

Abstract
The Moonwalker (Mwk) mouse is a model of dominantly inherited cerebellar ataxia caused by a gain-of-function mutation in the transient receptor potential (TRP) channel TRPC3. Here, we report impairments in dendritic growth and synapse formation early on during Purkinje cell development in the Mwk cerebellum that are accompanied by alterations in calcium signaling. To elucidate the molecular effector pathways that regulate Purkinje cell dendritic arborization downstream of mutant TRPC3, we employed transcriptomic analysis of developing Purkinje cells isolated by laser-capture microdissection. We identified significant gene and protein expression changes in molecules involved in lipid metabolism. Consistently, lipid homeostasis in the Mwk cerebellum was found to be disturbed, and treatment of organotypic cerebellar slices with ceramide significantly improved dendritic outgrowth of Mwk Purkinje cells. These findings provide the first mechanistic insights into the TRPC3-dependent mechanisms, by which activated calcium signaling is coupled to lipid metabolism and the regulation of Purkinje cell development in the Mwk cerebellum.
Introduction
Cerebellar ataxia defines a heterogeneous group of neurological disorders that affect the cerebellum and its afferent and efferent connections and manifest primarily as progressive gait and limb incoordination. The inherited forms of cerebellar ataxia encompass a growing list of genetically diverse disorders with more than 35 genetic loci known to cause the autosomal dominant ataxias alone, which are also referred to as spinocerebellar ataxias (SCAs) (1,2). Despite this genetic heterogeneity, common pathological mechanisms that may be shared among the different forms of cerebellar ataxia are beginning to emerge (2,3). Notably, disruption of intracellular calcium homeostasis and signaling in Purkinje cells has been proposed as a key mechanism in the pathogenesis of SCAs (1,4,5).
The transient receptor potential (TRP) channel TRPC3 is a non-selective cation channel that is highly expressed in the Purkinje cells of the cerebellum (6,7). TRPC3 is required for metabotropic glutamate receptor subtype 1 (mGluR1)-dependent synaptic transmission in Purkinje cells (6). Both genetic loss of Trpc3 and the dominant Moonwalker (Mwk) gain-of-function point mutation in Trpc3 result in cerebellar ataxia in the mouse (6,8), highlighting the importance of Purkinje cell calcium homeostasis for proper cerebellar function. Recently, we have identified the first functionally pathogenic variant (R672H) in the human TRPC3 gene in a patient with adult-onset cerebellar ataxia (9). The human R672H mutation likely acts through a toxic gain-of-function mechanism similar to the Mwk mutation. Moreover, TRPC3 signaling has been linked to several other genetic forms of cerebellar ataxia in human and mouse including SCA1, SCA14, SCA15 and mutations in the GluD2 receptor (1) (reviewed in (7)). Thus, the disruption of the TRPC3 signaling pathway might be a common pathological mechanism underlying cerebellar ataxia in mouse and human.
Interestingly, besides adult-onset Purkinje cell loss, the Mwk mutation in TRPC3 causes impairments in Purkinje cell function and dendritic arborization during cerebellar development (8,10). In contrast, no dendritic phenotype and a milder form of ataxia have been observed in the Trpc3 knockout mice (6,11). Hence, it is the sustained activation of TRPC3 that provides an important regulatory effect on dendritic growth in cerebellar Purkinje cells. This model is consistent with the observation that overexpression of SCA14-associated PKCγ mutants that fail to inhibit TRPC3 (12) causes impairments in the dendritic development of Purkinje cells (13). Similarly, related studies have shown the inhibition of Purkinje cell growth upon chronic activation of mGluR1 or PKC activation (14,15). However, the effector pathways that mediate the inhibition of dendritic growth downstream of activated TRPC3 have remained enigmatic.
Here, we report impairments in dendritic growth and synapse formation early on during Purkinje cell development in the Mwk cerebellum that are accompanied by alterations in key calcium signaling pathways. We also identify Purkinje cell-specific gene expression changes that result in abnormal lipid homeostasis in the mutant mice. Together, our study provides mechanistic insights into the TRPC3-dependent processes that link activated calcium signaling to lipid metabolism and the regulation of Purkinje cell development in the Mwk cerebellum.
Results
Dendritic and synaptic impairments in the developing Mwk cerebellum
Previously, we demonstrated abnormal Purkinje cell dendritic morphology in 3-week-old Mwk mice (8). To investigate whether the mutant phenotype results from a failure of the dendritic tree to arborize or whether it is a consequence of atrophy following normal outgrowth, we carried out a time course experiment in organotypic cerebellar slice cultures of Mwk mice compared with their wild-type littermates. The initial development of the Purkinje cell dendritic tree was similar in wild-type and mutant mice based on the extent of the Purkinje cell dendritic area (Fig. 1A). However, starting from postnatal day 15 (P9+DIV6), impairments in arborization were observed in the Mwk Purkinje cells (Fig. 1A). Although wild-type Purkinje cells continued to grow their dendritic arbors, mutant Purkinje cells failed to further expand the area of their dendritic tree (Fig. 1A and B), suggesting that it is the late phase of dendritic outgrowth that is specifically impaired in Mwk Purkinje cells.
Figure 1.

Mwk TRPC3 impairs Purkinje cell dendritic arborization. (A) Time course of Purkinje cell dendritic growth in organotypic slice cultures prepared from wild-type (WT) and mutant (Mwk) littermates. Cultures were fixed at the indicated days in vitro and stained with an anti-calbindin antibody to visualize dendritic morphology. Initially, Mwk dendritic arbors were indistinguishable from wild-type littermates but starting at around two weeks postnatally, Mwk Purkinje cells exhibited a significantly decreased size of their dendritic trees (P < 0.0001, ANOVA, n = 128). (B) Representative images of Purkinje cell dendritic morphology in organotypic slice cultures fixed at P9+DIV9 and immunostained for calbindin demonstrate the impaired arborization in Mwk Purkinje cells. The outlines of the dendritic arbors have been traced. (C) Size of the dendritic area in organotypic slice cultures (P9+DIV12) prepared from wild-type (WT) and mutant (Mwk) littermates, fixed and immunostained with an anti-calbindin antibody. Treatment with P/Q block significantly increased the dendritic area of the Mwk but not WT Purkinje cells (P < 0.0001, ANOVA, n = 218).
