’ families (7,8), making it the most frequent cause of retinal degeneration in addition to rhodopsin (9). Because of extensive phenotypic variations, both rods and cones should be considered as the target for treatment when developing a therapy for the RPGR disease.
In addition to a constitutively expressed transcript, RPGR1–19, multiple RPGR transcripts have been detected in the retina and are likely generated by alternative splicing and/or polyadenylation (2,10–12). A majority of the disease-causing mutations have been identified in a variant isoform RPGR-ORF15, which is abundant in the retina and includes a unique long stretch of purine-rich region in its last exon, called ORF15 (2,5). RPGR-ORF15 protein interacts with a number of centrosome-cilia proteins (13) and localizes to the connecting cilia in both rod and cone photoreceptors (14,15). Although its precise physiological role has not been delineated, RPGR-ORF15 seems to participate in regulating protein trafficking along the photoreceptor cilium (13,16). An early hallmark of RPGR-ORF15-related photoreceptor abnormality is opsin mis-localization in the retinal photoreceptors (14,17–19).
Gene replacement therapy using adeno-associated virus (AAV) vectors for gene delivery has proven efficacious in rescuing retinal function and structure in a number of animal models of retinal degeneration (20). This preclinical success has been translated into clinical effectiveness with minimal side effects in dozens of patients with Leber congenital amaurosis (LCA) caused by RPE65 deficiency (21–28) and in six patients with choroideremia (29). As a number of retinal gene therapy protocols are entering clinical stage (30–33), the RPGR disease has gained attention because of its severity and a relatively large patient population for this ‘orphan’ disease. Hence, several groups have characterized naturally occurring or genetically engineered animal models carrying RPGR mutations (14,17,18,34,35). Until recently, gene replacement for RPGR disease was hampered by the slow disease progression in mouse models (14,17,18) and the difficulty in obtaining an intact and stable full-length RPGR-ORF15 cDNA owing to the complexity of the purine-rich region in the ORF15 exon. Notably, germ-line transmission of an abbreviated (36) or full-length (37) version of Rpgr-ORF15 cDNA was able to rescue photoreceptor degeneration in Rpgr-knockout (Rpgr-KO) mice. More recently, AAV type 5 (AAV5) vectors carrying a full-length human RPGR-ORF15 cDNA have been shown to be efficacious for therapy in four dogs with different rates of retinal degeneration resulting from two distinct RPGR mutations (38); however, a detailed characterization of these therapeutic vectors, including examination of their genome integrity and ability to produce full-length RPGR-ORF15 protein, has not been reported (38).
A fully characterized therapeutic vector with detailed safety and dose-efficacy profiles in a disease model is a prerequisite for clinical testing of gene therapy. In order to advance the RPGR gene replacement to clinical stage, we constructed and characterized new vectors carrying either a mouse or a human full-length RPGR-ORF15-coding sequence. The vectors were packaged into AAV type 8 or 9 capsids because these serotypes exhibited excellent tropism for mouse and non-human primate photoreceptors (39–41). A short-term toxicity and a long-term dose-efficacy study of these vectors were carried out in Rpgr-KO mice, a slowly progressing disease model that develops obvious rod and cone photoreceptor degeneration in a 12- to 24-month period (14,36). In addition to providing vector dose-efficacy/toxicity relationship, our study demonstrates that a single optimal dose of vector administration is able to sustain long-term RPGR-ORF15 expression and rescue retinal structure and function in the Rpgr-KO mice. Our study thus represents a step forward toward clinical trials in patients with RPGR mutations.
Results
Generation of mouse and human RPGR-ORF15 AAV vectors
Disease-causing mutations occur in the common region of RPGR-ORF15 and RPGR1–19 isoforms and in the unique region of RPGR-ORF15 isoform. No mutation associated with retinal disease has yet been identified in exons 16 to 19 that are unique to RPGR1–19 isoform. We thus hypothesized that RPGR-ORF15 protein rather than the constitutively expressed RPGR1–19 should be able to rescue the RPGR disease. To develop a gene replacement therapy, we constructed AAV vectors carrying either mouse or human RPGR-ORF15 expression cassette (Fig. 1A). Efforts to obtain a full-length RPGR-ORF15 cDNA using reverse-transcription PCR have not been successful owing to the unique purine-rich region of the terminal ORF15 exon (data not shown). To overcome this problem, we conducted regular PCR using genomic DNA as a template to amplify the 3′ part of the ORF15 exon containing the purine-rich region and then ligated it to a synthetic DNA fragment encoding the upstream exons (Supplementary Material, Fig. S1). This strategy was adopted for obtaining both mouse and human RPGR-ORF15-coding sequences. A human rhodopsin kinase (RK) promoter, which shows rod and cone cell specificity (42), was used to drive RPGR-ORF15 expression (Fig. 1A). These two vectors were packaged into AAV type 8 and are hereafter referred to as AAV8-mRpgr and AAV8-hRPGR, respectively. The mouse Rpgr-ORF15 vector was also packaged into AAV type 9 (hereafter referred to as AAV9-mRpgr), a serotype that transduces cones of non-human primate efficiently (41).
Figure 1.

Construction and characterization of mouse and human RPGR-ORF15 AAV vectors. (A) Schematic representation of the vectors. (B) Examination of the vector integrity. PCR amplification of the region spanning the repetitive glutamic acid-glycine coding sequence in the mouse or human RPGR-ORF15 produced the expected 1.3-kb or 1.6-kb fragment in the vector plasmids and all vector preparations. Arrows indicate fragments with minor deletions in two vector preparations. (C) Immunoblot analysis using C100 anti-RPGR antibody that recognizes the C-terminal of mouse RPGR-ORF15 protein. The retinal lysate from an Rpgr-KO mouse injected subretinally with 1 × 109 vg AAV8-mRpgr vector revealed a ∼200-kDa protein band corresponding to the full-length RPGR-ORF15 protein, which is identical to that detectable in a WT C57/Bl6 mouse retina. (D) Immunoblot analysis using an antibody that recognizes the region upstream of the ORF15 exon of human RPGR. The retinal lysate from an Rpgr-KO mouse injected subretinally with 1 × 109 vg AAV8-hRPGR vector revealed a ∼200-kDa protein band corresponding to the full-length RPGR-ORF15 protein (closed arrowheads), identical to that from a commercially sourced human retinal lysate. A set of proteins with lower molecular weights indicating the truncated or alternatively spliced forms of the protein were also detected (open arrowhead). No signal was detected in the lane that included one-tenth amount of retinal lysate from the vector-injected eye, revealing the sensitivity limits of the assay. (E and F) Immunostaining of retina sections from Rpgr-KO mice that received subretinal injection of AAV8-mRpgr (E) or AAV8-hRPGR (F) at 16 weeks post-administration using an antibody against mouse or human RPGR. The magnified images of the marked areas are shown in (E). WT C57/Bl6 and vehicle-injected Rpgr-KO retina sections were used as positive and negative controls, respectively. Vector-expressed RPGR protein mainly localizes to the connecting cilia region of the retina as does the WT protein. RPGR staining is shown in red, and nuclei are stained blue by DAPI. RPE, retinal pigment epithelium; CC, connecting cilia; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer. Scale bars: 50 µm.
The vector plasmids containing the purine-rich region of RPGR-ORF15 and two AAV inverted terminal repeats (ITRs) are prone to deletions or rearrangements when the plasmid clones are propagated in commonly used bacterial strains (data not shown). After testing various Escherichia coli strains, we observed that the vector plasmids maintained their integrity in XL10 Gold cells, although the underlying mechanism is not totally clear. PCR assay did not identify visible deletion in most AAV vector preparations (Fig. 1B). Minor deletions were detected in two vector preparations, which reinforced the importance of integrity examination of the RPGR-ORF15-containing vectors and offered a note of caution on subsequent data interpretation.
