

Abstract
Recent studies identified a previously uncharacterized gene C5ORF42 (JBTS17) as a major cause of Joubert syndrome (JBTS), a ciliopathy associated with cerebellar abnormalities and other birth defects. Here we report the first Jbts17 mutant mouse model, Heart Under Glass (Hug), recovered from a forward genetic screen. Exome sequencing identified Hug as a S235P missense mutation in the mouse homolog of JBTS17 (2410089e03rik). Hug mutants exhibit multiple birth defects typical of ciliopathies, including skeletal dysplasia, polydactyly, craniofacial anomalies, kidney cysts and eye defects. Some Hug mutants exhibit congenital heart defects ranging from mild pulmonary stenosis to severe pulmonary atresia. Immunostaining showed JBTS17 is localized in the cilia transition zone. Fibroblasts from Hug mutant mice and a JBTS patient with a JBTS17 mutation showed ciliogenesis defects. Significantly, Hug mutant fibroblasts showed loss of not only JBTS17, but also NPHP1 and CEP290 from the cilia transition zone. Hug mutants exhibited reduced ciliation in the cerebellum. This was associated with reduction in cerebellar foliation. Using a fibroblast wound-healing assay, we showed Hug mutant cells cannot establish cell polarity required for directional cell migration. However, stereocilia patterning was grossly normal in the cochlea, indicating planar cell polarity is not markedly affected. Overall, we showed the JBTS pathophysiology is replicated in the Hug mutant mice harboring a Jbts17 mutation. Our findings demonstrate JBTS17 is a cilia transition zone component that acts upstream of other Joubert syndrome associated transition zone proteins NPHP1 and CEP290, indicating its importance in the pathogenesis of Joubert syndrome.
Introduction
Cilia are extracellular projection(s) comprising microtubules encapsulated by a ciliary membrane. Cilia are known to play important roles in signaling, including mechanosensory transduction, chemosensory reception as well as mediating signaling transduction, such as in mediating sonic hedgehog (Shh) signaling (1). Ciliogenesis is initiated with formation of a basal body template derived from the mother centriole and entails recruitment of proteins for cilia assembly regulated by vesicular trafficking, BBSome interactions and the intraflagellar transport proteins (2). The further regulation of protein trafficking in and out of the cilium is mediated by a specialized structure at the base of the cilium known as the cilia transition zone (1,3). Recent studies have shown many of the mutations causing human diseases known as ciliopathies involve genes encoding proteins in the cilia transition zone.
Ciliopathies are human diseases arising from mutations that disrupt ciliogenesis or perturb cilia structure or function. Ciliopathies can cause a wide spectrum of structural birth defects that can include eye defects, craniofacial and brain malformations, limb polydactyly, kidney cysts, heart defects and abnormal left–right patterning (4). These defects are observed in various combinations in different human diseases known as Meckel–Gruber syndrome (MKS), nephronophthisis (NPHP), Bardet–Biedl syndrome (BBS), Joubert syndrome (JS), Jeune syndrome, short-rib polydactyly, Senior-Løken syndrome, Leber congenital amaurosis, oral–facial–digital syndrome (OFD) and others (5,6). Genetic studies have indicated these are largely recessive disorders, and over the last decade, rapid progress has been made in elucidating the underlying genetic etiology for many of these ciliopathies. Such studies have identified pathogenic mutations in genes encoding cilia protein components.
Joubert Syndrome is a ciliopathy associated with a characteristic ‘molar tooth sign’ on brain imaging. This reflects a complex malformation of the midbrain–hindbrain junction and is associated with developmental malformation of the cerebellum vermis (7,8). Joubert syndrome patients, like those of other ciliopathies, also can have a host of other heterogeneous phenotypes that can include eye, cardiac and renal abnormalities (cystic renal disease), as well as craniofacial defects, skeletal dysplasia and limb polydactyly (9). Mutations in 21 genes have been found to cause Joubert syndrome and most are known to encode cilia proteins localized in the cilia transition zone or basal body. These genetic findings suggest defects in cilia assembly may drive the pathophysiology of Joubert syndrome (10–12).
A recent study in Canada reported mutations in C5ORF42 as a common cause for Joubert syndrome (OMIM: #614615) (13), including the very first family described to have Joubert Syndrome in 1969 (14,15). Previous proteomic analysis by mass spectrometry has shown that C5ORF42 is associated with the NPHP1-4-8 complex in the cilia transition zone (16). In this study, we report the first mutant mouse model of Joubert syndrome with a mutation in the mouse homolog of C5ORF42 (JBTS17), 2410089e03rik. This mouse model recapitulates all of the phenotypes observed in Joubert syndrome patients, including defects in the cerebellum. Using this mouse model, we showed JBTS17 is a cilia transition zone protein, and demonstrated the Hug mutation caused disruption of the cilia transition zone, resulting in the perturbation of ciliogenesis and cilia transduced Shh signaling that underlies the cerebellar defects in Joubert Syndrome.
Results
We recovered a mutant named Hug from a large-scale mouse forward genetic screen with ethylnitrosourea (ENU) mutagenesis. The screen was conducted using inbred C57BL/6J (B6) mice using non-invasive fetal ultrasound biomicroscopy (UBM) to interrogate for recessive mutations causing congenital heart disease (CHD). With fetal ultrasound screening, mutants that died prenatally could be recovered, as was the case with Hug. Hug mutants were diagnosed with CHD and multiple other anomalies that caused prenatal lethality. Some of the birth defects detected by ultrasound imaging included polydactyly and cleft palate (Fig. 1A versus B and C versus D), and these were also further analyzed by necropsy and/or histopathology examination. Ultrasound scanning with color flow Doppler imaging at E14.5 showed Hug mutants with a pattern of blood flow from the heart indicating a single outflow stream from the aorta (Ao in Fig. 1E). This was associated with regurgitation (Fig. 1F) and flow pattern indicating a ventricular septal defect (VSD in Fig. 1E and F). Some mutants also exhibited regurgitation of blood from the ventricle to the atrium, indicating an atrioventricular septal defect (AVSD, Fig. 1G, arrowhead). Subsequent histopathology examination with episcopic confocal microscopy (ECM) showed that compared with littermate controls (Fig. 1H–J), Hug mutants exhibited CHD including pulmonary atresia with a VSD (Fig. 1K), AVSD (Fig. 1L) and hypoplastic pulmonary artery (Fig. 1M).