P/Q-type calcium channels have been shown to contribute to the inhibition of dendritic growth following activation of mGluR1 (11). Moreover, TRPC3 signaling in cardiac myocytes has been linked to activation of voltage-gated calcium channels (VGCCs) (16). These observations raised the question of whether VGCCs might contribute to the dendritic phenotype of Purkinje cells in the Mwk mouse. To assess whether inhibition of VGCCs could rescue the dendritic phenotype in Mwk Purkinje cells, we used a combination of ω-agatoxin IVA and ω-conotoxin MVIIC to inhibit P/Q-type calcium channels (P/Q block) (11). Organotypic slice cultures (P9) were treated for 12 days with P/Q block, starting at DIV2. As seen previously, dendritic arborization of Mwk Purkinje cells was significantly impaired compared with wild-type cells (Fig. 1C). Interestingly, inhibition of P/Q-type calcium channels significantly increased the dendritic area of Mwk Purkinje cells when compared with untreated Mwk cultures by 50% (4457 ± 316 μm2 versus 2176 ± 135.2 μm2) (Fig. 1C). Treatment of wild-type slices with P/Q block had no significant effect (Fig. 1C) (11). Together, these results suggest that P/Q-type calcium channels are involved in the dendritic growth inhibition of Purkinje cells that is caused by mutant Mwk TRPC3.
We next examined whether the dendritic changes observed in the Mwk mice were accompanied by changes in the placement of excitatory synaptic terminals onto Purkinje cell dendritic trees. The distal part of the Purkinje cell dendritic tree receives excitatory input from the parallel fibers (PFs) of the granule cells, whereas the proximal portion of the dendritic tree is innervated by inputs from climbing fibers (CFs). The innervation areas of PFs and CFs are intimately linked, and stabilization of PF synapses restricts the innervation territory of CFs on the Purkinje cell dendritic tree and vice versa (17,18). First, we investigated the PF innervation pattern of wild-type and Mwk Purkinje cells by their immunoreactivity to the vesicular glutamate transporter VGLUT1. We found no detectable differences in the immunostaining pattern for VGLUT1 in cerebellar sections from Mwk mice compared with their wild-type littermates (density VGLUT1 staining WT = 76.26 ± 7.093; Mwk = 74.54 ± 8.694; P = 0.7992; n = 3) (Fig. 2A). Both wild-type and mutant cerebellar sections exhibited strong VGLUT1-positive puncta representing PF inputs throughout the molecular layer. To determine the spatial distribution of CF terminals on Purkinje cell dendritic arbors, we carried out immunostainings for VGLUT2. In adult wild-type cerebellar sections, the distribution of VGLUT2 staining representing the extent of the CF territory extended to a mean relative height of 81 ± 0.9862% of the molecular layer (Fig. 2B and C), consistent with previous reports using wild-type mice (17,19,20). In contrast, in cerebellar sections of adult Mwk mice, the mean relative height was reduced to 63 ± 1.108%. We also assessed the extension of the CF territory in cerebellar sections of younger animals. A significant reduction in CF arborization along the Purkinje cell dendrites was observed in the mutants as early as postnatal day 14 (P14) (Fig. 2C), concurrent with the observed dendritic abnormalities (Fig. 1A). Together, these results suggest that Mwk mice exhibit profound dendritic and synaptic impairments during development as well as in adulthood.
Figure 2.

Altered distribution of climbing fiber (CF) terminals on Purkinje cell dendrites of Mwk mice. (A) Representative image of the distribution of VGLUT1-labeled parallel fiber (PF) terminals in the molecular layer (ML) of cerebellar sections from a 30-day-old Mwk mouse compared with a wild-type (WT) littermate. No difference in VGLUT1-positive puncta was noted (density VGLUT1 staining WT = 76.26 ± 7.093; Mwk = 74.54 ± 8.694; P = 0.7992; n = 3). (B) Representative image of the distribution of VGLUT2-labeledCF terminals along Purkinje cell (PC) dendrites in the ML of cerebellar sections from a 30-day-old Mwk mouse compared with a WT littermate. Arrows depict the extension of the CF territory. (C) At all timepoints tested (P14, P18, P30), Mwk mice show a significant reduction in the CF territory (P < 0.001, ANOVA, n = 126).
Altered calcium signaling pathways in the developing Mwk cerebellum
Having identified these dendritic and synaptic abnormalities, we next investigated the mechanism underlying Mwk TRPC3-mediated impairments in Purkinje cell development. As the Mwk phenotype results from a gain-of-function mutation in the calcium-permeable TRPC3 channel (8) and we have also shown that VGCCs are involved (Fig. 1C), we asked which calcium-sensitive downstream signaling pathways might be affected in the developing Mwk cerebellum. We prepared cerebellar extracts from Mwk mice and their wild-type littermates at several time points during postnatal development (P15–P46), followed by immunoblotting for key proteins of neuronal calcium signaling. We found that several calcium-sensitive signaling molecules were altered in the mutant cerebellum compared with wild-type controls. Changes were observed as early as 2 weeks postnatally. The phosphorylation status and thus the activity levels of both the extracellular-regulated kinases 1/2 (ERK1/2) and the transcription factor cAMP response element-binding protein (CREB) were increased in Mwk cerebellum consistent with activated calcium signaling (Fig. 3A). Levels of phosphorylated calcium/calmodulin-dependent protein kinase type II (CaMKII) were unchanged in Mwk cerebellum at all ages studied (Fig. 3B). In contrast, phosphorylation of calcium/calmodulin-dependent protein kinase type IV (CaMKIV) was reduced in the Mwk cerebellum (Fig. 3C). Noteworthy, phosphorylated CaMKIV is predominantly present in Purkinje cells of the cerebellum (Supplementary Material, Fig. S1). Hence, the changes in phosphorylated CaMKIV that were observed in total cerebellar extracts reflect specific changes in the Purkinje cells. Together, these findings demonstrate alterations in important calcium-sensitive signaling kinases and downstream transcription factors in the developing Mwk cerebellum.
Figure 3.

Activation of key calcium signaling pathways in Mwk cerebellum. Cerebellar extracts from 3-week-old mutant Mwk mice and wild-type (WT) littermates were subjected to immunoblotting for calcium-responsive proteins. (A) Phosphorylated ERK1/2 and phosphorylated CREB (Ser133) levels were increased in the Mwk cerebellum. (B) Levels of phosphorylated CaMKII remained unchanged in Mwk cerebellum. (C) Levels of phosphorylated CaMKIV were reduced in Mwk cerebellum. Actin levels are included as loading control. Representative blots are shown for 3-week-old animals. Densitometry quantifications are shown (right panels). Statistical significance was determined by one-way ANOVA (n = 3). *P< 0.05; **P< 0.01.