AAV vector-mediated expression of RPGR-ORF15 proteins
To test whether these vectors mediate full-length RPGR-ORF15 protein expression in mouse retina, we performed immunoblot analyses of the retinal lysates from vector-treated Rpgr-KO mice. Using an antibody against C-terminal of the mouse RPGR-ORF15, we identified the protein in the Rpgr-KO retina treated with the AAV8-mRpgr vector (Fig. 1C). This protein was identical in size to the wild-type (WT) RPGR-ORF15, suggesting the ability of the vector to produce a full-length mouse RPGR-ORF15 protein. Similarly, AAV8-hRPGR vector was able to generate the full-length human RPGR-ORF15 protein in the Rpgr-KO retina (Fig. 1D). A set of proteins with lower molecular weights were also detected in the AAV8-hRPGR-treated retina when an antibody against the epitopes upstream of the ORF15 exon was employed. Although we cannot rule out the possibility that these shorter proteins were caused by deletions in the ORF15 exon in AAV vector preparations (Fig. 1B), they could also represent alternatively spliced or C-terminal truncated forms of RPGR-ORF15, as observed in WT mouse retina (reference 12 and X. Sun, T. Li, unpublished data). Shorter proteins have also been identified in AAV8-mRpgr-treated Rpgr-KO retina when an antibody against the epitopes upstream of the ORF14/15 exon of mouse RPGR was used (data not shown).
Previous studies have shown that RPGR-ORF15 protein localizes to the connecting cilia of the photoreceptors in mouse and other mammalian species (14,15). To test whether the vector-expressed mouse and human RPGR-ORF15 also localize to the connecting cilia, a brief on-slide fixation of frozen retinal section using 1% formaldehyde, instead of the conventional 4% paraformaldehyde (PFA) fixation, was employed for immunofluorescence assay because the latter blocks antibody penetration to this region (15). Similar to the WT protein, the vector-expressed RPGR-ORF15 primarily appeared as dots between the inner (IS) and outer segments (OS) corresponding to the location of the connecting cilia (Fig. 1E, F and Supplementary Material, Fig. S2). In addition to connecting cilia, the vector-expressed RPGR-ORF15 was frequently localized to the IS and sometimes to the nuclei and the synaptic terminals of the photoreceptors when the conventional 4% PFA fixation was used (Supplementary Material, Fig. S2). This apparent mis-localization of RPGR-ORF15 seems to be vector related as it was also observed in AAV5 RPGR vector-treated canine retina (38) but not in the WT mouse retina (Supplementary Material, Fig. S2). Over-expression of the protein by using a relatively high vector dose (1 × 109 vector genomes, vg) and a strong RK promoter may account for this observation. No obvious RPGR-ORF15 expression was observed in other retinal layers owing to the tropism of AAV8 and the photoreceptor specificity of the RK promoter. The mouse Rpgr-ORF15 delivered by the AAV9 vector expressed the protein at a similar level as the AAV8 vector when same dose was used, and the protein was targeted to identical subcellular localization (data not shown).
Short-term dose-toxicity profile of the mouse and human RPGR-ORF15 vectors
To define the dose range for a long-term efficacy study, we first conducted a short-term vector toxicity study over a 4-month period. Eight-week-old Rpgr-KO mice were unilaterally injected with 1 × 1010 or 1 × 109 vg of AAV8-hRPGR or AAV8-mRpgr vector per eye through subretinal injection. The fellow eyes were used as controls by injecting the same volume of vehicle. Dark- and light-adapted electroretinogram (ERG) were recorded to evaluate responses from rod and cone photoreceptors at 4 months post-injection (PI) before sacrificing the mice for immunofluorescence analyses. Owing to the slow retinal degeneration in the Rpgr-KO mouse line, we did not expect an obvious therapeutic effect at 4 months after vector treatment.
No statistically significant difference was observed between the vector and the vehicle-treated eyes in ERG amplitudes of dark-adapted a-, b- and light-adapted b-waves in mice that received 1 × 109 vg AAV8-hRPGR or AAV8-mRpgr vector (Fig. 2A and Supplementary Material, Fig. S3). However, remarkably lower amplitudes of all three ERG measurements were observed in eyes receiving 1 × 1010 vg vectors, indicating the vector toxicity at the high dose. This observation was corroborated by immunofluorescence analyses of the vector-treated retinas (Fig. 2B). As the retina sections were fixed in 4% PFA before freezing, only the pool of mis-localized recombinant RPGR (mainly at IS instead of connecting cilia) was detected (as explained earlier). More intensive RPGR-ORF15 expression was observed in 1 × 1010 vg vector-treated retina, accompanied by a much thinner outer nuclear layer (ONL) and shorter IS (Fig 2B). In contrast, the ONL thickness of the 1 × 109 vg vector-treated retina did not reveal marked difference from the vehicle-treated retina. Both the ERG and immunofluorescence analyses indicate that the dose of 1 × 109 vg per eye is well tolerated, whereas 1 × 1010 vg is toxic to the mouse retina. A combinational effect of overexpressing the RPGR-ORF15 protein, the large amount of AAV capsid protein and vector DNA that exceeds the processing capacity of the retinal cells might account for the toxicity of the high vector dose. Therefore, the dose of 1 × 1010 vg per eye was not included in the subsequent long-term efficacy study.
Figure 2.

Dose toxicity of AAV8-hRPGR vector at 4 months after subretinal injection into Rpgr-KO mice. Mice received unilateral injections of the vector and contralateral injections of the vehicle (control). (A) Full-field ERG at 4 months PI. Eyes receiving 1 × 1010 vg vector displayed significantly lower ERG amplitudes than eyes receiving vehicle, whereas 1 × 109 vg/eye vector administration did not cause significant ERG change. Error bars show SEM, and the significance was calculated using two-tailed paired t-test. *P < 0.05; **P < 0.01; ***P < 0.001. (B) Immunostaining of retinal sections with an antibody against human RPGR. More intensive RPGR-ORF15 expression was observed in 1 × 1010 vg vector-treated retina, accompanied by a much thinner ONL and shorter inner segments (IS). The ONL thickness of the 1 × 109 vg vector-treated retina did not show an obvious difference compared with the vehicle-treated eye. RPGR staining is shown in red, and nuclei are stained blue by DAPI. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar: 50 µm.
Treatment effect in the Rpgr-KO mice following gene delivery of mouse Rpgr-ORF15
To test whether the mouse Rpgr-ORF15-coding sequence delivered by AAV8 or AAV9 vector was efficacious, we injected the vectors in the subretinal space of 6- to 8-week-old mice at doses ranging from 1 × 108 to 1 × 109 vg per eye. Unilateral vector injection was performed on each mouse, and the contralateral eye was injected with the vehicle. Owing to the slow progression of retinal degeneration in the Rpgr-KO mice (14,36), a longitudinal ERG monitoring was performed during an 18-month follow-up period. Given the large variation in ERG amplitudes among individual mice, paired t-test was employed throughout the study to compare the vector- and the vehicle-treated eyes. Among all cohorts, mice receiving 3 × 108 vg AAV9-mRpgr displayed the strongest therapeutic effect in vector-treated eyes (Fig. 3 and Supplementary Material, Fig. S4). Although only a slightly better ERG response was observed in the vector-treated eyes at 12 months PI, the therapeutic effect became more pronounced at 18 months PI (Fig. 3A), in which significantly larger amplitudes were observed for dark-adapted a-wave and light-adapted b-wave. These eyes also displayed significantly larger dark-adapted b-wave amplitude, which was not seen at 12 months PI, reflecting a better preservation of visual signaling to the bipolar cells. In all seven mice that survived 18-month monitoring, each individual animal exhibited greater dark-adapted a-, b- and light-adapted b- wave amplitudes in the vector-treated eye (Fig. 3B). ERG waveforms from a representative mouse at 18 months PI are shown in Figure 3C. Cohorts receiving other vector doses displayed suboptimal rescue at 18 months PI compared with the one receiving 3e8 vg AAV9-mRpgr vector (Supplementary Material, Fig. S4).