Figure 1.

Fetal ultrasound imaging and episcopic confocal microscopy phenotyping of cardiovascular anomalies in Hug mutants. (A and B) Color flow imaging of ultrasound biomicroscopy (UBM) showed a single out flow tract (A, aorta) with regurgitation (B) and ventricular septal defect (A and B, VSD, arrowhead) suggesting pulmonary atresia with VSD. (C) Color flow imaging of UBM exhibited a regurgitation (Reg, arrowhead) of common atrioventricular septal defect (AVSD, arrowhead) indicating AVSD. (D and E) 2D imaging of UBM showed a limb of a normal mouse fetus with five digits (asterisks) (D). In contrast, (E) showed a limb of a mutant fetus with eight digits (asterisks) suggesting polydactyly. (F and G) 2D imaging of UBM displayed intact palate (arrow) of a normal mouse fetus (F), whereas, a cleft palate (arrowhead) was visualized by UBM for a mutant fetus in (G). (H–M) Imaging with episcopic confocal microscopy showed homozygous (m/m) Hug mutants had structural heart defects. Images from a wild-type heart are shown in (H–J). In (K–M) are images from a Hug mutant, showing double outlet right ventricle with the aorta positioned above the right ventricle (Ao in K), an AVSD in (L) and pulmonary stenosis (M). AVSD, atrioventricular septal defects; LA, left atrium; RA, right atrium; RV, right ventricle; LV, left ventricle; PA, pulmonary artery.
A spectrum of structural birth defect phenotypes
Necropsy examination of Hug mutant embryos showed a wide spectrum of phenotypes involving rib-cage abnormalities (Table 1), with the most severely affected mutants dying at mid-gestation with a transparent chest wall due to complete failure to form the rib cage (Fig. 2A). In such fetuses, the heart can be directly observed through the chest wall, as if viewing it under glass, hence the name, Heart under glass or Hug (Fig. 2A). Hug mutants surviving to late gestation show growth restriction (Fig. 2B) and while the rib cage is present in older surviving mutants, the sternovertebrae may be unfused along the midline, or are malaligned in a zipperlike pattern (Fig. 2H versus G), the latter being one of the most common rib-cage phenotype observed (Table 1).
Table 1.
Prevalence of congenital heart defects and rib cage anomalies
| Phenotypes | Hug mutants (n = 29), % |
|---|---|
| Congenital heart defects | |
| Double outlet right ventricle | 62 |
| Persistant truncus arteriosis | 34 |
| Pulmonary atresia/Stenosis | 51 |
| AVSDs | 34 |
| VSDs | 65 |
| Muscular ventricular septal defects | 7 |
| Rib cage | |
| Absent | 14 |
| Unfused sternum | 31 |
| Malaligned sternovertebrae | 52 |
| Normal | 3 |
Figure 2.

Hug mutants exhibit broad spectrum of structural birth defects. (A) The rib cage failed to form in this E14.5 Hug mutant, allowing the heart to be seen through the translucent chest wall. Note severe hydrops (see white arrow in A) and also craniofacial dysmorphology. (B) A normal mouse at P0 is pictured on the left while mutant mouse which is runted with polydactyly and micrognathia is seen on the right. (C and D) Necropsy examination of two Hug mutants, showed one stillborn (D) pup exhibited a single outflow tract (see arrowhead in D) and only a short segment of the small intestine (C in D) with no colon, while the other (C) mutant was born alive, exhibiting no evidence of cardiovascular anomalies. Ao, aorta; H, heart; K, kidney; Lg, lung; L, liver; PA, pulmonary artery; S, stomach. (E) The heart exhibits side-by-side positioning of the aorta (Ao) and pulmonary artery (PA), indicating possible overriding aorta or double outlet right ventricle (arrowheads in E). (F) An E17.5 Hug mutant exhibit abnormal right sided aortic arch (RAA) with a single outflow tract (white arrowhead), which was shown by histopathology to be pulmonary atresia (data not shown). The superior vena cava (SVC), inferior vena cava (IVC) and atria are blood congested. (G and H) Malalignment of sternovertebrae is seen in almost all mutants (H). Normal sternovertebrae are seen in G. (I) Mutants typically exhibit central polydactyly that is sometimes associated with preaxial digit duplication or syndactyly. (J–O) Skeletal staining of newborn wild-type (J, L and M) and Hugm/m (K, N and O) embryos reveals micrognathia (K), shorter diaphysis in the long bones (N) compared with control (J) and polydactyly (N). Hug mutants also show congenital vertebral anomalies like hemivertebrae seen in (O).
Necropsy examination showed congenital heart defect consistent with the fetal ultrasound and histopathology findings. They ranged from mutants with completely normal cardiac anatomy (Fig. 2C; data not shown) to the most severe phenotype comprising pulmonary atresia and persistent truncus arteriosus (Fig. 2D) and milder phenotypes in between comprising varying degree of pulmonary hypoplasia (Fig. 2E and F) (Table 1). Also observed were AVSDs and VSDs (Table 1). While there was no evidence of laterality defects, some Hug mutants exhibited intestinal atresia with only a short segment of the small intestine formed, but no colon (Fig. 2D). Central polydactyly was observed, sometimes accompanied by preaxial digit duplication (Fig. 2I).