Mwk Purkinje cells display significant gene expression changes
Little is known about the specific targets regulated by calcium-mediated signaling pathways that control Purkinje cell development. On the basis of our findings, the Mwk mouse represents an ideal system to investigate the intrinsic factors that link altered calcium signaling and Purkinje cell development. To identify gene expression changes specifically in Mwk Purkinje cells, we carried out laser capture microdissection (LCM) of Purkinje cells, followed by RNA isolation and microarray analysis using the Affymetrix Mouse Gene 1.0 ST array. This was done in 18-day-old Mwk mice and their wild-type littermates, thus around the critical period and the onset of molecular and behavioral changes in the mutant mice. Samples were obtained from four sets of Mwk and wild-type littermates, and 1000 Purkinje cells were collected per cerebellum. Applying a stringent detection threshold (1.5-fold change) and cut-off P-value of ≤0.5, we identified statistically significant changes in the expression levels of 634 genes between wild-type and Mwk Purkinje cells. Fifty-one percent (323) of genes were upregulated and 49% (311) of genes were downregulated (Fig. 4A and Supplementary Material, Table S1). The results were reproducible among the four biological replicates (Fig. 4A).
Figure 4.

Gene expression changes in Mwk Purkinje cells. (A) Heat map shows statistically significant changes in the expression of 634 genes between WT and Mwk Purkinje cells at P18 (P ≤ 0.05). Differentially expressed genes are listed in Supplementary Material, Table S1. Each column represents one of four RNA samples/biological replicates for each genotype. (B) Significant enrichment in genes differentially expressed in Mwk Purkinje cells in IPA top functions and disease categories. The number of changed genes is indicated. (C) Functional annotation of differentially expressed genes associated with lipid metabolism that are listed in Table 1. Only categories with two or more genes are listed. Annotations were made using a combination of pathway analysis tools including IPA, Kyoto Encyclopedia of Genes and Genomes (KEGG) and SuperPaths (http://www.genecards.org/info.shtml#pathways_interactions).
To verify the microarray results, we selected several genes that showed significant expression changes between Mwk and wild-type Purkinje cells for independent validation by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using LCM-isolated Purkinje cell RNA from an independent set of four Mwk mice and their wild-type littermates at P18. We detected similar changes in the expression levels of these genes by qRT-PCR as by microarray (Supplementary Material, Fig. S2 and Table S2). We also investigated the expression levels of these genes at earlier timepoints to determine how early we could detect changes in the mutant cerebellum. We found three of the selected genes (Ipo5, Opn3, Sv2c) upregulated in Mwk Purkinje cells as early as P11. Others were downregulated in the mutant Purkinje cells from P14 (Stk17b, Car2) (Supplementary Material, Table S2). Consistent with the expression changes observed by microarray and qRT-PCR, differences in mRNA expression levels between Mwk Purkinje cells and wild-type littermates were also detected by in situ hybridization (ISH) (Supplementary Material, Fig. S3). Together, these data show that significant gene transcription changes occur in Mwk mice early on in Purkinje cell development, concurrent with the observed dendritic and synaptic abnormalities.
Mwk Purkinje cells exhibit gene expression changes linked to lipid metabolism
To gain insights into the potential molecular pathways affected in Mwk Purkinje cells, we next analyzed the differentially expressed genes using Ingenuity® Pathway Analysis (IPA). This analysis revealed several biological pathways and functions that were significantly enriched in dysregulated genes of the Mwk cerebellum including cellular assembly and organization (P = 2.28E-04–4.38E-02) and nervous system development and function (P = 8.40E-04–4.21E-02), consistent with the developmental phenotype of the mice (Fig. 4B, Supplementary Material, Tables S3 and S4). Moreover, genes associated with neurological disease were significantly affected (P = 4.65E-04–4.34E-02) (Fig. 4B). Notably, included in this category were movement disorders (50 gene changes; P = 1.70E-03) and ataxia (13 gene changes; P = 1.59E-02) (Supplementary Material, Table S5).
Interestingly, the top enriched category for genes altered in Mwk Purkinje cells was lipid metabolism, and one of the top altered canonical pathways in Mwk Purkinje cells was sphingolipid metabolism (Fig. 4B, Supplementary Material, Tables S6 and S7). We identified 27 differentially expressed genes that are associated with lipid metabolism, of which 10 were upregulated in Mwk Purkinje cells and 17 were downregulated (Table 1). We classified these genes further into sub-categories of lipid metabolism using a combination of pathway analysis tools. Consistent with the results described above, we found that a large number of affected genes encode proteins that are key enzymes in sphingolipid metabolism, particularly the synthesis of ceramide (Fig. 4C). Notably, we observed reciprocal expression changes in the enzymes sphingosine kinase 2 (SPHK2) and sphingosine-1-phosphate phosphatase 1 (SGPP1) as well as changes in the expression of acid ceramidase (ASAH1), sphingomyelin synthase 1 (SGMS1) and sphingomyelin phosphodiesterase 1 (SMPD1), suggesting a coordinated deregulation of ceramide biosynthesis (Fig. 5A). To validate these gene expression changes at the protein level, we prepared cerebellar extracts from three-week-old Mwk mice and their wild-type littermates and subjected these to immunoblotting. Consistent with the observed microarray changes, we found the protein levels of ASAH1 and SPHK2 upregulated in the Mwk cerebellum (Fig. 5B). Conversely, SGPP1, SMPD1 and SGMS1 levels were reduced in the mutant cerebellum (Fig. 5B). Together, these enzyme expression changes point towards a reduction in the pool of available ceramide within the Mwk Purkinje cells. In addition, we observed expression changes in UDP-glucose ceramide glycosyltransferase (UCGC) and GM2 ganglioside activator protein (GM2A), two enzymes that are involved in the synthesis and degradation of complex glycosphingolipids that are linked to the ceramide pathway (Fig. 5A).
Table 1.