Figure 3.

Rescue of retinal function in Rpgr-KO mice that received 3 × 108 vg AAV9-mRpgr vector treatment. Mice received unilateral injections of the vector and contralateral injections of vehicle at 6–8 weeks of age. (A) ERG analysis at 12 months and 18 months PI. Although only a slight improvement was observed in the vector-treated eyes at 12 months PI, significantly larger amplitudes of dark-adapted a-, b-wave and light-adapted b-wave were obtained in response to increasing intensities of flash stimuli in these eyes at 18 months PI. The dotted lines represent the average ERG amplitudes of age-matched C57 Bl/6 mice (n = 12). (B) Each individual animal (as represented by each line) exhibited higher ERG amplitudes elicited from the highest flash intensity in the vector-injected eye at 18 months PI (n = 7). (C) Representative ERG waveforms from a single Rpgr-KO mouse at 18 months PI. Error bars show SEM and the significance between the vector-injected and vehicle-injected eyes was calculated using two-tailed paired t-test. *P < 0.05; **P < 0.01.
Functional rescue of the vector-treated retinas was correlated with their structural preservation. Much thicker ONL was maintained in 3 × 108 vg AAV9-mRpgr-treated eyes than the control eyes, as revealed by optical coherence tomography (OCT) retinal imaging at 18 months PI (Fig. 4A). A greater whole retina thickness in the vector-treated eye was also observed within a ∼1.0 mm2 field of view, except for the central area where optic nerve head (ONH) was located (Fig. 4B). The AAV-mediated RPGR expression spanned roughly half of the cross-sections, as revealed by immunofluorescence analyses of treated mouse retinas subsequent to OCT (Fig. 4C). Concomitant with the RPGR expression was the preservation of significantly more rows of photoreceptors in the vector-treated eyes than control eyes, which was also consistent with the OCT findings. Seven to ten rows of photoreceptors were maintained in a majority of the vector-treated eyes, compared with four to six rows in the control eyes. The measurements of ONL thickness at 500 µm of intervals along the vertical (dorsal–ventral) meridian on retinal sections further corroborated these findings (Fig. 4D). The average ONL thickness at different locations of the vector-treated retinas ranged between 31.7 and 43.5 µm, whereas it ranged between 19.0 and 28.3 µm in the control retinas. The treatment effect appeared to be even more pronounced at 24 months PI in one group of mice receiving 1 × 109 vg AAV8-mRPGR injection (Supplementary Material, Fig. S5). While the ONL of the control retina almost disappeared in the superior portion and only one to three rows of photoreceptors remained in the inferior retina, six–eight rows of photoreceptors survived in inferior areas of the vector-treated retina where RPGR expression was detected.
Figure 4.

Rescue of retinal structure in Rpgr-KO mice treated with 3 × 108 AAV9-mRpgr vector. (A and B) Representative OCT scans of vehicle-injected and vector-injected eyes of a single Rpgr-KO mouse at 18 months PI. (A) retina sections across the ONH. Substantially thicker ONL was observed in the vector-injected eye than the vehicle-injected eye. (B) Whole retinal thickness. The vector-injected eye displayed greater whole retina thickness within a ∼1.0 mm2 field of view centered on the ONH. (C) Immunostaining of retinal sections across the ONH with an antibody against mouse RPGR at 18 months PI. More rows of photoreceptors were preserved in RPGR-expressed area in the vector-injected eye. Inset: the magnified image of the marked area. RPGR staining is shown in red, and nuclei are stained blue by DAPI. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. (D) Quantitative analysis of ONL thickness from superior–inferior retinal sections across the ONH. Two-tailed paired t-test was used for statistics analysis (n = 5). *P < 0.05. Error bars, SEM.
Opsin mis-localization (because of altered transport/targeting to OS) was detectable in animal models and in a human carrier with RPGR mutations (14,17–19). We performed immunostaining on the 3 × 108 vg AAV9-mRpgr-treated retina at 18 months PI (Fig. 5A) to assess whether the opsin mistrafficking could be corrected by Rpgr gene delivery. In the WT retina, M-cone opsin is found exclusively in the OS of cone cells. In the vehicle-treated Rpgr-KO retina, M-opsin was detected in IS as well as in the perinuclear and synaptic regions in addition to the OS, consistent with previous findings (14). More M-opsin was present in photoreceptor IS in the superior retina compared with the inferior retina, probably due to the superior to inferior gradient of M-opsin expression. This M-opsin transport to OS was partially restored in the vector-treated retina at 18 months PI.
Figure 5.
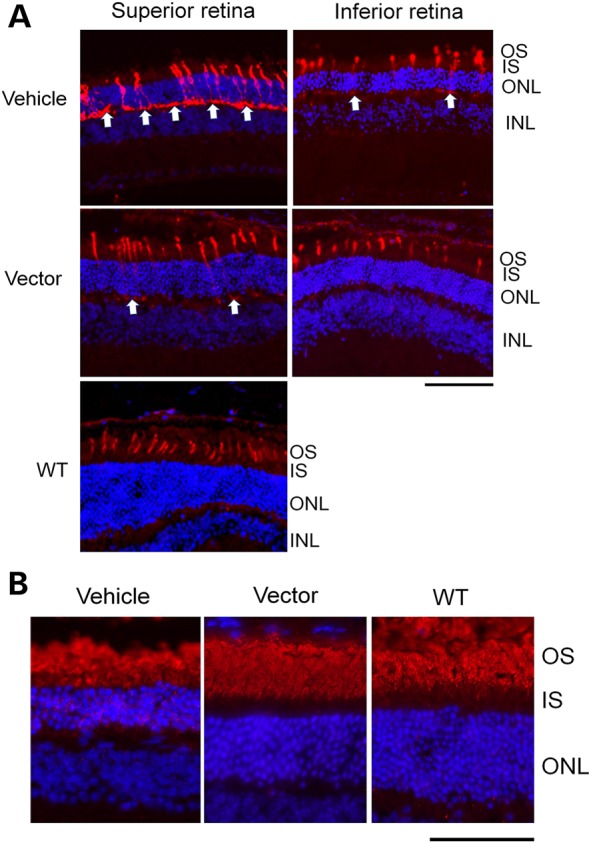
Correction of opsin mis-localization in Rpgr-KO mice treated with 3 × 108 vg AAV9-mRpgr vector. (A) M-cone opsin staining at 18 months PI. In the WT C57/Bl6 retina, M-opsin was localized to the outer segments (OS) of the cone cells. In the vehicle-treated Rpgr-KO retina, the M-opsin was additionally observed at the inner segments (IS), perinuclear and synaptic regions. This mis-localization was almost completely reversed in the inferior retina and partially reversed in the superior retina in the vector-injected eye. Arrows indicate mis-localized M-opsin at the synaptic regions. (B) Rhodopsin staining. Rhodopsin was only localized to the rod OS in WT retina but was additionally observed at the IS and perinuclei in the vehicle-injected KO retina. This mis-localization was not seen in the RPGR-expressed area in the vector-injected KO retina. Cone opsin or rhodopsin staining is shown in red, and nuclei are stained blue by DAPI. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer. Scale bar: 50 µm.
Rhodopsin is localized only to the rod OS in WT retina (Fig. 5B). In the young Rpgr-KO mouse retina, rhodopsin was appropriately localized [data not shown (14)]; however, rhodopsin immunostaining was detected in the IS and perinuclear region in 20-month-old vehicle-injected Rpgr-KO retina. Rhodopsin localization was corrected in the areas expressing RPGR-ORF15 in the vector-treated retina of Rpgr-KO mice.