Analysis of skeletal preparations showed hypoplasia of the mandible, shortening of the long bones in the limbs and butterfly vertebrae (Fig. 2J–O). Craniofacial anomalies are also observed with cleft lip, and cleft palate (Fig. 3B versus A and D versus C). In some mutants, bilateral clefting (Fig. 3D) was observed, a reflection of the failure in fusion between the maxillary and median nasal process. 3D histopathological reconstructions showed the tongue was abnormal in shape and position (Fig. 3F versus E, H versus G and J versus I). The nasal turbinates are deformed with an abnormal branching pattern (white arrow in Fig. 3L).
Figure 3.

Hug mutants show facial clefts and craniofacial abnormalities. (A–D) Newborn Hug mutants show facial clefting (B) and cleft palate (D) when compared with normal controls (A,C). (E–L) 3D reconstruction of the heads of wild-type (E,I,G,K) and Hug mutant (F,J,H,L) mice generated from 2D serial image stacks of the specimens obtained by ECM imaging. The nasal turbinate in the mutants (F and L) show more complex pattern of branching when compared with that of wild-type littermate (E and K). Cleft palate is observed in the mutant (F,H and J), when compared with the complete closure of the palatal shelves in the wild-type littermate (E,G and I). The nasal septum and tongue are also abnormally formed in the mutant (J and L) when compared with the wild-type littermate (I and K).
Hug mutants also exhibited duplex kidneys (Supplementary Material, Fig. S1B and D). Hematoxylin and eosin staining (H&E) showed numerous small cysts (Supplementary Material, Fig. S1D). Immunofluorescent staining showed tubule dilations in the nephrons (Supplementary Material, Fig. S1D and H). Staining of the cilia with acetylated tubulin indicated possibly fewer and shorter cilia in the kidney tubules from the Hug mutant (Supplementary Material, Fig. S1F versus. E), suggesting abnormal kidney development associated with a ciliogenesis defect.
Hug mutation recovery
To recover the pathogenic mutation in the Hug mutant, we conducted whole exome sequencing analysis at 50× coverage using DNA from one homozygous mutant (Supplementary Material, Table S2). By comparing the sequencing data obtained versus the C57BL/6J reference genome, three homozygous coding mutations were identified (Supplementary Material, Table S3), one being a missense mutation in 2410089e03rik, the C5ORF42 homolog, a gene also known as JBTS17 and recently identified to cause Joubert syndrome (Fig. 4). Genotyping of multiple Hug mutants showed this gene was the only one consistently homozygous in all of the mutant animals recovered from our screen, indicating it is the pathogenic mutation. This was additionally confirmed with the mapping of the mutation using traditional genome scanning with outcrossing of the Hug mutation into C57BL/10J (B10) mice. The hybrid animals generated were used to generate mutants for mapping analysis using B6/B10 polymorphic DNA markers (Supplementary Material, Table S1). This mapped the mutation to a 4 Mb region of chromosome 15, an interval that contained the T757C missense mutation in 2410089e03rik (Supplementary Material, Fig. S2). The Hug S253P missense mutation is expected to be damaging, as it is a highly conserved residue situated in a highly conserved region of the protein (Fig. 4B).
Figure 4.

Recovery and analysis of Hug mutation in 2410089e03rik gene. (A) Schematic of the mouse 2410089e03rik gene, containing 52 exons, with the mutation in the Hug mutant indicated above the gene. Previously reported mutations in the human homolog of 2410089e03rik are indicated below the gene. The two mutations outlined in boxes are biallelic mutations in a Joubert syndrome patient from whom fibroblasts were obtained for cilia analysis. (B) Multiple alignment of 2410089e03rik homologs from different species showing high degree of sequence conservation in the mutant residue.
Hug mutation causes ciliogenesis and cilia transition zone defects
To determine whether the Jbts17Hug mutation may affect ciliogenesis, mouse embryonic fibroblasts (MEFs) were generated from Hug mutant and wild-type littermate control embryos. Primary cilia formation after serum starvation was then assayed (Fig. 5A and B). This analysis showed a marked reduction in ciliation in the mutant MEFs, suggesting Jbts17 plays an important role in ciliogenesis (Fig. 5C). Similar analysis of fibroblast cells from a Joubert syndrome patient with compound heterozygous JBTS17 mutations [c.4006C>T/p.Arg1336Trp and c.7400+1G>A; individual II-1 from family 480 in (13)] showed no cilia, when compared with ∼90% ciliation in normal human fibroblast cells obtained from a healthy control subject (Fig. 5U and V).
Figure 5.

Ciliogenesis defects in Hug mutants. (A–C) Cilia immunostaining were carried out in wild-type (A) and mutant (B) MEFs using acetylated-α-tubulin, (ac-tub, red) and γ- tubulin, (green) antibodies. MEFs were obtained from two different mutant embryos (MUT1 and MUT2). Scoring for cilia showed fewer ciliated cells in Hug mutant MEFs when compared with wild-type control (C, asterisk denotes P < 0.0003 determined by t-test). Further analysis showed the percentage of cilia with JBTS17 immunolocalization as significantly reduced in the mutant versus control MEFs (P < 0.0001, χ2 test). (D) Analysis of ciliated wild-type (n = 100) versus ciliated Hug mutant (n = 100) MEFs showed significant decrease in JBTS17 localization in cilia in the Hug mutant MEFs (P-value determined by χ2 test). (E–H) Immunostaining of control IMCD3 cells with antibodies to acetylated tubulin (E), γ-tubulin (F) and a monoclonal antibody raised against JBTS17 (G). Merged image (H) suggests JBTS localization in the cilia transition zone (TZ). (I–L) JBTS17 is localized to the TZ of control MEFs as shown by immunostaining for TZ protein NPHP1 (I), acetylated tubulin (J), JBTS17 (K) and merged image (L). (M and N) IFT88 was normally localized to the cilia in both control and ciliated mutant MEFs. (O and P). Control and Hug mutant MEFs immunostained for IFT88 (green) and JBTS17 (red). Mutant MEFs do not have JBTS17 at the base of cilia. This is quantified as shown in (D). (Q–T) Transition zone proteins NPHP1 and CEP290 are present in the cilia of control MEFs (Q and S), but absent in the mutant (R and T), respectively. (U–Z) Fibroblasts obtained from Joubert syndrome patient were immunostained for various ciliary proteins. While IFT88, NPHP1 and CEP290 are present in cilia from fibroblast from healthy control subject (U, W and Y), the Joubert Syndrome patient fibroblasts failed to form cilia. Instead, IFT88 and CEP290 localization was observed at the centrosome, but NPHP1 was not detectable (V, X and Z). Scale bar is 10 µm. Insets are ×2 enlargements.
