

Abstract
Increasing evidence suggests that formation and propagation of misfolded aggregates of 42-residue human amyloid β (Aβ(1–42)), rather than the more abundant Aβ(1–40), provokes the Alzheimer’s cascade. To date, structural details of misfolded Aβ(1–42) have remained elusive. Here we present the atomic model of Aβ(1–42) amyloid fibril based on solid-state NMR (SSNMR) data. It displays triple parallel-β-sheet segments that are different from reported structures of Aβ(1–40) fibrils. Remarkably, Aβ(1–40) is not compatible with the triple-β motif, as seeding with Aβ(1–42) fibrils does not promote conversion of monomeric Aβ(1–40) into fibrils via cross-replication. SSNMR experiments suggest that the Ala42 carboxyl terminus, absent in Aβ(1–40), forms a salt-bridge with Lys28 as a self-recognition molecular switch that excludes Aβ(1–40). The results provide insight into Aβ(1–42)-selective self-replicating amyloid propagation machinery in early-stage Alzheimer’s disease.
INTRODUCTION
Fatal neurodegenerative diseases like Alzheimer’s (AD) and prion diseases are linked to misfolding of disease-specific amyloidogenic proteins.1 These proteins misfold into toxic amyloid fibrils, which self-replicate in vitro and in vivo,1–4 acting as pathogenic seeds. Plaques formed by misfolded amyloid-β (Aβ) are a hallmark of AD. Since cytotoxicity is triggered by misfolding of Aβ, intensive efforts have focused on elucidating the structures of amyloid fibrils2,4–12 and other aggregates.1,13–17 Among the Aβ species present in AD, the 42-residue Aβ(1–42) is generally considered to be the most pathogenic species.18,19 The Aβ(1–42) exhibits notably higher toxicity and aggregation propensity than the more abundant 40-residue Aβ(1–40),20–22 even though the sequences differ only slightly. The Aβ(1–42) fibril is the initial and predominant constituent of amyloid plaques23–25 despite the higher plasma Aβ(1–40) level. Increased production of Aβ(1–42) relative to Aβ(1–40) has been reported for numerous pathogenic mutants of γ-secretase linked with early onset of AD.26 For the less aggregation-prone Aβ(1–40), a handful of high-resolution structural models have been proposed by SSNMR methods.4,7–9 Most of these structures are characterized by a U-shaped stand–loop–stand (β–loop–β) or “β-arch” motif,27 where two parallel β-sheets are connected by a short curved loop region (between residues Asp23 and Gly29), with many stabilized by a salt-bridge between Asp23 and Lys28 side-chains.4,7–9,28 In contrast, for the more pathogenic Aβ(1–42) fibril, the structural details are poorly defined despite intensive efforts.5,6,10,11,14,28,29 Due to its high misfolding propensity, Aβ(1–42) fibrils typically show structural and morphological heterogeneity,10,11 limiting subsequent analyses. There are only a few low-resolution or computational models for Aβ(1–42) amyloid fibrils, and experimental conformational details and tertiary structures remain elusive.5,10,11,28,29 Another key question in AD is the interaction between Aβ(1–42) and Aβ(1–40) amyloid states. A lower ratio of Aβ(1–42) to Aβ(1–40) in the patient’s plasma is a known indicator of AD,30,31 which presumably suggests depletion of soluble Aβ(1–42) by selective aggregation of Aβ(1–42) species. However, it has been unclear why misfolded Aβ(1–42) does not trigger misfolding of Aβ(1–40) via cross seeding at an early stage of Alzheimer’s. Beyond in-vitro kinetics studies32 and recent studies on mouse models,33 there has been no mechanistic or structural understanding of these prion-like cross-propagation properties between Aβ isoforms.
Here, we have elucidated the first atomic model, to our knowledge, for a structurally homogeneous Aβ(1–42) fibril based on SSNMR measurements, a powerful structural tool for amyloid and other non-crystalline proteins.2,34–37 The molecular-dynamic (MD) based structural modeling unveils distinctive structural features of the Aβ(1–42) fibril, which were not identified in previous studies of Aβ(1–40) fibrils. The results provide the first direct evidence that Aβ(1–42) can misfold into amyloid fibril along a different path from that of Aβ(1–40), indicating notable structural differences between misfolded Aβ(1–42) and Aβ(1–40) in AD. The structural features of the Aβ(1–42) fibril also provide insight into how tertiary folds of amyloid proteins can define prion-like cross-propagation properties in AD and other amyloid diseases through discrimination of similar amyloid proteins adopting alternative states.
RESULTS
Seeded Aβ(1–42) fibril displays structural homogeneity
We first established a protocol to prepare structurally homogenous amyloid fibril samples for Aβ(1–42) and observed the morphology of the Aβ(1–42) fibril sample using transmission electron microscopy (TEM) (Fig. 1a). The sample was prepared by incubating an Aβ(1–42) solution for 24 h with the addition of 5% (w/w) of seeded amyloid fibrils.2 Reproducible preparation of Aβ(1–42) fibril samples was made possible by careful optimization of the purification protocol, sample concentration and incubation times. The seeded fibrils in the fourth generation (G4) were obtained by repeating this protocol for three successive generations after an initial incubation without a seed (generation 1 or G1) (see Methods for details). The seeded fibrils showed elongated filament-like shapes with a diameter within 10 nm, with homogeneous morphology over the samples. Many of them appeared bundled together. We confirmed that samples collected after 24–72 h of incubation with the seeding protocol produce fibrils with nearly identical morphologies up to 13 generations.