Treatment effect in the Rpgr-KO mice following gene delivery of human RPGR-ORF15
As a potential vector candidate for future human trials, AAV8-hRPGR was tested for its efficacy in Rpgr-KO mice with four different doses; 3 × 109, 1 × 109, 3 × 108 and 1 × 108 vg per eye. Six- to eight-week-old mice were injected with the vector subretinally, and ERG was performed at 12 and 18 months PI. Among the four dose groups, optimal outcome was observed in 1 × 109 vg-treated group (Fig. 6). At this dose, the vector-treated eyes displayed significantly higher amplitudes for all three ERG measurements at 18 month PI (Fig. 6A), indicating the rescue of retinal function in the Rpgr-KO mouse following human RPGR-ORF15 gene delivery. All 11 mice that survived 18 months of monitoring period exhibited higher light-adapted b-wave amplitudes in vector-treated eyes; of these, 9 and 10 mice respectively displayed higher dark-adapted a-wave and dark-adapted b-wave (Fig. 6B). ERG waveforms are shown from a representative mouse at 18 months PI (Fig. 6C).
Figure 6.

Rescue of retinal function in Rpgr-KO mice that received 1 × 109 vg AAV8-hRPGR vector treatment. Mice received unilateral injections of the vector and contralateral injections of vehicle at 6–8 weeks of age. (A) ERG analysis of vector-injected and vehicle-injected eyes at 12 months and 18 months PI. Although improvement in the vector-injected eyes was not obvious at 12 months PI, significantly larger amplitudes of dark-adapted a-, b-wave and light-adapted b-wave were observed in response to increasing intensities of flash stimuli at 18 months PI. (B) Comparisons between vector-injected and vehicle-injected eye in each individual animal for ERG amplitudes elicited from the highest flash intensity (n = 11). (C) Representative ERG wave forms from a single Rpgr-KO mouse at 18 months PI. Error bars show SEM, and the significance between the vector-injected and vehicle-injected eyes was calculated using two-tailed paired t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Mice receiving 3 × 109 vg vector demonstrated much lower ERG amplitudes in the vector-treated eyes than control eyes at 18 month PI; however, this difference was not statistically significant at 12 months PI (Supplementary Material, Fig. S6) indicating the long-term toxicity at this dose. The 3 × 108 vg and 1 × 108 vg vector-treated eyes did not show difference from control eyes for ERG amplitudes (Supplementary Material, Fig. S7). To investigate whether the therapeutic effect was too small to be detected, ERG was performed again 6 months later when these mice were almost 26 months old. ERG improvement was still not observed in the vector-treated eyes in both dose groups, indicating that these two vector doses were too low to achieve functional rescue in the Rpgr-KO mice.
OCT retinal imaging was performed on the Rpgr-KO mice treated with 1 × 109 vg AAV8-hRPGR vector at 18 months PI. Much thicker ONL was observed in the vector-treated retinas than the controls (Fig. 7A), and the thickness of the whole retina in the vector-treated eyes was greater than the controls within ∼1.0 mm2 field of view (Fig. 7B). By immunofluorescence analyses, vector-treated retina revealed hRPGR expression in about half of the area of the cross section (Fig. 7C). Consistent with the OCT findings, more rows of photoreceptors were preserved in the vector-treated retina than those in the control (Fig. 7C), and measurements of ONL thickness across 4 mm of retina along the vertical (dorsal–ventral) meridian corroborated these observations (Fig. 7D). The average ONL thickness at different locations of the vector-treated retinas ranged between 21.2 and 33.4 µm, whereas vehicle-treated retinas had ONL between 14.3 and 24.1 µm. Immunofluorescence analyses of the retina receiving a lower vector dose (3 × 108 vg) revealed hRPGR staining in a much smaller area; however, more photoreceptors were preserved in this area compared with the adjacent region (Supplementary Material, Fig. S8). Therefore, preservation of photoreceptors in vector-transduced areas was still achieved in the lower-dose groups despite the lack of overall functional preservation as evaluated by full-field ERG (Supplementary Material, Fig. S7). Rhodopsin mis-localization was not detected in areas with appropriate hRPGR expression in the vector-treated retina, whereas it was apparent in the control retina (Supplementary Material, Fig. S9).
Figure 7.

Rescue of retinal structure in Rpgr-KO mice treated with 1 × 109 AAV8-hRPGR vector. Mice received unilateral injections of the vector and contralateral injections of vehicle at 6–8 weeks of age. (A and B) Representative OCT scans of vehicle-injected and vector-injected eyes of a single Rpgr-KO mouse at 18 months PI. (A) Retina sections across the ONH. Markedly thicker ONL was observed in the vector-injected eye. (B) Whole retinal thickness. The vector-injected eye displayed greater whole retina thickness within a ∼1.0 mm2 field of view centered on the ONH. (C) Immunostaining of retinal sections across the ONH with an antibody against human RPGR at 18 months PI. Remarkably more rows of photoreceptors were preserved in RPGR-expressed area in the vector-injected eye. Inset: the magnified image of the marked area. RPGR staining is shown in red, and nuclei are stained blue by DAPI. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. (D) Quantitative analysis of ONL thickness from superior–inferior retinal sections across the ONH. Two-tailed paired t-test was used for statistics analysis (n = 7). *P < 0.05. Error bars, SEM.
Rescue of retinal function and structure following Rpgr-ORF15 gene delivery to older Rpgr-KO mice
To assess whether retina with more substantial degeneration would still benefit from the treatment, 3 × 108 vg AAV8-mRPGR was subretinally injected into 1-year-old Rpgr-KO mice. No appreciable difference was observed between vector- and vehicle-treated eyes when tested at 5 months PI (data not shown). However, the ERG rescue became apparent in vector-treated eyes at 11 months PI, when mice were 23 months old (Fig. 8A). OCT imaging revealed much thicker ONL in the vector-injected retina than in the vehicle-injected control (Fig. 8B); this finding was subsequently confirmed by morphology analyses (Fig. 8C). Compared with the control retina, significantly more rows of photoreceptors were preserved in the vector-injected retina where RPGR expression was detected. The weaker RPGR signal than that observed in early-injected mice at 18 months PI (Fig. 4C) could be the result of fewer photoreceptors survived at 23 months of age following delayed vector administration; however, lower AAV transduction of the aged degenerating photoreceptors than younger cells cannot be ruled out. Nevertheless, our results suggest that the Rpgr-KO mouse could still respond favorably to Rpgr gene delivery even when treated at an advanced age with active degeneration in the retina.
Figure 8.

Rescue of retinal function and structure in Rpgr-KO mice after receiving 3 × 108 vg AAV8-mRpgr at an advanced age. Mice received unilateral injections of the vector and contralateral injections of vehicle at 1 year of age. (A) ERG analysis at 11 months PI when the mice were 23 months old (n = 8). The vector-injected eyes displayed significantly larger amplitudes of dark-adapted a-, b-wave and light-adapted b-wave than vehicle-injected eyes in response to increasing intensities of flash stimuli. Two-tailed paired t-test was used for statistics analysis. *P < 0.05; **P < 0.01. Error bars, SEM. (B) Representative OCT scans of vehicle and vector-injected eyes of a single Rpgr-KO mouse. Much thicker ONL was observed in the vector-injected eye. (C) Immunostaining of retinal sections from a treated Rpgr-KO mouse with an antibody against mouse RPGR. Substantially more rows of photoreceptors were observed in the area with RPGR expression in the vector-injected eye. RPGR staining is shown in red, and nuclei are stained blue by DAPI. IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Discussion
Gene replacement therapy is fast becoming a mature technology after two decades of development and recent human experiments, with potential to bring immediate benefits to patients with monogenic retinal dystrophies as exemplified by the success of the RPE65-LCA clinical trials (21–28). To date, over twenty gene therapy clinical trials have been completed or are ongoing for retinal diseases, with dozens of patients having received the treatment (20) (www.clinicaltrials.gov). While in RPE65-LCA, retinal pigment epithelium (RPE) cells are directly affected, photoreceptors are primarily involved in the majority of inherited retinal dystrophies, including those associated with RPGR mutations. Advances in AAV vector technology have overcome major limitations of photoreceptor gene delivery, thereby making gene therapy for retinal degenerative diseases feasible. A recent study demonstrated promising treatment for RPGR disease using AAV5 vectors delivering a full-length human RPGR-ORF15 cDNA, with prevention of retinal structural and functional loss accomplished in four dogs with RPGR deficiency (38). As ORF15 exon of the RPGR gene is prone to deletions and rearrangements owing to its purine-rich nucleotide composition, generation of a stable vector with the ability of expressing a full-length protein is crucial for clinical development. Here, we created new AAV RPGR-ORF15 vectors and validated their genome integrity. We showed that full-length RPGR-ORF15 proteins were produced by these vectors with correct subcellular localization in mouse photoreceptors. More importantly, these vectors had a remarkable therapeutic effect in a relatively large cohort of Rpgr-KO mice, validating the results of the canine study. Our study thus represents a significant step closer to the initiation of human clinical trials.