To examine the distribution of JBTS17, a monoclonal antibody against mouse JBTS17 was developed commercially and used for confocal imaging analysis. In IMCD3 cells, JBTS17 localization was observed in a region distal to the gamma-tubulin-stained basal body, the cilia transition zone along with nuclear staining (Fig. 5E–H). We specifically looked for JBTS17 localization in the transition zone by double immunostaining of JBTS17 along with antibody to NPHP1, a cilia transition zone protein. This confirmed the colocalization of JBTS17 with NPHP1 (Fig 5I–L). This same antibody was unable to detect the JBTS17 protein on western blots. Examination of Hug mutant MEFs with cilia using IFT88 antibody showed a marked reduction in cilia localization of JBTS17 (Fig. 5O and P). Quantification showed only 34% of the cilia in Hug mutant MEFs had JBTS17 when compared with 96% in wild-type MEFs (P < 0.0001) (Fig. 5D). Immunostaining for transition zone proteins NPHP1 and CEP290 revealed both are reduced or absent from the cilia transition zone in the Hug mutant MEFs (Fig. 5R and T versus Q and S). In Hug mutant MEFs without cilia, JBTS17 localization was not observed in the centrosome. Similar analysis of the JBTS patient fibroblast cells (Fig. 5U–Z) showed no cilia, although IFT88 and Cep290 remain localized to the centrosome. Together these observations suggest JBTS17 plays an important role in ciliogenesis.
Cerebellar defects in Hug mutants
Midbrain and hindbrain abnormalities are hallmarks of Joubert syndrome, especially cerebellar vermis hypoplasia. We used 3D reconstruction with ECM histopathology to analyze the newborn Hug mutant brain (Fig. 6). This analysis showed the Hug mutant brains (Fig. 6B) are smaller (a reflection of the growth restriction), but grossly normal. However, defect in the cerebellum was noted with a reduction in the number of fissures in the developing cerebellum (Fig. 6E,F versus C,D) (17). This was shown to be statistically significant (P < 0.02, Fig. 6I). As previous studies have shown primary cilia in the cerebellum are required for mediating Shh signaling to promote proliferation of granule cell progenitors (GCPs) in the cerebellum (11), we carried out immunostaining to examine the degree of ciliation in the cerebellum. Using an antibody to ACIII to label the ciliary axoneme and γ-tubulin antibody to visualize the basal body, we examined the distribution of cilia in sections of the brain obtained from near term Hug mutant and littermate control embryos. This analysis showed a marked reduction in primary cilia in the region containing the presumptive GCPs in the Hug mutant cerebellum (Fig. 6G and H), with only 20% ciliation seen in the mutant versus 65% in the wild-type cerebellum (Fig. 6J; P = 0.0009).
Figure 6.

Analysis of cerebellar and ciliogenesis defects in Hug mutant brain and defective Shh signaling in mutant Hug cerebellum and MEFs. (A–F) Sagittal sections of wild-type (A) and mutant (B) brains showing abnormal cerebellum (Cb) and Choroid plexus (Cpx). Cerebellum from mutant brains (E and F) when compared with the wild-type control (C and D) showed decreased number of cerebellar fissures (red asterisk in E) compared with control (red asterisks in C). C and E are ECM images, and D and F are the same sample after staining with cresyl violet. (G and H) Wild-type (G) and Hug mutant (H) brain sections stained with ACIII (green), gamma-tubulin (red) and DAPI (blue). Significantly less ciliated cells are seen in the mutant (H) when compared with the wild-type control (G). (I) Quantitative analysis revealed significant difference in the number of cerebellar fissures in the Hug mutant versus wild-type cerebellum (based on analysis of four mutant embryos and three control embryos). (J) Quantitative analysis showed a significant reduction in ciliation in the cerebellum of Hug mutants (based on analysis of 100 cells from the mutant versus 100 cells from wild-type littermate control). (K and L) Quantitative PCR analysis showed significant increase in Ptch1 and Gli1 transcript expression in wild-type MEFs after treatment with the Shh agonist SAG, but Hug mutant MEFs failed to respond, showing only low level increase in Patch1 and Gli1 expression after SAG treatment (P < 0.05, t-test).
Shh signaling defect in Hug mutants
To further assess the effect of the Jbts17Hug mutation on Shh signaling, we assayed the responsiveness of the Hug mutant MEFs to serum starvation and stimulation with SAG, a Shh agonist. Real-time PCR quantitation showed only a low level of increase in Ptch1 and Gli1 expression, indicating the perturbation of Shh signaling (Fig. 6K and L). We also examined Shh-dependent dorsoventral patterning of the neural tube with analysis of Islet1 and Nkx6.1 expression in the neural tube, but observed no change in the Hug mutant embryos (Supplementary Material, Fig. S4).