Figure 1.
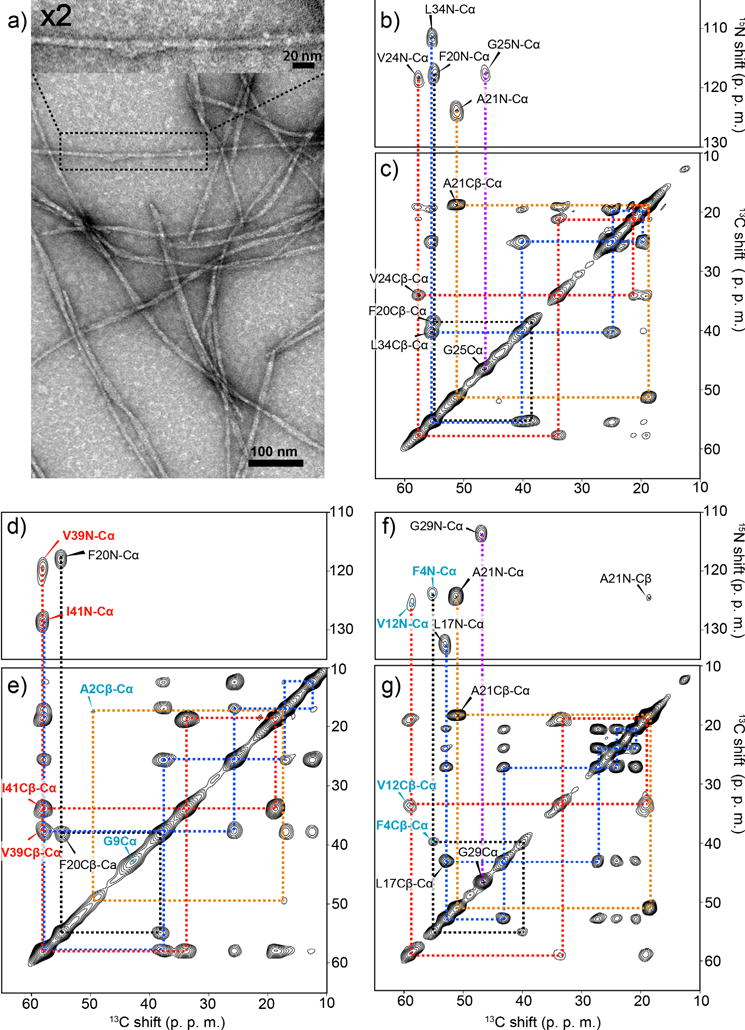
Structural homogeneity and morphologies analysis of Aβ(1–42) amyloid fibril. (a) Transmission electron microscopy (TEM) images of seeded Aβ(1–42) fibrils. The sample was obtained 24 h after the 4th generation (G4) incubation of an Aβ(1–42) solution with seed Aβ(1–42) fibrils (5% in weight). (b, d, f) 2D 15N–13C correlation SSNMR spectra and (c, e, g) 2D SSNMR 13C–13C correlation spectra of seeded fibril samples labeled with uniformly 13C-, 15N-labeled at (b, c) Phe20, Ala21, Val24, Gly25, Leu34, (d, e) Ala2, Gly9, Phe20, Val39, Ile41, and (f, g) Phe4, Val12, Leu17, Ala21, Gly29. In (b, d, f) 2D DARR spectra with a mixing time of 50 ms present single intra-residue cross peaks for each 13C-13C pair, indicating a single conformer. The base contour levels were set to 4–6 times the root-mean-square (RMS) noise level. The contour levels in the 2D 13C–13C correlation spectra were set to (b) 5%, (d) 7%, and (f) 10% of the diagonal signals of (b, f) 13Cα of Ala21 or (d) Ile41.
In order to examine atomic-level structures and heterogeneities, we performed SSNMR for 13C- and 15N-isotope labeled Aβ(1–42) in fibrils prepared according to the seeding protocol. By observing chemical shifts, which sensitively reflect conformations, site-specific structural heterogeneity can be monitored from the NMR spectra of the fibrils.38 We collected 2D 13C–15N chemical-shift correlation SSNMR spectra (Fig. 1b, d, f) and 2D 13C–13C SSNMR spectra (Fig. 1c, e, g) for three Aβ fibril samples in which uniformly 13C and 15N-labeled amino acids were introduced at several different residues (see the caption for labeling schemes). The data indicated the presence of a single conformer in the seeded fibril. For example, the spectra for Sample 1 (Fig. 1b, c) show a single set of cross peaks for all the directly bonded 13C–15N or 13C–13C pairs for Phe20, Ala21, Val24, Gly25, Leu34 except for a few very weak minor peaks. As chemical shifts are sensitive indicators of protein conformations, a single set of chemical shifts for each residue implies that Aβ(1–42) in the fibril had mostly a single conformer (see Table S1). Similar trends were observed for Sample 2 and Sample 3 (Fig. 1d–g), respectively. In contrast, Aβ(1–42) samples prepared without the seeding protocol exhibited two or more sets of cross peaks (Fig. S1a black), suggesting the presence of polymorphs.2,39,40 Neglecting the polymorphs in a structural analysis by H/D exchange solution NMR5,6 or other methods may result in a misleading structure. The homogeneous Aβ(1–42) fibril which we exploited for the structural analysis is equivalent to a pure Aβ(1–42) “amyloid strain”.4 Thus, the system can be also used as a model to study self-propagation and cross-propagation of Aβ(1–42) as discussed below.