The preclinical safety and efficacy profiles of a therapeutic vector are critical when defining the dose range for a clinical trial. We evaluated the efficacy and/or toxicity of the mouse and human RPGR-ORF15 AAV vectors with 100-fold dose range (1 × 108 to 1 × 1010 vg per eye) in the Rpgr-KO mice, and the results clearly revealed a dose-dependent pattern of retinal response. The mouse Rpgr-ORF15 vectors (in both AAV8 and AAV9) demonstrated optimal treatment outcome at the dose of 3 × 108 vg/eye, whereas the dose of 1 × 109 vg/eye was optimal for the AAV8 human RPGR-ORF15 vector. At these doses, the expression of the RPGR-ORF15 proteins spanned roughly half of the retina sections (Figs. 4 and 7) at 18 months PI, resulting in remarkable preservation of retinal structure and function. The difference in optimal dose between the mouse and the human RPGR-ORF15 vector may reflect different expression levels and/or distinct interactions of the mouse or human RPGR-ORF15 protein in the KO retina. Vectors at lower doses were not able to produce rescue of global retinal function as examined by ERG (Supplementary Material, Figs S4 and S7); however, partial rescue of retinal structure was achieved where expression of RPGR protein was detected (Supplementary Material, Fig. S8). Administration of higher doses of the vectors was apparently toxic to the retina (Fig. 2 and Supplementary Material, Fig. S6), although mouse photoreceptors could reportedly tolerate ∼40-fold higher than normal level of RPGR-ORF15 protein (37). This retinal toxicity was probably caused by an even higher level of RPGR-ORF15 protein accumulated from sustained expression, a result of the high copy number of AAV vector genomes persisted in the efficiently transduced photoreceptors. In addition, high doses of vector administration could also impose excessive loads of AAV capsid protein and vector DNA into the retina, thereby exceeding the processing capacity of the retinal cells. Based on the results of dose-efficacy/toxicity, the determination of the vector doses in future human trials requires careful balancing of the risks and benefits for patients. Considering vast genetic and clinical heterogeneity of patients with RPGR mutations, a universal vector dose is unlikely to achieve satisfactory therapeutic effect to all patients. Thus, the treatment dose may have to be individualized according to patient's condition.
Several preclinical retinal gene therapy studies indicate a successful treatment outcome only when conducted prior to the onset of photoreceptor degeneration (43,44). It was believed that once the degeneration has progressed to a ‘critical’ threshold, gene replacement therapy is not likely to rescue the ‘dying’ photoreceptors. A recent RPE65-LCA clinical study further validates this assertion because the photoreceptors continued to degenerate despite observed visual improvement after treatment (45). However, early intervention is often difficult in patients with retinal neurodegeneration. In the present study, we tested whether the Rpgr-KO mice could benefit from the Rpgr-ORF15 gene delivery when the treatment was provided at 1 year of age, when a substantial portion of photoreceptors had begun to degenerate. Our results revealed that eyes receiving late vector administration still preserved significantly better retinal function and structure than the control eyes (Fig. 8), consistent with those observed in the XLPRA2 canine model (38). Future investigations may be necessary to determine an effective window of disease status for treatment design in individual patients. Regardless, our findings suggest the feasibility of beneficial gene therapy treatment even in RPGR patients with advanced stage disease.
In the present study, we did not perform experiments to evaluate whether viral DNA persisted in transduced mouse photoreceptors. It should, however, be noted that AAV vector genomes are able to persist as episomal circular monomer or concatemer in post-mitotic cells in adult animals and in humans (46–48). Although an age- and AAV serotype-matched comparison between early and late intervention was not included in our experimental design, we show that at 18 months PI (when mice are ∼20 months old), almost 50% of the photoreceptors in the retinal vertical meridian sections displayed vector-mediated RPGR expression in mice receiving early intervention (Figs. 4C and 7C). We observed relatively weak RPGR expression and less-pronounced structural and functional rescue of the retina in late intervention studies (Fig. 8C). It is difficult to ascertain whether low RPGR expression is the cause or the consequence of the less-pronounced rescue. We suggest that lower RPGR expression likely results from lesser number of fully functional photoreceptors, which are available for rescue when intervention is given at a later stage; however, lower efficiency of viral transduction in older retina cannot be ruled out. A better rescue might be achieved by vector dose optimization or by using combinatorial treatment strategies in which neuroprotection is included as well.
In contrast to the rapid disease progression in patients with RPGR mutations, retinal degeneration in the Rpgr-KO mice is relatively slow. We wondered whether the role of RPGR-ORF15 in mouse photoreceptors is distinct from that in human, and consequently whether the vector dose-efficacy/toxicity data obtained in the mouse model can be used as reference for future human trials. The connecting cilia localization of RPGR-ORF15 proteins from different species (14,15), together with opsin trafficking deficiency observed in a human carrier of RPGR mutation and in the existing murine and canine models (14,17–19,38), implicates a conserved role of RPGR-ORF15 in facilitating protein trafficking along photoreceptor cilia. As protein trafficking in photoreceptors requires numerous cilia-associated proteins and multi-protein complexes (13), interactions among ciliary proteins could be distinct in different species. It is therefore possible that depletion of RPGR in mouse has less severe impact on ciliary interactome and outer segment renewal compared with that in human. Despite slower disease progression, the Rpgr-KO mice share many common features with RPGR patients in retinal structure and function. Though the two canine models carrying RPGR mutations appear to recapitulate the human disease better than the existing mouse models (34,38), the requirement for a large number of animals for efficacy/toxicity studies makes mouse models equally valuable.
Our current vector dose-efficacy/toxicity profile obtained in the Rpgr-KO mice can be extrapolated to patients with predicted null alleles. Given the vast genetic and clinical heterogeneity in patients with RPGR mutations, it will be of interest to determine whether the vector dose responses are different in mouse models with other Rpgr mutations (e.g. missense, truncation or splicing mutations) or other genetic background (17,18,35). In particular, the naturally occurring Rd9 mouse deserves evaluation because it carries a mutation within ORF15 exon (18), mimicking a majority of patients with RPGR mutations. Additionally, since rods and cones appear to be equally affected in the Rpgr-KO mice, the vector should also be tested in models with rod- or cone-dominant phenotypes (17) to accommodate patients with different clinical manifestations.
Therapy development for RPGR disease is critical because this ‘orphan’ disease afflicts a relatively large patient population and manifests a more devastating clinical outcome. The present report represents the first comprehensive vector dose-efficacy/toxicity study for gene replacement therapy of the RPGR disease. Our studies, together with another recent report in canine models (38), offer a framework for the design of future clinical experiments in human patients. While the AAV8 human RPGR-ORF15 vector we developed warrants further testing in animal models with other RPGR mutations, initial phase I clinical trial could be planned for patients with predicted null mutations, with a starting dose cautiously determined based on our present data set, the patient's disease history and the number and nature of residual photoreceptors in the retina. We concur with the previous report on RPGR gene therapy in canine models (38) and suggest that initial human clinical experiments should focus on patients at late stages of disease but when some photoreceptors are still available for rescue.