Significant expression changes (≥1.5-fold) in Mwk Purkinje cells compared with wild-type littermates in genes associated with lipid metabolism (P ≤ 0.05)
| Gene symbol | Gene product | Fold change |
|---|---|---|
| Ctsd | Cathepsin D | 5.7 |
| Ptplb | Protein tyrosine phosphatase-like (proline instead of catalytic arginine), member b | 4.3 |
| Prdx6 | Peroxiredoxin 6 | 3.5 |
| Scd | Stearoyl-Coenzyme A desaturase 1 | 2.8 |
| Asah1 | N-acylsphingosine amidohydrolase 1 (acid ceramidase) | 2.4 |
| Ugcg | UDP-glucose ceramide glucosyltransferase | 2.0 |
| Dab1 | Disabled homolog 1 | 1.8 |
| Lpin2 | Lipin 2 | 1.7 |
| Sphk2 | Sphingosine kinase 2 | 1.5 |
| Crh | Corticotropin releasing hormone | 1.5 |
| Scp2 | Sterol carrier protein 2 | −5.7 |
| Ptpla | Protein tyrosine phosphatase-like (proline instead of catalytic arginine), member a | −4.9 |
| Gdpd1 | Glycerophosphodiester phosphodiesterase domain containing 1 | −3.2 |
| Acot1 | Acyl-CoA thioesterase 1 | −3.1 |
| Far2 | Fatty acyl CoA reductase 2 | −2.8 |
| Fgf7 | Fibroblast growth factor 7 | −2.5 |
| Sgpp1 | Sphingosine-1 phosphate phosphatase 1 | −2.2 |
| Smpd1 | Sphingomyelin phosphodiesterase 1, acid lysosomal | −1.9 |
| Ggps1 | Geranylgeranyl diphosphate synthase | −1.9 |
| Etnk1 | Ethanolamine kinase 1 | −1.8 |
| Osbpl1a | Oxysterol binding protein-like 1A | −1.8 |
| Ncoa4 | Nuclear receptor coactivator 4 | −1.6 |
| Gm2a | GM2 ganglioside activator protein | −1.5 |
| Sgms1 | Sphingomyelin synthase 1 | −1.5 |
| Agpat4 | 1-acylglycerol-3-phosphate O-acyltransferase 4 | −1.5 |
| Gfra2 | Glial cell line derived neurotrophic family receptor alpha 2 | −1.5 |
| Acat2 | Acetyl-Coenzyme A acetyltransferase 2 | −1.5 |
Figure 5.

Protein expression changes in enzymes involved in ceramide homeostasis. (A) Schematic diagram of mammalian ceramide biosynthesis. Differentially expressed enzymes in Mwk Purkinje cells are indicated in red (upregulated) and blue (downregulated). (B) Cerebellar extracts from 3-week-old mutant Mwk mice and WT littermates were subjected to immunoblotting for lipid metabolism enzymes. ASAH1 and SPHK2 levels were increased in the Mwk cerebellum (left panel), whereas levels of SGPP1, SMPD1 and SGMS1 were reduced in Mwk cerebellum (middle panel). Actin levels are included as loading control. Densitometry quantifications are shown (right panel). Statistical significance was determined by one-way ANOVA (n = 3). **P< 0.01; ***P< 0.001.
Having identified that the expression levels of enzymes involved in lipid metabolism are significantly changed in the Mwk mutant, we next investigated whether levels of lipids were changed as a consequence in the Mwk cerebellum. We carried out lipid analysis by mass spectrometry in the cerebella of three Mwk mice and their wild-type littermates at P18. Consistent with the expression changes observed in Mwk Purkinje cells, we identified alterations of lipid levels in the mutant cerebellum including a reduction in distinct ceramides (Fig. 6A). We also observed changes in other lipids in the Mwk cerebellum, including fatty acyls and sterol lipids (data not shown). Interestingly, changes in lipid metabolism were not only evident in our analysis of the Mwk cerebellum, but serum levels of free fatty acids, triglycerides and cholesterol have also been found to be significantly changed in the Mwk mice in the EuroPhenome mouse phenotyping project (Supplementary Material, Fig. S4) (21).
Figure 6.
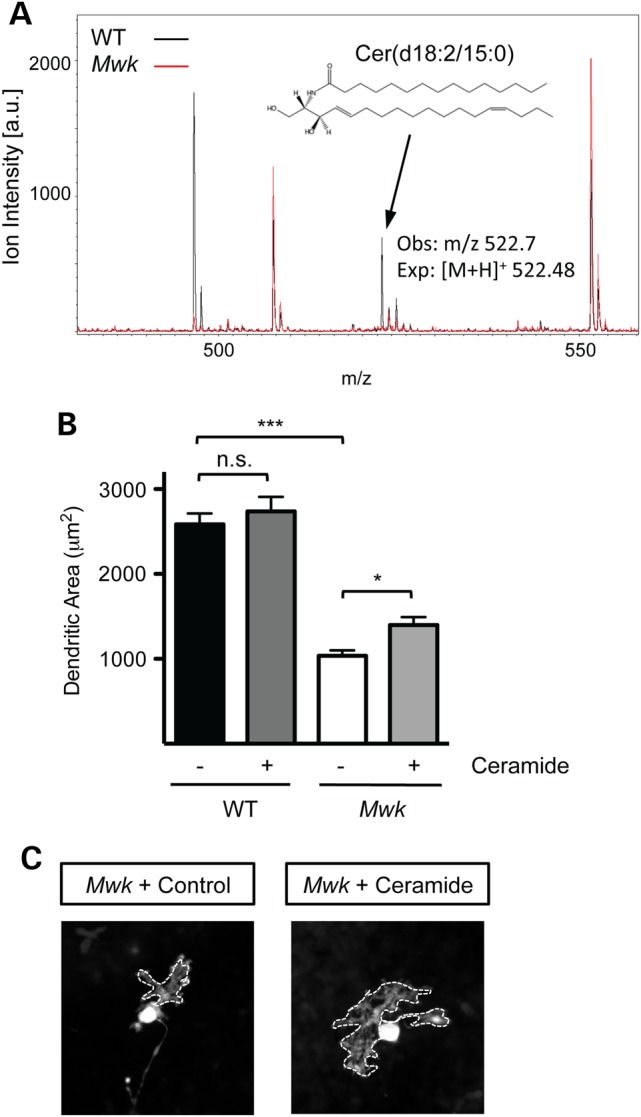
Altered ceramide homeostasis in Mwk Purkinje cells. (A) MALDI-TOF mass spectrometry analysis of cerebellar lipid extracts from Mwk mice and wild-type (WT) littermates at P18. Following mass spectrometry, spectrum peaks were annotated using the METLIN metabolite database (http://metlin.scripps.edu/index.php). Mass spectrometry spectra overlay of lipid profiles from Mwk (red) and WT cerebellum (black) show a reduction of ceramide Cer(d18:2/15:0), depicted by an arrow (observed m/z 522.7; expected [M+H]+ 522.5). The graph is a representative of the results obtained from three independent biological replicates. (B) The size of the dendritic area in organotypic slice cultures (P9+DIV12) prepared from wild-type (WT) and mutant (Mwk) littermates, fixed and immunostained with an anti-calbindin antibody. Treatment with C6-ceramide (Cer) but not inactive control (−) significantly increased the dendritic area of the Mwk but not WT Purkinje cells (P < 0.05, ANOVA, n = 202). (C) Representative images of Purkinje cell dendritic morphology in organotypic slice cultures fixed at P9+DIV12 and immunostained for calbindin demonstrate the increased arborization in Mwk Purkinje cells treated with C6-ceramide compared with inactive control. The outlines of the dendritic arbors have been traced.