Aβ(1–42) fibril forms a triple parallel β-sheet structure
Analysis of the signal intensities in the 13C SSNMR spectra offers information on dynamics and structural homogeneity as mobility and structural heterogeneity typically reduce signals in a SSNMR scheme using cross polarization.13,39 For the seeded fibril sample, we observed strong cross peaks for directly bonded 13C–13C and 13C–15N pairs for most of the inspected residues (residues 17–42), which include the hydrophobic core and the C-terminal region. For example, the two isotope-labeled residues at the C-terminus (Val39 and Ile41) showed single sets of strong cross peaks (red labels in Fig. 1d, e), indicating high structural order and lack of mobility at the C-terminus. It is also noteworthy that many of the cross peaks are weak or missing for the residues located at the N-terminal region at Ala2, Phe4, Gly9, and Val12 (cyan labels in Fig. 1e–g) and at His13 and His14 (data not shown). Thus, we only inspected a handful of residues in the N-terminus region in the analysis. These results establish an overall structural homogeneity of the obtained fibril sample with well-defined conformations at the hydrophobic core and C-terminus residues and dynamic N-terminus residues.
On the basis of the assigned 13C and 15N chemical shifts of the Aβ(1–42) fibril from Fig. 1 and other data (Table S1), the secondary structure analysis by TALOS-N software41 indicated the presence of three extended β-strand regions at Val12–Phe20, Asn27–Ile32, and Val36–Ile41 connected by two loop regions at Ala21–Ser26 and Gly33–Met35 (Figure S2a, b). Additionally, inter-strand 13CO–13CO distance measurement for Aβ fibril samples selectively labeled at 13CO of Ala30 and Leu34 indicated the CO–CO distances of 5.0 Å ± 0.1 Å at both residues (Figure S2c, d). The finding reveals a fibril made of three stretches of in-register parallel β-sheet regions. Although early SSNMR studies of Aβ(1–42) fibril also reported in-register parallel β-sheet formation,14,42 major structural differences between Aβ(1–42) and Aβ(1–40) fibril were not identified. In previous studies for in-vitro prepared Aβ(1–40) fibrils, the fibril structures were commonly characterized by a β-loop-β motif, where two stretches of parallel β-strands are connected with a single curved non-β-strand region near Asp23–Gly29.2,8,9 As will be discussed below, Aβ(1–40) fibril seeded with brain amyloid from atypical AD inherits a U-shaped β-arch motif,4 which is different from the motif of the Aβ(1–42) fibril. Thus, importantly, the triple-β motif indicated for the fibril structure of Aβ(1–42) is markedly different from those of Aβ(1–40).
S-shaped triple-β motif is stabilized of by a salt bridge
In order to elucidate the packing of the multiple β-strands in amyloid fibril, we examined long-range inter-residue contacts by 2D 13C dipolar-assisted-rotational-resonance (DARR)43 SSNMR experiments using an extended 13C–13C mixing period of 200 ms (red spectra in Fig. 2a–c) with an additional 13C–15N distance measurement (Fig. 2d), which will be discussed below. Multiple long-range inter-residues 13C–13C contacts within a distance of ∼6 Å were observed. Note that correlation only within residues or adjacent residues are observed with a shorter mixing time of 50 ms (black spectra in Fig. 2a–c) using the same mixing condition as in Fig. 1. Superimposed SSNMR spectra with 200 ms mixing and 50 ms mixing highlight long-range cross peaks between Phe19 or 20 side chains and other amino acids. The observed inter-residue contacts are as follows: Phe20–Ala21, Phe20–Val24 (Fig. 2a), Phe19–Ala30 (Fig. 2b, c), Phe19–Ile32 (Fig. 2b), and Phe19–Ile31 (Fig. 2c). We confirmed that these are intra-molecular contacts by experiments using isotope labeled Aβ mixed with unlabeled Aβ (Fig. S1b–d provides an example).
Figure 2.