Materials and Methods
Mouse line and husbandry
The Rpgr-KO mice (14) were maintained in NIH animal care facilities in controlled ambient illumination on a 12 h light/12 h dark cycle. Studies conform to ARVO statement for the Use of Animals in Ophthalmic and Vision Research. Animal protocols were approved by NEI Animal Care and Use Committee.
AAV vector construction and production
Strategy of assembling the full-length human and mouse RPGR-ORF15 coding sequences is illustrated in Supplementary Material, Figure S1. To construct an AAV vector plasmid carrying the human RPGR-ORF15-coding sequence, a 2.2-kb DNA fragment containing the sequence from start codon to exon ORF15 upstream of the purine-rich region (corresponding to 60 to 2208 of human RPGR-ORF15 mRNA, GenBank accession No. BK005711.1) was synthesized and sequence-verified (DNA2.0, Menlo Park, CA, USA). The fragment also contains the GCCACC Kozak sequence upstream of the start codon and is flanked by Cla I and Xho I sites at 5′ and 3′ ends, respectively. The fragment was digested with Cla I and Xho I and placed downstream of a human RK promoter (42) and a chimeric CMV/human β-globin intron (49), and upstream of a human β-globin polyadenylation site in an existing AAV shuttle plasmid maintained in the lab. The resulting plasmid was designated pV4.7-RK-hRPGR5′. The downstream part of RPGR-ORF15 exon including the purine-rich region was PCR-amplified from genomic DNA of a healthy adult male donor, using the following primers:
hRPGR F: 5′ AGTGGGAGAAGCAGAGGATGGGCCTG 3′; hRPGR R: 5′ GTTTTCTCGAGTGACTGGCCATAATCGGGTCACATTTAAGGTTTGT 3′.
PCR was performed with PrimeSTAR HS DNA Polymerase (Clontech Laboratories, Inc., Mountain View, CA, USA). The PCR conditions were 94°C for 1 min followed by 30 cycles at 98°C for 10 s and 72°C for 80 s followed by 7 min of extension at 72°C and hold at 4°C. The 1.6-kb PCR product was digested with Sap I and Xho I and was ligated with the 5.3-kb Xho I-Mfe I fragment and the 534-bp Mfe I-Sap I fragment of pV4.7-RK-hRPGR5′, to form the vector plasmid carrying the full-length hRPGR-coding sequence. This plasmid was designated pV4.7-RK-hRPGR-ORF15.
To construct an AAV vector plasmid carrying the mouse RPGR-ORF15-coding sequence, a 2.9-kb DNA fragment was synthesized, which contains the sequence from start codon to exon 14/15 (corresponding to 103 to 2668 of the mouse RPGR transcript variant 2, NCBI Reference Sequence: NM_001177951.1) and the rest of exon ORF15 upstream of the purine-rich region (corresponding to 48 843 to 49 190 of GenBank AJ318464.1). The fragment also contains the GCCACC Kozak sequence upstream of the start codon and is flanked by Cla I and Xho I sites at 5′ and 3′ ends, respectively. The sequence-verified fragment was digested with Cla I and Xho I and inserted into the same location of the AAV shuttle plasmid as the human counterpart as described earlier. The resulting plasmid was designated pV4.7-RK-mRpgr5′. The downstream part of the mouse RPGR-ORF15 exon including the purine-rich region was PCR-amplified from genomic DNA of a male C57/Bl6 mouse, using the following primers: mRpgr F: 5′ GGACGGATCTGAGGGTGACGGGGA 3′; mRpgr R: 5′ GTTTTTCTCGAGACGCATCAGGCATGGAGATGACTTCCCT 3′.
PCR conditions were the same as those for the human sequence. The 1.3-kb PCR product was digested with Sap I and Xho I and was ligated with the 6.5-kb Xho I-Sap I fragment of pV4.7-RK-mRpgr5′, to form the vector plasmid carrying the full-length mouse RPGR-ORF15-coding sequence. This plasmid was designated pV4.7-RK-mRpgr-ORF15. The purine-rich regions of both human and mouse RPGR-ORF15 exons were verified by sequencing (Sequetech, Inc., Redwood City, CA, USA). Schematic of the two vector plasmids is shown in Figure 1A. The plasmids were propagated in XL10-Gold bacterial strain (Agilent Technologies, Inc., Santa Clara, CA, USA).
AAV vectors were produced by triple-plasmid transfection to HEK293 cells, as described previously (50). The human RPGR-ORF15 AAV construct was packaged into AAV8, whereas the mouse Rpgr-ORF15 construct was packaged into both AAV8 and AAV9. The vectors were purified by polyethylene glycol precipitation followed by cesium chloride density gradient fractionation, as described earlier (50). Purified vectors were formulated in 10 mm Tris–HCl, 180 mm NaCl, pH 7.4, quantified by real-time PCR using linearized plasmid standards and stored at −80°C until use. Integrity of the vectors was examined each time after purification by amplifying the purine-rich region of the RPGR-ORF15.
Subretinal injections
AAV vectors were injected subretinally, as previously described (43) but with some modifications. Briefly, mice were anesthetized by intra-peritoneal injection of ketamine (80 mg/kg) and xylazine (8 mg/kg). Pupils were dilated with topical atropine (1%) and tropicamide (0.5%). Surgery was performed under an ophthalmic surgical microscope. A small incision was made through the cornea adjacent to the limbus using 18-gauge needle. A 33-gauge blunt needle fitted to a Hamilton syringe was inserted through the incision while avoiding the lens and pushed through the retina. All injections were made subretinally in a location within the nasal quadrant of the retina. Each animal received 1 μl of AAV vector at the concentration of 1 × 1011 to 1 × 1013 vector genomes per milliliter. Treatment vectors were given in the right eye, and control vehicle was injected in the fellow eye. Visualization during injection was aided by addition of fluorescein (100 mg /ml AK-FLUOR, Alcon, Fort Worth, TX, USA) to the vector suspensions at 0.1% by volume.
Immunoblot analysis
Mouse retinas were homogenized in RIPA lysis buffer containing 1 × proteinase inhibitor by brief sonication. The tissue debris was removed by a brief centrifugation. Retinal protein was separated on SDS–polyacrylamide gel by electrophoresis and transferred to nitrocellulose membranes. After pre-adsorption with 5% nonfat dry milk for 1 h at room temperature, the membrane blots were incubated overnight at 4°C with the primary antibody. The blots were then washed with Tris-buffered saline with the Tween® 20 (TBST: 137 mm sodium chloride, 20 mm Tris, 0.1% Tween-20, pH 7.6), incubated for 1 h at room temperature with the secondary antibody-horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Jackson Immunoresearch, West Grove, PA, USA) and developed by SuperSignal West Pico Chemiluminescent (Thermo Fisher Scientific, Inc., Rockford, IL, USA). The primary antibodies used in this study were as follows: rabbit anti-mouse RPGR-ORF15 antibody C100 and rabbit anti-human RPGR antibody 643 (T. Li, unpublished), which recognize the C-terminal of the mouse RPGR-ORF15 and a common region of human RPGR-ORF15 and RPGR1–19 isoforms, respectively. Mouse monoclonal anti-β-actin antibody (Sigma) was used for loading controls.