We next tested whether treatment of Mwk organotypic slice cultures with ceramide might alleviate the dendritic phenotype observed in the mutant Purkinje cells. Sphingolipids have previously been shown to be important for Purkinje cell dendritic differentiation (22,23). Moreover, treatment of organotypic cerebellar cultures with sphingomyelinase was demonstrated to enhance dendritic branching complexity of Purkinje cells (23). In mice sphingomyelinase is encoded by the gene Smpd1, the expression of which is downregulated in Mwk cerebellum (Fig. 5A and B, Table 1). We prepared organotypic cerebellar slice cultures from Mwk mice and their wild-type littermates at P9 and treated these for 12 days in vitro with C6-ceramide or its inactive form dihydroceramide C6 as control. We found that treatment of the Mwk cerebellar slices with C6-ceramide significantly improved Purkinje cell dendritic outgrowth compared with control (Fig. 6B and C). Together, these findings suggest that the Mwk gain-of-function mutation in TRPC3 results in aberrant calcium signaling and gene expression changes in Purkinje cells that lead to alterations in lipid homeostasis and impairments in dendritic growth.
Discussion
In this study, we describe early dendritic and synaptic impairments and altered calcium-activated signaling pathways during Purkinje cell development in the Mwk mouse, an ataxic mouse model in which we previously identified a novel gain-of-function mutation in the non-selective cation channel TRPC3 (8). Combining Purkinje cell-specific transcriptomic and cerebellar lipid analysis, this study provides the first mechanistic insights into the molecular pathways underlying the Mwk phenotype. Together, our findings support the model that activated TRPC3 leads to aberrant calcium signaling in Purkinje cells and subsequent gene expression changes that result in altered lipid homeostasis and consequently abnormal dendritic development.
Purkinje cells form one of the most elaborate dendritic arbors among all neurons in the brain. Although the morphological stages of Purkinje cell dendritic development have extensively been characterized, relatively little is known about the molecular mechanisms that underlie these processes (24–26). Previous work has demonstrated that chronic activation of Purkinje cell post-synaptic signaling including mGlur1 (14), PKC (15,27) and TRPC3 (8) causes inhibition of dendritic growth. Consistent with this hypothesis, Purkinje cells in the lurcher mouse mutant, which harbors a gain-of-function mutation in the δ2 glutamate receptor (GluD2) resulting in chronic depolarization, also exhibit impaired dendritic growth (28). These signaling events may provide a negative feedback mechanism for limiting the size of the dendritic tree once Purkinje cells have established appropriate synaptic contacts with the parallel fibers in the developing cerebellum. However, the downstream effector pathways mediating this inhibitory effect remain elusive. Calcium signaling is thought to regulate dendritic growth and patterning through distinct mechanisms that are dependent on calcium entry (local/global), timing (acute/chronic), neuronal cell type and developmental stage (29,30). Our study points to specific calcium-sensitive pathways that may restrict dendritic growth in Purkinje cells upon sustained TRPC3 activation and chronic Purkinje cell depolarization. Interestingly, we found that CaMKIV is hypophosphorylated in the Mwk cerebellum compared with wild-type controls (Fig. 3B). Our findings of ataxia, impaired dendritic Purkinje cell growth and reduced CaMKIV activity in Mwk mutants are consistent with the phenotype observed in CaMKIV-deficient mice. The latter also display cerebellar ataxia and reduced dendritic arborization of Purkinje cells (31). CaMKIV is predominantly nuclear and is thought to regulate gene transcription either directly or through co-factors (32). Notably, one of the candidate transcription factors regulated by CaMKIV is RORα, an orphan member of the nuclear hormone-receptor superfamily (33,34). RORα is abundantly expressed in Purkinje cells (35) and strongly implicated in Purkinje cell development (36) as well as maintenance (37). Moreover, RORα has been linked to gene expression regulation that controls lipid metabolism (36). We have performed an initial bioinformatics-based analysis to identify transcription factor enrichment among the genes that are differentially expressed in Mwk Purkinje cells, which identified RORα among others (Supplementary Material, Table S8). In view of our findings, these observations raise the interesting possibility that RORα might link TRPC3 and CaMKIV signaling to gene expression changes in the Mwk cerebellum. Interestingly, staggerer mice with reduced RORα expression display decreases in triglycerides and cholesterol levels similar to the changes we observed in the Mwk mice (38).
Deficiencies in sphingolipid metabolism have been implicated in a range of neurological diseases, many of which are associated with cerebellar ataxia. Examples include autosomal-recessive cerebellar spasticity, which was recently been found to be caused by mutations in the GBA2 gene (39,40) and many lysosomal storage disorders (41). Notably, mice that are deficient in enzymes that we found to be downregulated in the Mwk cerebellum also display cerebellar dysfunction including Scp2 knockout mice (42), Smpd1-deficient mice, a model of Niemann-Pick disease (43), and Gm2a knockout mice, a model of GM2-gangliosidosis (44). Moreover, a deficiency of ceramide biosynthesis caused by mutations in the Lass1 gene encoding ceramide synthase 1 (CerS1) (Fig. 5A) has been shown to cause cerebellar ataxia and Purkinje cell degeneration (45). Interestingly, Purkinje cell arborization is also greatly reduced in the Lass1 mutants (45). The overlap in affected proteins and cerebellar phenotypes between the Mwk mouse and models of lysosomal storage diseases might point to converging pathological pathways underlying the cerebellar dysfunction in these models. It will be interesting to determine in future studies whether lipid-related pathways are also affected in other genetic forms of cerebellar ataxia and contribute to pathology more generally. This might be particularly relevant for those cerebellar ataxias that are linked to TRPC3 signaling such as SCA1, SCA14 and SCA15 (1) (reviewed in (7)).
Although long implicated in neuronal differentiation, the precise role of sphingolipids in dendritic development is unclear. Membrane traffic is required to supply growing dendrites with building material and hence, abnormalities in the production of required lipids might impede this process. It is also conceivable that ceramide and other sphingolipids control dendritic differentiation through their role in the formation of lipid rafts and the organization and activation of signaling receptors (46). Finally, ceramides have been demonstrated to act as a second messenger in signal transduction where they participate in growth inhibition, induction of differentiation and programmed cell death (47).
Although this study is focused on altered lipid homeostasis in the Mwk cerebellum, changes in other pathways are likely to contribute to the Mwk phenotype. The observed gene expression changes in several genes associated with vesicle fusion and exocytosis including Sv2c (Supplementary Material, Figs S2 and S3) point towards alterations in the secretory pathway in the Mwk cerebellum, which has a known role in dendritic arborization in many neuronal cell types (48). Of interest are also gene changes that have been observed in other ataxic mouse models such as the downregulation of the Car2 gene (Supplementary Material, Fig. S2), encoding carbonic anhydrase II, which has also been reported in a mouse model of spinocerebellar ataxia type 7 (SCA7) (49). Moreover, protein levels of carbonic anhydrase II are decreased in the ataxic lurcher mouse mutant (50). Carbonic anhydrase II is highly and specifically expressed in developing Purkinje cells and has been postulated to be involved in differentiation and synaptogenesis of Purkinje cells (50). These findings highlight another potential converging pathological mechanism in the Mwk mouse and other models of cerebellar ataxia that will be interesting to explore further.