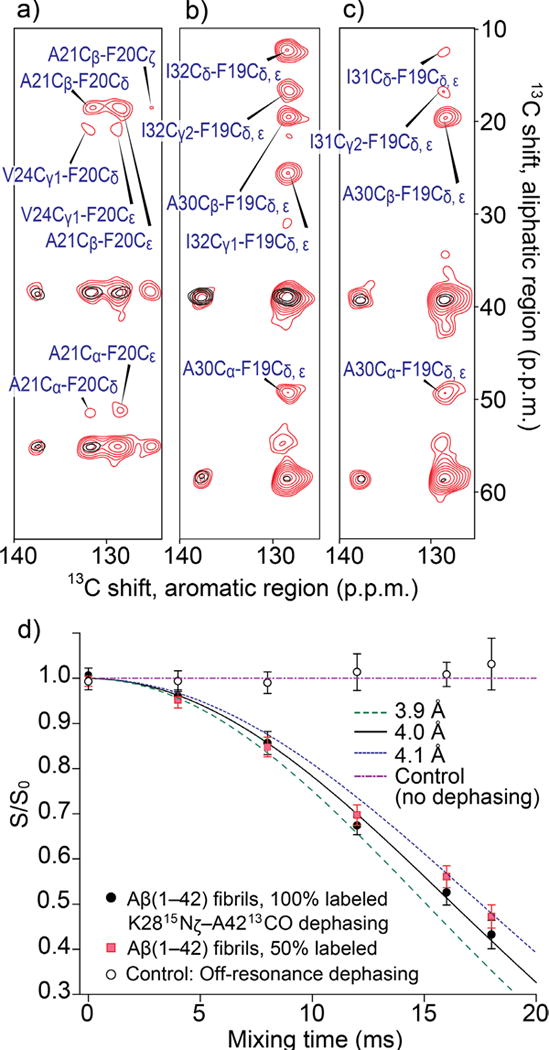
SSNMR-based structural constraints for the Aβ(1–42) fibril. (a–c) Superimposed aromatic-aliphatic cross peaks in 2D 13C–13C SSNMR spectra of the same fibril samples obtained with 200-ms (red) and 50-ms (black) mixing times. The observed inter-residue long-range contacts are (a) Phe20–Ala21, Phe20–Val24, (b) Phe19–Ala30, Phe19–Ile32, and (c) Phe19–Ala30, Phe19–Ile31. The samples were labeled with uniformly 13C-, 15N-labeled at (a) Phe20, Ala21, Val24, Gly25, Leu34, (b) Phe19, Ala30, Ile32, Gly38, Val40, and (c) Phe19, Ala30, Ile31, Gly33, and Val36. The base-contour levels were at 4–5 (red) and 6–8 times (black) the RMS-noise levels. (d) Dephasing curves by frequency-selective REDOR45 for measurement of the distance between Ala42 13CO and Lys28 15Nζ for a 100%-labeled sample (black filled circles) and a 50%-labeled sample obtained by mixing with unlabeled Aβ sample (red filled squares), in comparison with simulated dephasing curves obtained with Spinevolution software45 for 13C-15N distances of 3.9 Å (olive dashed line), 4.0 Å (black line) and 4.1 Å (blue dashed line). The best-fit data were obtained for the simulated result for 4.0 Å. The carrier frequency for the selective 15N pulse45 was set to 35 ppm near the Lys28 15Nζ resonance. Open black circles represent control experiments in which 15N was irradiated at off-resonance at 200 ppm. No dephasing was observed for the data, confirming that there were no effects due to 13CO and neighboring amide 15N groups. The errors bars were estimated from the noise level of the spectra.
Based on the chemical shifts, dihedral angles predicted from the 13C and 15N shifts (Table S1), and long-range distance restrains, we elucidated a multi-β-segment atomic model with the aid of molecular-dynamics (MD) simulations (Fig. 3a–c). The structural model (Fig. 3a) is characterized by S-shaped three β-strand regions that are connected by major coil- and turn-regions at residues 21–23 and 34–35; the results are largely consistent with the above mentioned secondary structure prediction. Moreover, we identified a novel contact between Lys28 and Ala42, as discussed below. The identified side-chain contacts (Fig. 3b) not only show good agreement with experimentally observed long-range distance restrains, but also explain unobserved long-range contacts for distances beyond 5 Å, which were also used as constraints (Table S2). The undetected contacts include those for Phe19–Leu34, Phe19–Val36, Phe19–Gly38, Phe19–Val40, and Asp23–Lys28, many of which were reported for Aβ(1–40) fibrils with similar β–loop–β motifs2,8 or Aβ(1–42) fibrils.5,14 The structure meets nearly all the structural restrains, including those for unobserved contacts, with a few minor violations (Table 1) and well reproduced the 13C and 15N chemical shifts by the ShiftX2 software44 (Table S4) at a level comparable to a previous study for the Het-s prion fibrils35 (see Method). More interestingly, our initial efforts of MD-optimized modeling suggested that with the SSNMR distance constraints, Lys28 cannot maintain a salt bridge with Asp23, which was observed for many of the models for Aβ(1–40) fibrils. Rather, a contact between Ala42 and Lys28 was suggested as shown in Fig. 3a, b. Thus, we performed an additional long-range distance measurement between the 13CO2− terminus of Ala42 and 15NH3+ side chain of Lys28 by monitoring 13C signal dephasing in frequency-selective rotational-echo-double-resonance (REDOR) experiments45 (Fig. 2d). The measured intra-molecular 13C–15N distance was 4.0 Å ± 0.1 Å, which suggests the formation of a unique salt bridge between Lys28 and Ala42. The distance was unaffected (4.1 ± 0.1 Å) in the same experiment for a sample in which labeled and unlabeled Aβ(1–42) samples were mixed in 1: 1 ratio. This confirmed that the salt bridge was formed primarily via an intra-molecular contact. From a separate long-range DARR experiment, we also observed contact between Gly29 and Ile41, which was assigned to intra- and inter-molecular contacts. With the intra-molecular contact between Lys28 and Ala42, we attributed the inter-molecular contacts to contacts of Gly29 with Ile41 from the neighboring Aβ chain, but did not include them for the structural calculations. The model shown in Fig. 3 was reoptmized from the preliminary model with the new restraints, including the contact between Lys28 and Ala42 (see Methods). The stabilization by this salt bridge between Lys28 and Ala42 explains why the unique S-shaped triple- or multi-β sheet motif is only observed for Aβ(1–42) fibrils. As Ala42 does not exist in Aβ(1–40), such a structure is not likely to be stable for Aβ(1–40). The structure also exhibits Gly29–Ile41 contacts. This evidence suggests the possibility that Aβ(1–42) constitutes a distinct amyloid strain, which has different propagation and structural properties from that of Aβ(1–40).