Tissue processing, immunofluorescence and morphometric analysis
After euthanasia, mouse eyes were harvested. A blue dye was used to mark the orientation of the eye before enucleation to ensure that immunostaining was performed on equivalent areas on vector-treated and vehicle-treated eyes. For fixation, eyes were immediately placed in 4% paraformaldehyde for 1 h. The fixed tissues were soaked in 30% sucrose/PBS overnight, quickly frozen and sectioned at a thickness of 10 µm using cryostat. A previously described alternative protocol was used to detect RPGR localization to the connecting cilia (15). Briefly, eyes were embedded in optimal cutting temperature compound without fixation and quick-frozen in liquid nitrogen. Cryosections were cut at 10 μm and collected on pretreated glass slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA, USA). Sections were stored at −80°C and used within 2 to 3 days. Just before use, sections were fixed on slides for 2 min with 1% formaldehyde in phosphate-buffered saline (PBS) at pH 7.0. If sections were stored for longer than 1 week, an additional treatment was performed in 0.1% 2-mercaptomethanol (in PBS) for 5 min, followed by 1% formaldehyde fixation for 5 min. Sections were then washed once in PBS and carried through to immunofluorescence staining.
For immunofluorescence staining, the cryosections were pre-adsorbed in 5% goat serum in PBS containing 0.1% Triton X-100 (PBST) for 1 h and then incubated overnight at 4°C with primary antibody diluted in 5% goat serum, as described (15). Sections were washed three times in PBST and incubated with fluorochrome-conjugated secondary antibodies and 0.2 µg/ml DAPI for 1 h. Sections were washed again and mounted in Fluoromount-G (SouthernBiotech, Birmingham, AL, USA). Images were captured using a fluorescence microscope Axio Imager Z1 or a confocal scanning microscope LSM700 (Zeiss, Germany).
The primary antibodies included the poly-clonal rabbit anti-human RPGR-ORF15 antibody 636 (T. Li, unpublished) and rabbit anti-mouse RPGR-ORF15 antibody S1 (12), which recognize the common region of RPGR-ORF15 and RPGR1–19 isoforms in human and mouse, respectively. Other primary antibodies used in this study include monoclonal antibody for rhodopsin (1D4, Santa Cruz Biotechnology, Dallas, TX, USA) and M-cone opsin (Millipore, Billerica, MA, USA). Secondary antibodies included goat anti-rabbit and anti-mouse antibodies conjugated with Alexa Fluor 555 and 568 (Life Technologies, Grand Island, NY, USA).
For morphometric analyses of ONL thickness, measurements were made along the vertical meridian at four locations to each side of the ONH separated by 500 μm each. Measurements began at ∼500 μm from the ONH itself.
Electroretinogram
Mice were dark-adapted overnight. Anesthesia and pupil dilation were conducted as described earlier. A computer-based system (E2, Diagnosys LLC, Lowell, MA, USA) was used for ERG recordings in response to flashes produced with LEDs or Xenon bulbs. Corneal ERGs were recorded from both eyes using gold wire loop electrodes with a drop of 2.5% hypromellose ophthalmic demulcent solution. A gold wire loop placed in the mouth was used as reference, and a ground electrode was on the tail. The ERG protocol consisted of recording dark-adapted ERGs using brief flashes of −2 to +3 log sc cd.s.m–2/flash. Responses were computer-averaged and recorded at intervals of 3 to 60 s depending upon the stimulus intensity. Light-adapted ERGs were recorded after 2 min of adaptation to a white 32 cd.m–2 rod-suppressing background. ERGs were recorded for stimulus intensities of −0.52 to +2 log sc cd.s.m–2.
Optical coherence tomography
OCT volume scan images were acquired with a spectral domain OCT system (SPECTRALIS®, Heidelberg Engineering, Carlsbad, CA, USA). Mice were anesthetized and pupils were dilated as described earlier. The ONH was centered within ∼1.0 mm2 field of view. Retinal thickness maps were generated by Heidelberg Eye Explorer software.
Statistical analysis
Two-tailed paired t-test was used to compare outcomes in vector-treated versus vehicle-treated eyes. GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis.
Supplementary Material
Funding
This work was supported by the intramural research program of the National Eye Institute.
Supplementary Material
Acknowledgements
We thank Yi-Sheng Chang, Oleg Bulgakov, Rivka Rachel, Yichao Li, Yide Mi, Jacob Nellissery and Xun Sun for assistance.
Conflict of Interest statement. None declared.
References
- 1.Hartong D.T., Berson E.L., Dryja T.P. (2006) Retinitis pigmentosa. Lancet, 368, 1795–1809. [DOI] [PubMed] [Google Scholar]
- 2.Vervoort R., Lennon A., Bird A.C., Tulloch B., Axton R., Miano M.G., Meindl A., Meitinger T., Ciccodicola A., Wright A.F. (2000) Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet., 25, 462–466. [DOI] [PubMed] [Google Scholar]
- 3.Sharon D., Sandberg M.A., Rabe V.W., Stillberger M., Dryja T.P., Berson E.L. (2003) RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet., 73, 1131–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelletier V., Jambou M., Delphin N., Zinovieva E., Stum M., Gigarel N., Dollfus H., Hamel C., Toutain A., Dufier J.L., et al. (2007) Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum. Mutat., 28, 81–91. [DOI] [PubMed] [Google Scholar]
- 5.Shu X., McDowall E., Brown A.F., Wright A.F. (2008) The human retinitis pigmentosa GTPase regulator gene variant database. Hum. Mutat., 29, 605–608. [DOI] [PubMed] [Google Scholar]
- 6.Branham K., Othman M., Brumm M., Karoukis A.J., Atmaca-Sonmez P., Yashar B.M., Schwartz S.B., Stover N.B., Trzupek K., Wheaton D., et al. (2012) Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci., 53, 8232–8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Churchill J.D., Bowne S.J., Sullivan L.S., Lewis R.A., Wheaton D.K., Birch D.G., Branham K.E., Heckenlively J.R., Daiger S.P. (2013) Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 54, 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan L.S., Bowne S.J., Reeves M.J., Blain D., Goetz K., Ndifor V., Vitez S., Wang X., Tumminia S.J., Daiger S.P. (2013) Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 54, 6255–6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowne S.J., Sullivan L.S., Koboldt D.C., Ding L., Fulton R., Abbott R.M., Sodergren E.J., Birch D.G., Wheaton D.H., Heckenlively J.R., et al. (2011) Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest. Ophthalmol. Vis. Sci., 52, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan D., Swain P.K., Breuer D., Tucker R.M., Wu W., Fujita R., Rehemtulla A., Burke D., Swaroop A. (1998) Biochemical characterization and subcellular localization of the mouse retinitis pigmentosa GTPase regulator (mRpgr). J. Bio. Chem., 273, 19656–19663. [DOI] [PubMed] [Google Scholar]
- 11.Kirschner R., Rosenberg T., Schultz-Heienbrok R., Lenzner S., Feil S., Roepman R., Cremers F.P., Ropers H.H., Berger W. (1999) RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Genet., 8, 1571–1578. [DOI] [PubMed] [Google Scholar]
- 12.Hong D.H., Li T. (2002) Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Invest. Ophthalmol. Vis. Sci., 43, 3373–3382. [PubMed] [Google Scholar]
- 13.Rachel R.A., Li T., Swaroop A. (2012) Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia, 1, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong D.H., Pawlyk B.S., Shang J., Sandberg M.A., Berson E.L., Li T. (2000) A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl Acad. Sci., 97, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong D.H., Pawlyk B., Sokolov M., Strissel K.J., Yang J., Tulloch B., Wright A.F., Arshavsky V.Y., Li T. (2003) RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest. Ophthalmol. Vis. Sci., 44, 2413–2421. [DOI] [PubMed] [Google Scholar]
- 16.Hosch J., Lorenz B., Stieger K. (2011) RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet., 32, 1–11. [DOI] [PubMed] [Google Scholar]
- 17.Brunner S., Skosyrski S., Kirschner-Schwabe R., Knobeloch K.P., Neidhardt J., Feil S., Glaus E., Luhmann U.F., Ruther K., Berger W. (2010) Cone versus rod disease in a mutant Rpgr mouse caused by different genetic backgrounds. Invest. Ophthalmol. Vis. Sci., 51, 1106–1115. [DOI] [PubMed] [Google Scholar]