Wound closure assay shows defect in orienting cell polarity
Given defects in planar cell polarity (PCP) and PCP signaling are known to contribute to some of the phenotypes associated with ciliopathies, such as abnormalities in hair cell patterning in the cochlea, we examined the Hug mutant cochlea. We observed Hug mutant cochlea exhibited the normal stereotypical patterning of stereocilia and kinocilia as in wild-type cochlea (Supplementary Material, Fig. S5). Using the Hug mutants MEFs, we also conducted a wound scratch assay to assess whether Jbts17Hug may affect cell polarity and polarized cell migration as observed with other cilia transition mutations (18). Normally, a wound gap created in a monolayer of wild-type MEFs will cause cells to reorient their polarity to allow directional cell migration towards the opposing wound edge for rapid closure of the wound gap. This reorienting of cell polarity can be observed by immunostaining for Golga2 to delineate the Golgi, which is normally situated at the cell's leading edge and directed towards the direction of cell migration (Fig. 7). Our analysis showed in Hug mutant MEFs, cell orientation was randomized and not aligned with the direction of wound closure (Fig. 7B and C). Binning the direction of Golgi orientation relative to the direction of wound closure showed a significant increase in the proportion of mutant MEFs oriented 60°–120° (χ2 P-value = 0.017) and 120°–180° (χ2 P-value = 0.003) (Fig. 7B and C) from the direction of wound closure when compared with that observed in wild-type MEFs (Fig. 7A). These results show the Jbtshug mutation disrupted the ability of cells to establish cell polarity required for directional cell migration.
Figure 7.
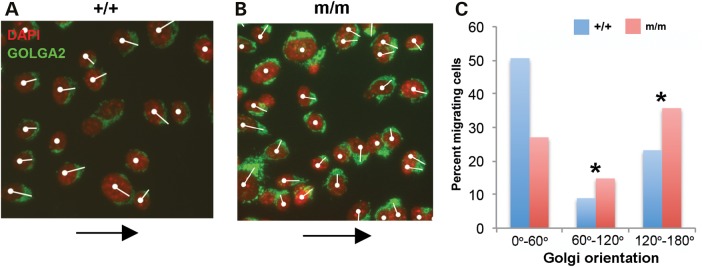
Wound scratch assay showing planar cell polarity defects in Hug mutants. (A and B) Wound scratch assay in control and mutant MEFs where cell migration towards the wound (black arrow) is indicated by Golgi orientation, which is shown by a white line drawn from the cell nucleus through the center of the Golgi. (A) Control MEFs were well aligned with the direction of wound closure (white arrow) compared with disorganized distribution shown by the mutant MEFs (B). Quantitative analysis of the Golgi orientation is shown in (C). χ2 test was used to test if the GOLGA2 orientation was similar among the three groups and parallel to the direction of migration.
Discussion
In this study, we report the first JBTS17 mouse model of Joubert syndrome. This mutant, Hug, recovered from a prenatal fetal ultrasound screen of ENU mutagenized mice, harbored a S263P missense mutation in Jbts17, a predicted pathogenic mutation situated in a highly conserved region at the N terminus of the protein. Joubert syndrome patients with mutations in JBTS17 have been identified in two different populations (10,13,19), and is now thought to account for a large fraction of Joubert syndrome cases. The human JBTS17 mutations are mostly loss of function, and none are in the N-terminus of the protein. Perhaps mutations in the N-terminus may have more deleterious effects incompatible with term survival and thus not represented in the clinical population.
Hug mutants die prenatally with multiple developmental defects consistent with the spectrum of MKS–BBS–Joubert syndrome phenotypes. This included not only skeletal dysplasia, but also craniofacial defect, polydactyly, cystic kidney and cerebellar hypoplasia. In some mutants, congenital heart defects of varying degrees of severity were observed. We note cardiac defects are not commonly seen in Joubert syndrome patients, but this could reflect ascertainment bias, with human conceptuses with deleterious CHD phenotypes not clinically represented. In fact, the constellation of structural birth defect phenotypes observed in Hug mutants is collectively of the more severe spectrum, consistent with its prenatal lethality. In the most severely affected mutants, we observed agenesis of the rib cage. Such mutant embryos typically died by E12.5–13.5. Hug mutants surviving to later gestation often show malformed rib cage with malaligned sternovertebrae, consistent with the asphyxiating thoracic dystrophy (JATD) phenotype observed in some Joubert syndrome patients (20).
JBTS17 is a protein previously identified as a component of the NPHP1-4-8 multiprotein complex in the cilia transition zone (16). Proteomic studies have shown many proteins responsible for the pathophysiology of Joubert syndrome, Meckel syndrome and nephronophthisis are colocalized in large multiprotein complexes associated with the basal body and transition zone (12,16). Using antibody staining, we showed JBTS17 is indeed colocalized with NPHP1 in the cilia transition. In the Hug mutant MEFs, there was marked reduction in ciliation. In cilia formed in the Hug mutant MEFs, not only was JBTS17 localization lost, but NPHP1 and CEP290, two other Joubert syndrome associated transition zone proteins, were also absent. We also observed a profound ciliogenesis defect in fibroblast cells derived from a Joubert syndrome patient harboring JBTS17 mutations. This was associated with the complete absence of NPHP1 immunostaining. Together these findings suggest JBTS17 acts upstream of NPHP1 in the assembly of the NPHP1-4-8 complex in the cilia transition zone.