Figure 3.
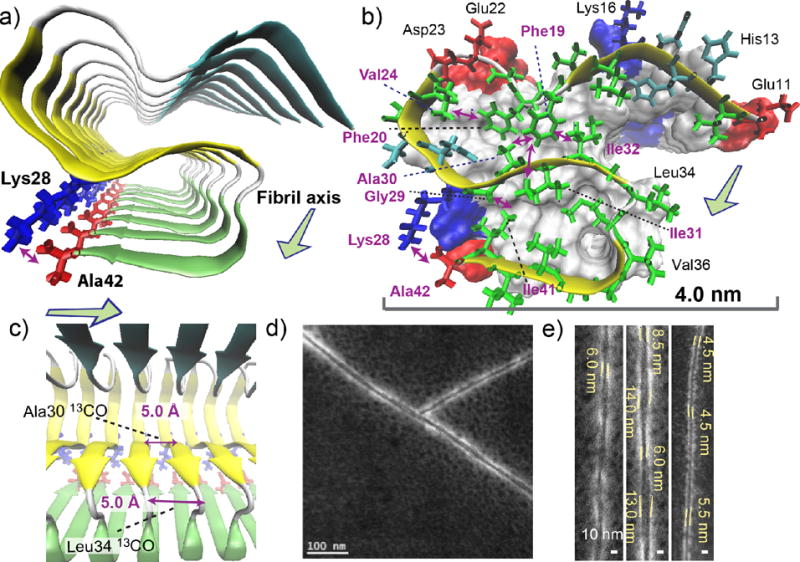
Structural details of the Aβ(1–42) fibril revealed by the SSNMR analysis. (a–c) A structural model of the amyloid fibril of Aβ(1–42). Disordered residues 1–10 were omitted for clarity. (a) View from the fibril axis shows three β-strand regions (arrows) connected by short coil (white) or turn (silver) regions (tube); the β-strands are represented by color-coded arrows in cyan (resides 12–18), yellow (24–33), and green (36–40). The unique salt bridge between Lys28 (blue) and Ala42 (red) is shown. (b) Side chain contacts for a single Aβ chain in a skeletal and a ribbon diagram with a van der Waals surface and polarity diagram for the rest of the Aβ chains. Hydrophobic, polar, acidic, and basic residues are represented by green, cyan, red, and blue, respectively. Observed long-range side-chain intra-molecular contacts (purple arrows) and inter-molecular contacts (blue arrow) are shown. All β-sheet regions are presented in yellow. The surface plot indicates positively charged (Lys; blue) and negatively charged (Glu, Asp; red) side chains, and Ala42 that has a negatively charged carboxyl group (red). (c) The side view in ribbon diagram. The in-register parallel β-sheet arrangement was confirmed by measurements of intermolecular 13CO-13CO distances of ∼4.8 Å at Ala30 and Leu34 (purple arrows). (d, e) Scanning TEM (STEM) images of seeded fibril filaments. (e) The diameters of the fibril filaments are ranged between 4.5 and 6.0 nm for thinner filaments (left and right) and between 6.0 and 14.0 nm for wider filaments (middle).
Table 1.