Our study identifies molecular alterations in the Mwk cerebellum that are common to other mouse models of cerebellar ataxia. Future studies will reveal whether these alterations pinpoint to converging pathways that could be targeted to modulate or monitor the pathogenesis of genetically different cerebellar diseases.
Materials and Methods
Animals and reagents
The generation of Mwk mice has been described elsewhere (8). The Mwk colony was maintained by backcrossing to C3H/HeH. All animal work was approved by the University of Oxford Ethics Panel and in accordance with UK Home Office regulations. All reagents were purchased from Sigma unless otherwise noted.
Organotypic slice cultures
Organotypic slice cultures were prepared as previously described (8). The medium was changed every 2–3 days. For experiments using P/Q block, 100 nM ω-agatoxin IVA (Bachem) and 1 μM ω-conotoxin MVIIC (Bachem) were added from day 2 onwards (11). For ceramide treatment, 50 μM of C6-ceramide (N-hexanoyl-d-sphingosine) or its inactive form dihydroceramide C6 were added from day 2 onwards. At days-in vitro (DIV) 5–12, slice cultures were fixed in 4% paraformaldehyde followed by brief methanol permeabilization. Slices were blocked in PBS containing 0.3% Triton-X and 3% normal goat serum for 1 h at room temperature and incubated with rabbit anti-calbindin D-28K (Swant; 1:5000) for two nights at 4°C, followed by incubation with Alexa Fluor 594 goat anti-rabbit antibody (Invitrogen; 1:2000). For the quantification of Purkinje cell dendritic arbors, the size of the dendritic area was measured by tracing the outline of the dendritic tree using Axiovision 4.3 software (Zeiss) in a blinded manner. Cells were acquired from three independent experiments. Data were analyzed using GraphPad Prism. All data are represented as mean ± SEM. Statistical significance was assessed by one-way ANOVA followed by Bonferroni's multiple comparison test (*P< 0.05; **P< 0.01; ***P< 0.001).
Biochemical analysis
Three-week-old mice were sacrificed and cerebella were dissected out and kept on ice. Protein extracts were prepared by 10-s sonication in an RIPA buffer (50 mm Tris pH 8, 150 mm sodium chloride, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 100 μM sodium vanadate, 10 mm sodium fluoride, 1× Complete Protease Inhibitors [Roche]) followed by 10-min incubation on ice and 15-min centrifugation at 14 000g at 4°C. Fifty micrograms of protein extracts were analyzed by SDS–polyacrylamide gel electrophoresis and immunoblotting. The following antibodies were used: p44/42 MAPK (Erk1/2) (Cell Signaling; 1:1000), phospho-p44/42 MAPK (Erk1/2) (Tyr202/Tyr204) (Cell Signaling; 1:1000), CaMKIV (Abcam; 1:1000), p-CaMKIV (Thr196) (Santa Cruz; 1:1000), CaMKII (Cell Signaling, 1:500), phospho-CaMKII (Cell Signaling, 1:1000), CREB (Cell Signaling, 1:500), phospho-CREB (Ser133) (Cell Signaling, 1:1000), ASAH1 (Santa Cruz, 1:200), SPHK2 (Novus, 1:500), SGPP1 (Novus, 1:1000), SMPD1 (Santa Cruz, 1:200), SGMS1 (Santa Cruz, 1:200), Actin (Abcam, 1:1000). Antibody binding was detected by enhanced chemoluminescence (ECL, GE Healthcare). The intensity of the bands was quantified using ImageJ software (NIH). Data were normalized to actin levels and wild-type bands and analyzed using GraphPad Prism. All data are represented as mean ± SEM. Statistical significance was assessed by one-way ANOVA (*P< 0.05; **P< 0.01; ***P< 0.001).
Immunohistochemistry
Freshly dissected cerebella from Mwk mice and wild-type littermates were fixed overnight in 4% paraformaldehyde, cryoprotected in 30% sucrose, embedded in OCT compound (TissueTek) and frozen. 10-μm sagittal sections were incubated for 30 min in 0.1 M glycine buffer (pH7.4), blocked for 1 h (10% goat serum, 0.3% Triton-X100 in PBS) and incubated with primary antibodies in block solution overnight at 4°C. The following antibodies were used: VGLUT1 (Synaptic Systems, 1:500), VGLUT2 (Synaptic Systems; 1:200), p-CaMKIV (Thr196) (Santa Cruz; 1:50) and Calbindin D28k (Synaptic Systems; 1:200). Sections were incubated with Alexa Fluor-labeled secondary antibodies (Invitrogen; 1:2000) for 3 h at room temperature before being mounted using DAPI-containing Vectashield medium (Vector Labs).
LCM and RNA preparation
Freshly dissected cerebella from Mwk mice and wild-type littermates were embedded in OCT compound (TissueTek) and stored at −80°C until sectioning. 10-μm sagittal midline sections were cut and mounted onto PEN membrane-coated slides (Carl Zeiss), before fixation in a graded series of ice-cold ethanol solutions (95, 75 and 50%) (containing ProtectRNATM RNase inhibitor (Sigma)) for 30 s each, and staining for 1 min with 1% cresyl violet acetate. Sections were rinsed in 50% ethanol solution and dehydrated in 75, 95 and 100% ethanol solutions for 30 s each, and dehydrated twice in xylene for 30 s. Slides were left to air-dry briefly before proceeding with the LCM. Purkinje cells were microdissected from all cerebellar lobes of the sections using the P.A.L.M. Micro Beam system in combination with Robo software (Zeiss) and directly catapulted into a tube cap containing RLT lysis buffer with β-mercaptoethanol (RNeasy Micro kit (Qiagen)). One thousand Purkinje cells were collected for microarray analysis and 300–500 cells were collected for qRT-PCR. Total RNA was extracted using the RNeasy Micro Kit (Qiagen) and RNA quality was assessed on a 2100 BioAnalyzer using the RNA 6000 Pico Assay (Agilent Technologies). All samples used for microarray and qRT-PCR analysis had an RNA Integrity Number (RIN) of ≥6.
Microarray analysis
Fragmented and labeled cDNA was generated from 1 ng of total RNA using the Ovation Pico WTA System, the WT-Ovation Exon Module and the Encore Biotin Module (all NuGEN) according to manufacturer's instructions and hybridized to the Affymetrix Mouse Gene 1.0 ST Array. Chips were processed on an Affymetrix GeneChip Fluidics Station 450 and Scanner 3000.