We note analysis of mutant mouse models of other cilia proteins have suggested hierarchical interactions of the cilia transition zone components. Thus the Hug mutant phenotypes, including ciliogenesis and Shh defects, share similarities to other mouse models with mutations in genes that code for transition zone proteins that form complexes, such as Cep290 (JBTS module) Tmem67 (MKS/MKSR module), Rpgrip1l (NPHP module), Tctn1 and Tctn3 (Tectonic module) (21–31). The Hug mutant mouse model further emphasizes the important role of hierarchical interaction of protein complexes in the cilia transition zone in the pathology of ciliopathies. Such protein complexes and their hierarchical interactions may work in concert to mediate the sorting of proteins involved in cilia assembly and signaling (32).
The cerebellar defects observed in Hug mutants are consistent with the cerebellar vermis hypoplasia that is a hallmark of Joubert syndrome, a phenotype also observed in Joubert syndrome mouse models with mutations in other cilia transition proteins such as Ahi, Tctn1 and Tctn3 (29–31). Hug mutants exhibited a ciliogenesis defect that would suggest Shh signaling is disrupted in the GCPs in the cerebellum. These cells will give rise to the external granule cell layer of the cerebellum and their proliferation and expansion are known to be Shh dependent. We also observed Hug mutant MEFs responded poorly to SAG stimulation, indicating Jbts17 is required for efficient Shh signaling. The perturbation of Shh signaling also likely accounts for the limb polydactyly phenotype. However, we noted Hug mutants have normal dorsoventral patterning of the neural tube, a Shh-dependent process. This is similar to findings in mice deficient for Tmem67, another Joubert syndrome associated cilia transition protein (24). Together these findings suggest the disruption of Shh signaling in Hug mutants may be tissue specific and is not a global phenomenon.
Our analysis for possible PCP defect by examining stereocilia patterning in the cochlea did not uncover any obvious PCP defect, but more subtle PCP-related defects cannot be excluded. In the bpck Tmem67 mouse mutant, very subtle stereocilia patterning defects were observed, but interestingly, this occurred without disruption of PCP signaling (33). Analysis of Hug mutant MEFs in a wound scratch assay indicated JBTS17 is required for establishing cell polarity. Thus in Hug mutant MEFs, the Golgi failed to reorient to the cell's leading edge, which is required to drive directional cell migration. Golgi orientation is specified by the microtubule organizing center or centrosome, which also templates cilia formation. The inability to properly reorient the Golgi to establish cell polarity may underlie some of the hedgehog-independent phenotypes, shortened long bones or malaligned sternovertebrae. Defect in cell polarity also has been observed in association with another cilia transition zone protein, Wdpcp (18). These findings suggest further studies are needed to investigate how cilia transition zone components regulate ciliogenesis, and their role in the specification of cell polarity required for normal development.
In summary, we showed C5ORF42, now known as JBTS17, is localized to the cilia transition zone. Analysis of the Hug mutant mouse model and patients with JBTS17 mutations showed this gene is required for assembly of the cilia transition zone complex upstream of other ciliopathy-associated transition zone components, CEP290 and NPHP1. This suggests JBTS17 may play a major role in the pathophysiology of Joubert syndrome and perhaps other ciliopathies. We note mutation in JBTS17 has recently been identified as a major cause of OFD syndrome type VI in nine families. In yet another study, compound heterozygous JBTS17 mutations were associated with cerebellar or vermis atrophy (34,35). The Hug mutant mouse model will be invaluable for future studies to investigate the mechanism by which this transition protein contributes to pathogenesis of Joubert syndrome and other ciliopathies.
Materials and Methods
Institutional approval for animal studies
All mouse experiments were carried out using protocols approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. The experiments involving fibroblasts from a patient with Joubert syndrome were approved by the ethics committee of the Sainte-Justine Hospital Research Center, and informed consent was obtained from his legal guardian.
Necropsy and skeletal staining
Pregnant heterozygous Hug females mated with heterozygous Hug males were sedated with isoflurane and ultrasound-scanned as mentioned in (36). Fetuses were collected in 10% formalin followed by necropsy to determine heart defects. After initial necropsy, fetuses were subjected to episcopic fluorescence image capture (EFIC) and 3D images were generated for the heart and the brain as described in (36). Skeletal staining was performed as previously described (37). Briefly, newborn mutants were fixed in 10% formalin followed by evisceration and dehydration in ethanol. After incubation in acetone, staining was carried out using 0.15% Alcian Blue, 0.05% Alizarin Red and 5% glacial acetic acid in ethanol. This was followed by multiple changes of water and clearing in 20% glycerol and 1%KOH until the skeleton was clearly visible.
Episcopic confocal microscopy histopathology
Fetus or newborn pup fixed in 10% formalin was paraffin embedded and imaged using ECM (36). Briefly, this entails the use of a Leica LSI scanning confocal microscope to capture the tissue autofluorescence visible at the paraffin block face as the paraffin block is being sectioned using a Leica SM2500 microtome. This results in production of a registered 2D serial image stack of the specimen that can be readily 3D reconstructed or digitally resectioned in any imaging plane using software such as ImageJ or Osirix. For analysis of the cerebellar folds, paraffin sections were collected during ECM imaging, and subsequently stained using 0.1% cresyl violet.
Recovery of the Hug mutation and mouse breeding
C57BL/6J(B6)/ C57BL/10J(B10) hybrid mutants were generated by intercrossing the original Hug mutant line with the B10 strain. DNA from these hybrid mutants was used for genome scanning to map the mutation using 62 polymorphic SNPs, and additional SNPs on mouse chromosome 15 to refine the map interval (38). Exome sequencing was performed on genomic DNA from a single Hug mutant, and captured using SureSelectXT Mouse All Exon kit (Agilent Technologies), then sequenced using SOLiD 5500XL (Life Technologies).