Solid-state NMR and refinement statistics for protein structures
| Best-fit model (Fig. 3) | Ensemble of 10 models (Fig. S3) | |
|---|---|---|
| NMR distance and dihedral constraintsa | ||
| Total constraints | 78 | 78 |
| Distance constraints | ||
| Total distance constraints | 40 | 40 |
| Intra-residueb | – | – |
| Inter-residue | 40 | 40 |
| Sequential (|i – j| = 1)b | ||
| Medium-range (2≤|i – j| ≤ 4) | – | – |
| Long-range (|i – j| ≥ 5) | 11 | 11 |
| Intermolecular | 2 | 2 |
| Hydrogen bonds | – | – |
| Unobserved long-range contact constraintsc | 27 | 27 |
| Total dihedral angle constraints | 38 | 38 |
| ϕ | 19 | 19 |
| ψ | 19 | 19 |
| Structure statistics | ||
| Violations (mean ± s.d.) | ||
| Distance constraints (Å) | 0.007 ± 0.050 | 0.011 ± 0.073 |
| Dihedral angle constraints (°) | 0.04 ± 0.13 | 0.05 ± 0.97 |
| Max. dihedral angle violation (°) | 0.90 | 67.63 |
| Max. distance constraint violation (Å) | 0.500 | 1.040 |
| Deviations from idealized geometry | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 2.10 | 2.10 |
| Average r.m.s. deviation from mean structure (Å)d | ||
| Heavy | N/A | 1.53 |
| Backbone | N/A | 1.08 |
Only includes experiment-based constraints per Aβ molecule. Constraints related to the molecular symmetry were excluded.
Intra-residue contacts were observed as listed in Supplementary Table 2, but not included in the structural calculations.
Only side-chain contacts were used for well ordered residues for which strong intra-residue cross peaks were observed.
The RMSD was calculated for the central 4 Aβ molecules in the 12-mer model.
The high-resolution negatively-stained scanning TEM (STEM) image (Fig. 3c, d) for fibrils gently washed with deionized water shows twisted single strands that exhibit a periodic modulation in diameter between 6 ± 1 nm and 13 ± 1 nm (Fig. 3d). We also observed thinner filaments that show a modulation approximately between 4.5 and 6.0 nm (Fig. 3d). The range agrees with the dimensions of the SSNMR-based structural model (Fig. 3b), which exhibits similar dimensions of 4.5 nm by 3.5 nm perpendicular to the fibril axis. An alternative model made of dimeric protofilament elements also explains well the morphological properties (data not shown), whereas the use of negative staining makes it difficult to elucidate the exact mass-per-length from the STEM data. The thicker filaments may be attributed to a hydrophobic assembly of multiple basic proto-filament units shown in Fig. 3b. Although further analysis by SSNMR and other complementary methods is needed to define the detailed protofilament arrangements of Aβ(1–42), the obtained atomic model reproduces well the morphological features of the amyloid fibril.
Aβ(1–42) fibril does not template Aβ(1–40) fibril formation
Previous in vitro kinetics studies and recent studies in mouse models suggested distinct propagation properties for Aβ(1–42) and Aβ(1–40) fibrils.32,33 However, these studies have utilized amyloid fibrils for which structural profiles and homogeneity were not well defined. More importantly, there has been no molecular-level mechanism that explains the differences in amyloid propagation of Aβ(1–40) and Aβ(1–42) fibrils, which mimic different amyloid strains. By taking advantage of the structurally homogeneous fibril of Aβ(1–42), which is equivalent to a pure Aβ(1–42) amyloid strain, we analyzed the propagation of amyloid formation from a “seed” Aβ(1–42) fibril to Aβ(1–40) fibril using Thioflavin T (ThT) fluorescence, which is an indicator of amyloid fibril formation. Incubation-time dependence of ThT fluorescence (Fig. 4) shows that fibril formation for a control sample containing only Aβ(1–40) monomer required a lag time of 13.0 h ± 0.1 h (black open circle in Fig. 4a, b) until the ThT fluorescence started to increase. This is explained by a multi-step misfolding mechanism in which monomeric Aβ requires time for conversion to fibril via oligomeric intermediate states.46 Substantially faster fibril growth was observed for another control experiment in which the Aβ(1–40) monomer sample was incubated with seed Aβ(1–40) fibril (Fig. 4a; black filled circle). The lag time became nearly zero when Aβ(1–40) fibril was added as “seed”. This is typically interpreted as evidence that monomers are directly converted to the fibril at the terminus of the seed fibril using the seed fibril as a template.2,46,47 Of particular interest is the fact that when the Aβ(1–42) fibril (G3 incubated for 3 days) was added as seed to an Aβ(1–40) monomer solution (Fig. 4b; red filled square), we found that the lag time (12.8 h ± 0.2 h) showed nearly no deviation from that for the control without any seeds. Our preliminary analysis showed that 2D 13C SSNMR spectra of Aβ(1–40) fibril sample prepared with and without Aβ(1–42) seed fibrils displayed little differences (data not shown). These results suggested that the fibril structure of Aβ(1–40) is not replicated from the cross-seeded Aβ(1–42) fibrils. Therefore, despite the high sequence similarity, monomeric Aβ(1–40) is incompatible with the distinct tertiary fold of the Aβ(1–42) fibril.
Figure 4.