Microarray data were normalized by Probe Logarithmic Intensity Error (PLIER) using GeneSpring GX11.0 (Agilent), and differentially expressed genes (fold change difference ≥1.5) were identified using a moderated t-test with a Benjamini and Hochberg multiple testing correction of 0.05. Differentially expressed genes were hierarchically clustered. Gene interaction networks and canonical pathways were analyzed using IPA (www.ingenuity.com). Further functional annotation was carried out using a combination of IPA, Kyoto Encyclopedia of Genes and Genomes (KEGG) and SuperPaths (http://www.genecards.org/info.shtml#pathways_interactions). Microarray data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-3517.
Lipid analysis by mass spectrometry
Freshly dissected cerebella from P18 Mwk mice and wild-type littermates were homogenized by 10-s sonication in an RIPA buffer (50 mm Tris pH 8, 150 mm sodium chloride, 1% Nonidet P40, 0.5% sodium deoxycholate). Subsequently, lipid material was extracted by methanol/chloroform (4:1). The aqueous and organic phases were isolated, dried down in vacuum and resuspended in acetone/methanol (2:1). The samples were then subjected to analysis by matrix-assisted laser desorption time-of-flight (MALDI-TOF) mass spectrometry (MS) using an Ultraflex II (Bruker Daltonics) mass spectrometer. Lipid profiles were analyzed using FlexAnalysis (Bruker Daltonics, v2.4) and annotated using the METLIN metabolite database (http://metlin.scripps.edu/index.php).
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by the UK Medical Research Council and the Royal Society. We thank the Wellcome Trust Integrative Physiology Initiative in Ion Channels and Diseases of Electrically Excitable Cells (OXION) for use of the microarray facility. E.B. is the recipient of a Research Fellowship from the Royal Society. B.M.K. was supported by the Biomedical Research Centre (NIHR), Oxford, and the Kennedy Trust Fund. Funding to pay the Open Access publication charges for this article was provided by the University of Oxford RCUK Open Access Block Grant.
Supplementary Material
References
- 1.Bettencourt C., Ryten M., Forabosco P., Schorge S., Hersheson J., Hardy J., Houlden H. and United Kingdom Brain Expression Consortium. (2014) Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol., 71, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matilla-Dueñas A., Ashizawa T., Brice A., Magri S., McFarland K.N., Pandolfo M., Pulst S.M., Riess O., Rubinsztein D.C., Schmidt J., et al. (2014) Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum, 13, 269–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson K.M., Andresen J.M., Orr H.T. (2009) Emerging pathogenic pathways in the spinocerebellar ataxias. Curr. Opin. Genet. Dev., 19, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schorge S., van de Leemput J., Singleton A., Houlden H., Hardy J. (2010) Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci., 33, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasumu A., Bezprozvanny I. (2010) Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum, 11, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartmann J., Dragicevic E., Adelsberger H., Henning H.A., Sumser M., Abramowitz J., Blum R., Dietrich A., Freichel M., Flockerzi V., et al. (2008) TRPC3 channels are required for synaptic transmission and motor coordination. Neuron, 59, 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker E.B.E. (2014) The Moonwalker mouse: new insights into TRPC3 function, cerebellar development, and ataxia. Cerebellum, 13, 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker E.B.E., Oliver P.L., Glitsch M.D., Banks G.T., Achilli F., Hardy A., Nolan P.M., Fisher E.M.C., Davies K.E. (2009) A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in Moonwalker mice. Proc. Natl Acad. Sci. USA, 106, 6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fogel B.L., Hanson S.M., Becker E.B.E. (2015) Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov. Disord., 30, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekerková G., Kim J.-A., Nigro M.J., Becker E.B.E., Hartmann J., Birnbaumer L., Mugnaini E., Martina M. (2013) Early onset of ataxia in Moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J. Neurosci., 33, 19689–19694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gugger O.S., Hartmann J., Birnbaumer L., Kapfhammer J.P. (2012) P/Q-type and T-type calcium channels, but not type 3 transient receptor potential cation channels, are involved in inhibition of dendritic growth after chronic metabotropic glutamate receptor type 1 and protein kinase C activation in cerebellar Purkinje cells. Eur. J. Neurosci., 35, 20–33. [DOI] [PubMed] [Google Scholar]
- 12.Adachi N., Kobayashi T., Takahashi H., Kawasaki T., Shirai Y., Ueyama T., Matsuda T., Seki T., Sakai N., Saito N. (2008) Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J. Biol. Chem., 283, 19854–19863. [DOI] [PubMed] [Google Scholar]
- 13.Seki T., Shimahara T., Yamamoto K., Abe N., Amano T., Adachi N., Takahashi H., Kashiwagi K., Saito N., Sakai N. (2009) Mutant gammaPKC found in spinocerebellar ataxia type 14 induces aggregate-independent maldevelopment of dendrites in primary cultured Purkinje cells. Neurobiol. Dis., 33, 260–273. [DOI] [PubMed] [Google Scholar]
- 14.Sirzen-Zelenskaya A., Zeyse J., Kapfhammer J. (2006) Activation of class I metabotropic glutamate receptors limits dendritic growth of Purkinje cells in organotypic slice cultures. Eur. J. Neurosci., 24, 2978–2986. [DOI] [PubMed] [Google Scholar]
- 15.Metzger F., Kapfhammer J.P. (2000) Protein kinase C activity modulates dendritic differentiation of rat Purkinje cells in cerebellar slice cultures. Eur. J. Neurosci., 12, 1993–2005. [DOI] [PubMed] [Google Scholar]
- 16.Onohara N., Nishida M., Inoue R., Kobayashi H., Sumimoto H., Sato Y., Mori Y., Nagao T., Kurose H. (2006) TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J., 25, 5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto K., Kano M. (2013) Synapse elimination in the developing cerebellum. Cell. Mol. Life Sci., 70, 4667–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosman L.W.J., Konnerth A. (2009) Activity-dependent plasticity of developing climbing fiber-Purkinje cell synapses. Neuroscience, 162, 612–623. [DOI] [PubMed] [Google Scholar]