Cell culture and reagents
Primary MEFs were isolated from E12.5 mutant mice and their wild-type littermates as previously described (37). Briefly, the E12.5 embryos were torn apart in 0.25% trypsin-EDTA with sterile needles or pipette tips. After incubation for 20 min in 10 cm tissue culture dishes, the remaining fragments were pipetted multiple times and incubated overnight in DMEM supplemented with 10% FBS and, penicillin and streptomycin. Early passage MEFs and human fibroblasts obtained from a Joubert syndrome patient and a control were all grown in DMEM supplemented with 10% FBS and, penicillin and streptomycin.
Wound scratch assay
MEFs were cultured to confluence and a wound gap was generated using a 10 μl micropipette tip. Cells were allowed cultured for an additional 18 h, then cells fixed and immunostained with DAPI and Golgin subfamily A member 2 (GOLGA2). Polarity was scored by quantitating the relative orientation of cell migration (0°–60°, 60°–120° and 120°–180°) relative to the wound gap.
Immunofluorescence analysis
MEFs grown on coverslips were fixed 4% paraformaldehyde for 10 min at room temperature followed by permeabilization in 0.1% Triton-X 100 (Sigma) in 3% bovine serum albumin for 15 min and primary antibody incubation overnight. After 1 h in secondary antibodies, the coverslips were mounted in Vectashield with DAPI. Primary antibodies used included anti-acetylated α tubulin (Sigma; 1:1000 dilution), anti-γ tubulin (Sigma-1:1000 dilution), anti-CEP290, anti-NPHP1 as described in (39), anti- acetylated α tubulin and anti-IFT88 as described in (40).
For quantitation of cilia, we tracked cilia with acetylated tubulin staining found in conjunction with γ-tubulin at the cilia base. All imaging analysis was carried out using confocal microscopy (Olympus FV1000).
To examine the distribution of JBTS17, custom JBTS17 mouse monoclonal antibody was commercially developed (Abmart, Shanghai, China). The specificity of the monoclonal antibody was assessed using Jbts17 siRNA knockdown, which showed significant loss of Jbts17 transcripts and also marked reduction in JBTS17 immunostaining in the cilia after siRNA knockdown (Supplementary Material, Fig. S3).
Cochlea staining: mutant and control cochlea were whole mount stained with Phalloidin (Sigma; 1:40 dilution) and anti-acetylated α tubulin (1:1000 dilution).
Cryosection analysis: cryosections were obtained from OCT embedded tissues, with 12 μm thick sections for cilia and basal body staining using antibodies to ACIII and γ tubulin (37).
SAG stimluation of Shh and quantitative RT-PCR analysis
MEFs from two Hug mutants and a control littermate were grown in serum starvation (0.1% FBS) for 24 h and were exposed to 400 nm SAG for an additional 24 h in serum Total RNA was isolated from MEFs using the RNeasy Plus Mini Kit (Qiagen). RNA was then converted to cDNA using High Capacity RNA-to-cDNA™ Kit (Life technologies) and qPCR for two experimental replicates with each reaction in triplicate were performed for Gli1, and Ptch1, with β-actin serving as internal control using the 7900HT Fast Real-Time PCR System (Life technologies).
Jbts17 siRNA knockdown analysis of JBTS monoclonal antibody specificity
Jbts17 siRNA pool were obtained from ON-TARGET and SMART pool from Dharmacon GE Healthcare. These five siRNA comprised: (i) GUGACUGGCUUGACGGAUA; (ii) GAGCAGUACACUCACACGUUA; (iii) GGGAAGUCAUGAUGCGAGU; (iv) GGGAAGUCAUGAUGCGAGU; (v) GCUUAGUGCCCGUCGACUU. Briefly, 30 pg of siRNA pool was transfected into IMCD3 cells using Lipofectamine® RNAiMAX Transfection Reagent (Life Technologies). After 24 h, the cells were subjected to serum starvation for 18 h, after which the cells were fixed and processed for immunostaining with antibodies to JBTS17, acetylated tubulin and γ-tubulin. Quantitative PCR was performed using two different mouse Jbts17 primers with RNA extracted from mock and Jbts17-siRNA transfected cells. Ciliated cells from the mock and JBTS-siRNA transfected cells were analyzed for JBTS17 localization in the cilia. The immunostain intensity was quantitatively assessed using ImageJ.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by National Institutes of Health grant U01HL098180 (C.W.L.), R37 GM030626 (G.B.W.) and by the Robert W. Booth Endowment (G.B.W.).