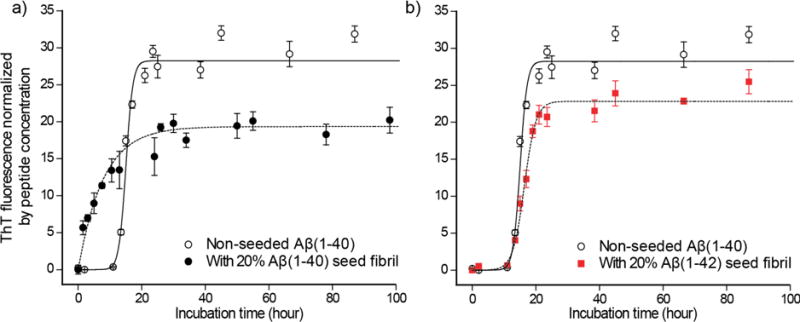
Cross-propagation kinetics of Aβ(1–40) monomers incubated with the seed Aβ(1–42) fibrils. Incubation-time dependence of ThT-fluorescence for 50 µM Aβ(1–40) solution incubated (a) with 10 µM Aβ(1–40) G1 seed fibrils (black filled circle) and (b) with 10 µM Aβ(1–42) G3 seed fibrils (red filled square) in comparison with (a, b) the control data for 50 µM Aβ(1–40) without any seed fibrils (black open circle). The identical control data are displayed in (a, b). The data for the Aβ(1–42)-seeded samples display very similar kinetic behaviors and lag times with those for the unseeded Aβ(1–40) solution. The fitting curves (dotted curves) using a sigmoidal equation46 (see Methods) respectively indicate lag times of 13.0 ± 0.1 h, and 12.8 ± 0.2 h for the unseeded and Aβ(1–42) seeded samples.46 The Aβ(1–40) seeded data show no lag time, and fits well with curve fitting using an equation that describes the first-order kinetic through a self-replicating reaction (see Methods). The error bars were estimated from the s.d. (n = 3).
DISCUSSION
In this work, we have established the first, to our knowledge, atomic structural model for structurally homogeneous Aβ(1–42) fibril samples, which have been hitherto unavailable. Despite the moderate resolution, the structure displays some remarkable features, which are summarized below with their biological significance. First, the Aβ(1–42) fibril structural model elucidated by this work shows a unique triple-β motif, which is made of three β-sheets encompassing residues 12–18 (β1), 24–33 (β2), and 36–40 (β3). The suggested structure is distinct from a β-loop-β motif, which commonly characterizes the reported high-resolution structural models of in-vitro Aβ(1–40) fibrils.2,7–9 This structure is also notably different from the recently reported structure of a brain seeded Aβ(1–40) fibril, which largely retains a U-shaped topology of the β-arch motif with a Asp23–Lys28 salt bridge, but involves greater non-β regions at residues 25–33 and 37–40.4 Our result clearly shows that despite the minimal sequence difference, Aβ(1–42) misfolds into fibril having a markedly different tertiary fold from those observed for Aβ(1–40) fibrils in the past studies (see Fig. S4). The formation of the Aβ(1–42)-specific amyloid fibril having a unique tertiary fold provides an innovative view in AD research, in which fibrils of Aβ(1–40) and Aβ(1–42) are often considered to be very similar. Second, we identified a salt bridge between Lys28 side chain and Ala42 carboxyl terminus in the Aβ(1–42) fibril structure. Major differences in the stabilizing interactions between Aβ(1–42) and Aβ(1–40) fibrils explain why Aβ(1–42) can misfold into fibrils in a distinct pathway from Aβ(1–40) while offering a mechanistic clue to early-stage misfolding of Aβ.48 Third, the obtained structural features explain well Aβ(1–42)-selective misfolding at an early AD stage and the lack of cross-propagation of Aβ(1–40) fibril from Aβ(1–42) fibril. Although recent developments made it possible to delineate the structures of Aβ(1–40) fibrils seeded from AD patients’ brains, no structural details have been provided even for synthetic Aβ(1–42) fibrils. This work suggests that cross-propagation barriers are likely caused by major tertiary structural differences between the Aβ(1–40) and Aβ(1–42) fibrils and the structural incompatibility of monomeric Aβ(1–40) and the Aβ(1–42) fibril, the latter of which utilizes Ala42 as a stabilizing salt-bridge contact. Such cross propagation behavior between slightly different amyloid proteins is considered to be critical in propagation of prion across different mammalian species.49 Indeed, recent studies showed that inoculation of synthetic Aβ(1–42) fibrils in mouse models prompted formation of plaque-like aggregates that were primarily comprised of Aβ(1–42) without involving Aβ(1–40) as major species.33 The present study has provided a stimulating initial example that explains how a tertiary fold of an amyloid fibril can be used as a self-recognition machinery and pose a structural barrier between amyloid or prion proteins even among those having high sequence similarity. Finally, it should be noted that Aβ is known to form various polymorphs as indicated in the present and previous studies.2,10,11 Indeed, some of the side-chain contacts, such as Phe19–Leu34, which were indicated in the previous SSNMR studies of Aβ(1–42) fibrils14 are missing in the present Aβ(1–42) fibril structures. Thus, this study represents only the first step toward revealing previously unknown structural details and structural variations of Aβ(1–42) fibrils, which are likely to be more relevant to the pathology of AD than well studied Aβ(1–40) fibrils.