- 19.Duvick L., Barnes J., Ebner B., Agrawal S., Andresen M., Lim J., Giesler G.J., Zoghbi H.Y., Orr H.T. (2010) SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron, 67, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenzetto E., Caselli L., Feng G., Yuan W., Nerbonne J.M., Sanes J.R., Buffelli M. (2009) Genetic perturbation of postsynaptic activity regulates synapse elimination in developing cerebellum. Proc. Natl Acad. Sci. USA, 106, 16475–16480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan H., Beck T., Blake A., Gates H., Adams N., Debouzy G., Leblanc S., Lengger C., Maier H., Melvin D., et al. (2010) EuroPhenome: a repository for high-throughput mouse phenotyping data. Nucleic Acids Res., 38, D577–D585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuya S., Ono K., Hirabayashi Y. (1995) Sphingolipid biosynthesis is necessary for dendrite growth and survival of cerebellar Purkinje cells in culture. J. Neurochem., 65, 1551–1561. [DOI] [PubMed] [Google Scholar]
- 23.Furuya S., Mitoma J., Makino A., Hirabayashi Y. (1998) Ceramide and its interconvertible metabolite sphingosine function as indispensable lipid factors involved in survival and dendritic differentiation of cerebellar Purkinje cells. J. Neurochem., 71, 366–377. [DOI] [PubMed] [Google Scholar]
- 24.Kapfhammer J. (2004) Cellular and molecular control of dendritic growth and development of cerebellar Purkinje cells. Prog. Histochem. Cytochem., 39, 131–182. [DOI] [PubMed] [Google Scholar]
- 25.Fujishima K., Horie R., Mochizuki A., Kengaku M. (2012) Principles of branch dynamics governing shape characteristics of cerebellar Purkinje cell dendrites. Development, 139, 3442–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metzger F. (2010) Molecular and cellular control of dendrite maturation during brain development. Curr. Mol. Pharmacol., 3, 1–11. [DOI] [PubMed] [Google Scholar]
- 27.Schrenk K., Kapfhammer J., Metzger F. (2002) Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cgamma-deficient mice. Neuroscience, 110, 675–689. [DOI] [PubMed] [Google Scholar]
- 28.Zanjani H.S., McFarland R., Cavelier P., Blokhin A., Gautheron V., Levenes C., Bambrick L.L., Mariani J., Vogel M.W. (2009) Death and survival of heterozygous Lurcher Purkinje cells in vitro. Dev. Neurobiol., 69, 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohmann C., Wong R.O.L. (2005) Regulation of dendritic growth and plasticity by local and global calcium dynamics. Cell Calcium, 37, 403–409. [DOI] [PubMed] [Google Scholar]
- 30.Hagenston A.M., Bading H. (2011) Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol., 3, a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ribar T.J., Rodriguiz R.M., Khiroug L., Wetsel W.C., Augustine G.J., Means A.R. (2000) Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J. Neurosci., 20, RC107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wayman G.A., Lee Y.-S., Tokumitsu H., Silva A., Soderling T.R. (2008) Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron, 59, 914–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kane C.D., Means A.R. (2000) Activation of orphan receptor-mediated transcription by Ca2+/calmodulin-dependent protein kinase IV. EMBO J., 19, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ermisch M., Firla B., Steinhilber D. (2011) Protein kinase A activates and phosphorylates RORα4 in vitro and takes part in RORα activation by CaMK-IV. Biochem. Biophys. Res. Commun., 408, 442–446. [DOI] [PubMed] [Google Scholar]
- 35.Ino H. (2004) Immunohistochemical characterization of the orphan nuclear receptor ROR alpha in the mouse nervous system. J. Histochem. Cytochem., 52, 311–323. [DOI] [PubMed] [Google Scholar]
- 36.Gold D.A., Gent P.M., Hamilton B.A. (2007) ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain. Res., 1140, 19–25. [DOI] [PubMed] [Google Scholar]
- 37.Chen X.R., Heck N., Lohof A.M., Rochefort C., Morel M.-P., Wehrlé R., Doulazmi M., Marty S., Cannaya V., Avci H.X., et al. (2013) Mature Purkinje cells require the retinoic acid-related orphan receptor-α (RORα) to maintain climbing fiber mono-innervation and other adult characteristics. J. Neurosci., 33, 9546–9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau P., Fitzsimmons R.L., Raichur S., Wang S.-C.M., Lechtken A., Muscat G.E.O. (2008) The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet-induced obesity. J. Biol. Chem., 283, 18411–18421. [DOI] [PubMed] [Google Scholar]
- 39.Hammer M.B., Eleuch-Fayache G., Schottlaender L.V., Nehdi H., Gibbs J.R., Arepalli S.K., Chong S.B., Hernandez D.G., Sailer A., Liu G., et al. (2013) Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet., 92, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin E., Schüle R., Smets K., Rastetter A., Boukhris A., Loureiro J.L., Gonzalez M.A., Mundwiller E., Deconinck T., Wessner M., et al. (2013) Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet., 92, 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platt F.M. (2014) Sphingolipid lysosomal storage disorders. Nature, 510, 68–75. [DOI] [PubMed] [Google Scholar]
- 42.Seedorf U., Raabe M., Ellinghaus P., Kannenberg F., Fobker M., Engel T., Denis S., Wouters F., Wirtz K.W.A., Wanders R.J.A., et al. (1998) Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein-2/sterol carrier protein-x gene function. Genes Dev., 12, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horinouchi K., Erlich S., Perl D.P., Ferlinz K., Bisgaier C.L., Sandhoff K., Desnick R.J., Stewart C.L., Schuchman E.H. (1995) Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat. Genet., 10, 288–293. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y., Hoffmann A., Grinberg A., Westphal H., McDonald M.P., Miller K.M., Crawley J.N., Sandhoff K., Suzuki K., Proia R.L. (1997) Mouse model of GM2 activator deficiency manifests cerebellar pathology and motor impairment. Proc. Natl. Acad. Sci. USA, 94, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao L., Spassieva S.D., Jucius T.J., Shultz L.D., Shick H.E., Macklin W.B., Hannun Y.A., Obeid L.M., Ackerman S.L. (2011) A deficiency of ceramide biosynthesis causes cerebellar Purkinje cell neurodegeneration and lipofuscin accumulation. PLoS Genet., 7, e1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen J.A., Halverson-Tamboli R.A., Rasenick M.M. (2007) Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci., 8, 128–140. [DOI] [PubMed] [Google Scholar]
- 47.Bieberich E. (2012) It's a lipid's world: bioactive lipid metabolism and signaling in neural stem cell differentiation. Neurochem. Res., 37, 1208–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jan Y.-N., Jan L.Y. (2010) Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci., 11, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou A.-H., Chen C.-Y., Chen S.-Y., Chen W.-J., Chen Y.-L., Weng Y.-S., Wang H.-L. (2010) Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem. Int., 56, 329–339. [DOI] [PubMed] [Google Scholar]
- 50.Nógrádi A., Jonsson N., Walker R., Caddy K., Carter N., Kelly C. (1997) Carbonic anhydrase II and carbonic anhydrase-related protein in the cerebellar cortex of normal and lurcher mice. Brain Res. Dev. Brain Res., 98, 91–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.