- 18.Thompson D.A., Khan N.W., Othman M.I., Chang B., Jia L., Grahek G., Wu Z., Hiriyanna S., Nellissery J., Li T., et al. (2012) Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PloS One, 7, e35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamian M., Pawlyk B.S., Hong D.H., Berson E.L. (2006) Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am. J. Ophthal., 142, 515–518. [DOI] [PubMed] [Google Scholar]
- 20.Boye S.E., Boye S.L., Lewin A.S., Hauswirth W.W. (2013) A comprehensive review of retinal gene therapy. Mol. Ther., 21, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M., et al. (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Eng. J. Med., 358, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bainbridge J.W., Smith A.J., Barker S.S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G.E., Stockman A., Tyler N., et al. (2008) Effect of gene therapy on visual function in Leber's congenital amaurosis. N. Eng. J. Med., 358, 2231–2239. [DOI] [PubMed] [Google Scholar]
- 23.Cideciyan A.V., Aleman T.S., Boye S.L., Schwartz S.B., Kaushal S., Roman A.J., Pang J.J., Sumaroka A., Windsor E.A., Wilson J.M., et al. (2008) Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl Acad. Sci., 105, 15112–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonelli F., Maguire A.M., Testa F., Pierce E.A., Mingozzi F., Bennicelli J.L., Rossi S., Marshall K., Banfi S., Surace E.M., et al. (2010) Gene therapy for Leber's congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther., 18, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maguire A.M., High K.A., Auricchio A., Wright J.F., Pierce E.A., Testa F., Mingozzi F., Bennicelli J.L., Ying G.S., Rossi S., et al. (2009) Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial. Lancet, 374, 1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett J., Ashtari M., Wellman J., Marshall K.A., Cyckowski L.L., Chung D.C., McCague S., Pierce E.A., Chen Y., Bennicelli J.L., et al. (2012) AAV2 gene therapy readministration in three adults with congenital blindness. Sci. Transl. Med., 4, 120ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobson S.G., Cideciyan A.V., Ratnakaram R., Heon E., Schwartz S.B., Roman A.J., Peden M.C., Aleman T.S., Boye S.L., Sumaroka A., et al. (2012) Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthal., 130, 9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cideciyan A.V., Hauswirth W.W., Aleman T.S., Kaushal S., Schwartz S.B., Boye S.L., Windsor E.A., Conlon T.J., Sumaroka A., Pang J.J., et al. (2009) Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum. Gene Ther., 20, 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLaren R.E., Groppe M., Barnard A.R., Cottriall C.L., Tolmachova T., Seymour L., Clark K.R., During M.J., Cremers F.P., Black G.C., et al. (2014) Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet, 383, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam B.L., Feuer W.J., Schiffman J.C., Porciatti V., Vandenbroucke R., Rosa P.R., Gregori G., Guy J. (2014) Trial end points and natural history in patients with G11778A Leber hereditary optic neuropathy: preparation for gene therapy clinical trial. JAMA Ophthal., 132, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lheriteau E., Petit L., Weber M., Le Meur G., Deschamps J.Y., Libeau L., Mendes-Madeira A., Guihal C., Francois A., Guyon R., et al. (2014) Successful gene therapy in the RPGRIP1-deficient dog: a large model of cone-rod dystrophy. Mol. Ther., 22, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai C.M., Estcourt M.J., Himbeck R.P., Lee S.Y., Yew-San Yeo I., Luu C., Loh B.K., Lee M.W., Barathi A., Villano J., et al. (2012) Preclinical safety evaluation of subretinal AAV2.sFlt-1 in non-human primates. Gene Ther., 19, 999–1009. [DOI] [PubMed] [Google Scholar]
- 33.Binley K., Widdowson P., Loader J., Kelleher M., Iqball S., Ferrige G., de Belin J., Carlucci M., Angell-Manning D., Hurst F., et al. (2013) Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest. Ophthalmol. Vis. Sci., 54, 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q., Acland G.M., Wu W.X., Johnson J.L., Pearce-Kelling S., Tulloch B., Vervoort R., Wright A.F., Aguirre G.D. (2002) Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet., 11, 993–1003. [DOI] [PubMed] [Google Scholar]
- 35.Huang W.C., Wright A.F., Roman A.J., Cideciyan A.V., Manson F.D., Gewaily D.Y., Schwartz S.B., Sadigh S., Limberis M.P., Bell P., et al. (2012) RPGR-associated retinal degeneration in human X-linked RP and a murine model. Invest. Ophthalmol. Vis. Sci., 53, 5594–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong D.H., Pawlyk B.S., Adamian M., Sandberg M.A., Li T. (2005) A single, abbreviated RPGR-ORF15 variant reconstitutes RPGR function in vivo. Invest. Ophthalmol. Vis. Sci., 46, 435–441. [DOI] [PubMed] [Google Scholar]
- 37.Wright R.N., Hong D.H., Perkins B. (2011) Misexpression of the constitutive Rpgr(ex1–19) variant leads to severe photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci., 52, 5189–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beltran W.A., Cideciyan A.V., Lewin A.S., Iwabe S., Khanna H., Sumaroka A., Chiodo V.A., Fajardo D.S., Roman A.J., Deng W.T., et al. (2012) Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl Acad. Sci., 109, 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allocca M., Mussolino C., Garcia-Hoyos M., Sanges D., Iodice C., Petrillo M., Vandenberghe L.H., Wilson J.M., Marigo V., Surace E.M., et al. (2007) Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J. Virol., 81, 11372–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vandenberghe L.H., Bell P., Maguire A.M., Cearley C.N., Xiao R., Calcedo R., Wang L., Castle M.J., Maguire A.C., Grant R., et al. (2011) Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci. Transl. Med., 3, 88ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandenberghe L.H., Bell P., Maguire A.M., Xiao R., Hopkins T.B., Grant R., Bennett J., Wilson J.M. (2013) AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS One, 8, e53463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khani S.C., Pawlyk B.S., Bulgakov O.V., Kasperek E., Young J.E., Adamian M., Sun X., Smith A.J., Ali R.R., Li T. (2007) AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest. Ophthalmol. Vis. Sci., 48, 3954–3961. [DOI] [PubMed] [Google Scholar]
- 43.Sun X., Pawlyk B., Xu X., Liu X., Bulgakov O.V., Adamian M., Sandberg M.A., Khani S.C., Tan M.H., Smith A.J., et al. (2010) Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther., 17, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan M.H., Smith A.J., Pawlyk B., Xu X., Liu X., Bainbridge J.B., Basche M., McIntosh J., Tran H.V., Nathwani A., et al. (2009) Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum. Mol. Genet., 18, 2099–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cideciyan A.V., Jacobson S.G., Beltran W.A., Sumaroka A., Swider M., Iwabe S., Roman A.J., Olivares M.B., Schwartz S.B., Komaromy A.M., et al. (2013) Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl Acad. Sci., 110, E517–E525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnepp B.C., Jensen R.L., Chen C.L., Johnson P.R., Clark K.R. (2005) Characterization of adeno-associated virus genomes isolated from human tissues. J. Virol., 79, 14793–14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimm D., Pandey K., Nakai H., Storm T.A., Kay M.A. (2006) Liver transduction with recombinant adeno-associated virus is primarily restricted by capsid serotype not vector genotype. J. Virol., 80, 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penaud-Budloo M., Le Guiner C., Nowrouzi A., Toromanoff A., Chérel Y., Chenuaud P., Schmidt M., von Kalle C., Rolling F., Moullier P., Snyder R.O. (2008) Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol., 82, 7875–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park T.K., Wu Z., Kjellstrom S., Zeng Y., Bush R.A., Sieving P.A., Colosi P. (2009) Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther., 16, 916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grimm D., Zhou S., Nakai H., Thomas C.E., Storm T.A., Fuess S., Matsushita T., Allen J., Surosky R., Lochrie M., et al. (2003) Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood, 102, 2412–2419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.