Supplementary Material
References
- 1.Fliegauf M., Benzing T., Omran H. (2007) When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell. Biol., 8, 880–893. [DOI] [PubMed] [Google Scholar]
- 2.Kim S., Dynlacht B.D. (2013) Assembling a primary cilium. Curr. Opin. Cell Biol., 25, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature, 426, 83–87. [DOI] [PubMed] [Google Scholar]
- 4.Ware S.M., Aygun M.G., Hildebrandt F. (2011) Spectrum of clinical diseases caused by disorders of primary cilia. Proc. Am. Thorac. Soc., 8, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waters A.M., Beales P.L. (2011) Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol., 26, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidts M., Vodopiutz J., Christou-Savina S., Cortes C.R., McInerney-Leo A.M., Emes R.D., Arts H.H., Tuysuz B., D'Silva J., Leo P.J., et al. (2013) Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet., 93, 932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valente E.M., Marsh S.E., Castori M., Dixon-Salazar T., Bertini E., Al-Gazali L., Messer J., Barbot C., Woods C.G., Boltshauser E., et al. (2005) Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann. Neurol., 57, 513–519. [DOI] [PubMed] [Google Scholar]
- 8.Juric-Sekhar G., Adkins J., Doherty D., Hevner R.F. (2012) Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta. Neuropathol., 123, 695–709. [DOI] [PubMed] [Google Scholar]
- 9.Doherty D. (2009) Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol., 16, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srour M., Hamdan F.F., Schwartzentruber J.A., Patry L., Ospina L.H., Shevell M.I., Desilets V., Dobrzeniecka S., Mathonnet G., Lemyre E., et al. (2012) Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet., 49, 636–641. [DOI] [PubMed] [Google Scholar]
- 11.Aguilar A., Meunier A., Strehl L., Martinovic J., Bonniere M., Attie-Bitach T., Encha-Razavi F., Spassky N. (2012) Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc. Natl Acad. Sci. USA, 109, 16951–16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romani M., Micalizzi A., Valente E.M. (2013) Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol., 12, 894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srour M., Schwartzentruber J., Hamdan F.F., Ospina L.H., Patry L., Labuda D., Massicotte C., Dobrzeniecka S., Capo-Chichi J.M., Papillon-Cavanagh S., et al. (2012) Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet., 90, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andermann F., Andermann E., Ptito A., Fontaine S., Joubert M. (1999) History of Joubert syndrome and a 30-year follow-up of the original proband. J. Child. Neurol., 14, 565–569. [DOI] [PubMed] [Google Scholar]
- 15.Joubert M., Eisenring J.J., Robb J.P., Andermann F. (1969) Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology, 19, 813–825. [DOI] [PubMed] [Google Scholar]
- 16.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M., et al. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell, 145, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sudarov A., Joyner A.L. (2007) Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neurl. Development., 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui C., Chatterjee B., Lozito T.P., Zhang Z., Francis R.J., Yagi H., Swanhart L.M., Sanker S., Francis D., Yu Q., et al. (2013) Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PLoS Biology, 11, e1001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaheen R., Faqeih E., Alshammari M.J., Swaid A., Al-Gazali L., Mardawi E., Ansari S., Sogaty S., Seidahmed M.Z., AlMotairi M.I., et al. (2013) Genomic analysis of Meckel-Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. Eur. J. Hum. Genet., 21, 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuz K., Bachmann-Gagescu R., O'Day D.R., Hua K., Isabella C.R., Phelps I.G., Stolarski A.E., O'Roak B.J., Dempsey J.C., Lourenco C., et al. (2014) Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet., 94, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murdoch J.N., Copp A.J. (2010) The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol., 88, 633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lancaster M.A., Gopal D.J., Kim J., Saleem S.N., Silhavy J.L., Louie C.M., Thacker B.E., Williams Y., Zaki M.S., Gleeson J.G. (2011) Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med., 17, 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norris D.P., Grimes D.T. (2012) Mouse models of ciliopathies: the state of the art. Dis. Model. Mech., 5, 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelhamed Z.A., Wheway G., Szymanska K., Natarajan S., Toomes C., Inglehearn C., Johnson C.A. (2013) Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet., 22, 1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caspary T., Larkins C.E., Anderson K.V. (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell, 12, 767–778. [DOI] [PubMed] [Google Scholar]
- 26.Cheung H.O., Zhang X., Ribeiro A., Mo R., Makino S., Puviindran V., Law K.K., Briscoe J., Hui C.C. (2009) The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal., 2, ra29. [DOI] [PubMed] [Google Scholar]
- 27.Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V., et al. (2008) THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet., 40, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C., et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet., 39, 875–881. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J., et al. (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet., 43, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas S., Legendre M., Saunier S., Bessieres B., Alby C., Bonniere M., Toutain A., Loeuillet L., Szymanska K., Jossic F., et al. (2012) TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet., 91, 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chih B., Liu P., Chinn Y., Chalouni C., Komuves L.G., Hass P.E., Sandoval W., Peterson A.S. (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell. Biol., 14, 61–72. [DOI] [PubMed] [Google Scholar]
- 32.Szymanska K., Johnson C.A. (2012) The transition zone: an essential functional compartment of cilia. Cilia, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leightner A.C., Hommerding C.J., Peng Y., Salisbury J.L., Gainullin V.G., Czarnecki P.G., Sussman C.R., Harris P.C. (2013) The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum. Mol. Genet., 22, 2024–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez E., Thauvin-Robinet C., Reversade B., Khartoufi N.E., Devisme L., Holder M., Ansart-Franquet H., Avila M., Lacombe D., Kleinfinger P., et al. (2013) C5orf42 is the major gene responsible for OFD syndrome type VI. Hum. Genet., 133, 367–377. [DOI] [PubMed] [Google Scholar]
- 35.Ohba C., Osaka H., Iai M., Yamashita S., Suzuki Y., Aida N., Shimozawa N., Takamura A., Doi H., Tomita-Katsumoto A., et al. (2013) Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics, 14, 225–232. [DOI] [PubMed] [Google Scholar]
- 36.Liu X., Francis R., Kim A.J., Ramirez R., Chen G., Subramanian R., Anderton S., Kim Y., Wong L., Morgan J., et al. (2014) Interrogating congenital heart defects with noninvasive fetal echocardiography in a mouse forward genetic screen. Circ. Cardiovasc. Imaging, 7, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui C., Chatterjee B., Francis D., Yu Q., SanAgustin J.T., Francis R., Tansey T., Henry C., Wang B., Lemley B., et al. (2011) Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis. Model. Mech., 4, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia Y., Won S., Du X., Lin P., Ross C., La Vine D., Wiltshire S., Leiva G., Vidal S.M., Whittle B., et al. (2010) Bulk segregation mapping of mutations in closely related strains of mice. Genetics, 186, 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schermer B., Hopker K., Omran H., Ghenoiu C., Fliegauf M., Fekete A., Horvath J., Kottgen M., Hackl M., Zschiedrich S., et al. (2005) Phosphorylation by casein kinase 2 induces PACS-1 binding of nephrocystin and targeting to cilia. EMBO. J., 24, 4415–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pazour G.J., Baker S.A., Deane J.A., Cole D.G., Dickert B.L., Rosenbaum J.L., Witman G.B., Besharse J.C. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell. Biol., 157, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.