In conclusion, the novel structural and kinetic features of Aβ(1–42) fibril achieved by the present study has offered a new perspective of how tertiary folds of amyloid fibrils critically influence amyloid propagation in AD and possibly in other neurodegenerative diseases. They also caution that drugs designed to optimally obstruct the Aβ(1–40) β-arch motif may not work as well against AD, which can be caused by the more toxic Aβ(1–42) fibrils having triple-β motif discovered here.
ONLINE METHODS
Sample preparation
Aβ(1–42) peptide (sequence DAEFR-HDSGY-EVHHQ-KLVFF-AEDVG-SNKGA-IIGLM-VGGVV-IA) was chemically synthesized by an Applied Biosystems (ABI) model 433A automated peptide synthesizer (Life Technologies, Carlsbad, CA) with Fmoc protected 13C- and 15N-labeled amino acids (Sigma Aldrich, St. Louis, MO, and Cambridge Isotope Laboratories, Andover, MA) at selected sites,13 and was purified by reversed-phase HPLC (Shimadzu Scientific Instruments, Columbia, MD), using an Agilent ZORBAX 300 Extend-C18 column.50 Fmoc protection of the labeled amino acids (Sigma-Aldrich, St. Louis, MO and Cambridge Isotope Laboratories, Andover, MA) was performed at the UIC Research Resource Center (RRC). Purity of the Aβ samples was determined to be approximately 85% and 95% before and after the HPLC purification, respectively, based on the mass analyses using an ABI 4700 MALDI TOF/TOF mass spectrometer at the UIC RRC. The lyophilized peptide after HPLC purification was weighted, and then completely dissolved at 2 mg/mL in an aquatic solution containing 30% acetonitrile (Fisher Scientific, Hanover Park, IL) and 0.1% of trifluoroacetic acid (TFA; American Bioanalytical, Natick, MA) at 4°C; the solution was subsequently lyophilized again. The lyophilized peptides were stored with drying reagents in a freezer at −20°C. Before each incubation, the peptide was warmed to room temperature and dissolved in hexafluoroisopropanol (HFIP) (Sigma-Aldrich) at a concentration of ∼2 mg/mL; after 1 h, the solution was subsequently lyophilized. This dissolution-lyophilization cycle was repeated twice following the previously published protocol.50
The HFIP-treated peptide was first dissolved in a 10 mM NaOH solution (Fisher Scientific) to 0.6 mM, and then the Aβ solution was diluted to 60 µM at pH 7.4 with a 10 mM phosphate buffer. The fresh Aβ(1–42) peptide solution was filtered by centrifugation using a 50-kDa molecular-mass-cutoff filter (EMD Millipore Amicon™ Ultra-15 filter with regenerated cellulose membrane, Hayward, CA) at 4.8 ×103 g for 3 min in order to remove any undissolved peptide or pre-formed aggregates. The final Aβ monomer concentration was typically ∼50 µM. It was confirmed by TEM analysis and ThT assay that no aggregated Aβ remains in the solution at the beginning of the incubation. The peptide solution was agitated by a continuous slow rotation at room temperature for 3 to 4 days. The generation-1 (G1) fibril sample was sonicated in an ice-water bath for 2 min, and then was seeded (5% w/w) to a newly prepared Aβ(1–42) solution that was dissolved and filtered as described above. The seeded solution (G2) was incubated for 3–4 days. Subsequently, Aβ(1–42) solution in generation n+1 (Gn+1) sample was seeded with 5% seed fibrils from generation n (Gn) and incubated for 3–4 days. The fibril morphology was monitored by TEM and STEM. As a result of optimization to achieve both improved structural homogeneity and experimental efficiency, 15N- and 13C-labeled Aβ fibril samples were typically harvested after incubation at G4 or at a later generation for 1 day to 1 week. The fibril samples were pelleted by centrifugation at 9,000 g for 45 min at 24°C, and subsequently lyophilized after removal of the supernatant. The lyophilized fibrils samples (5–10 mg) were packed into 2.5 mm SSNMR MAS rotors (10 μL volume) and subsequently rehydrated with ∼0.5 μL of water per mg of peptide. The samples used for the SSNMR analysis are listed in Table S4.
MALDI-TOF mass spectroscopy
The HPLC purified peptide, was dissolved in a 50% acetonitrile solution with 0.01% TFA (0.1 mg/10μL), and mixed (1:1 v/v) with a MALDI matrix solution (Sigma Aldrich) (5 mg in 200 μL of 70% acetonitrile solution with 2% TFA). The mixture of 0.5–1 μL was loaded onto a MALDI chip (model ABI 01-192–6-AB, Life Technologies) and air-dried before MALDI-TOF analysis. The peptides utilized in this study showed high purity (> 95%).
TEM analysis
Nano-scale morphologies of fibril samples was observed by TEM using JEOL 1220 (JEOL, Tokyo, Japan) operated at 80 kV and magnification of 120,000. For the grid preparation, 10 μL of a fibril sample, which was collected during 24–72 h of incubation time, was loaded on a 300 mesh copper formvar/carbon grid (Electron Microscopy Sciences, Hatfield, PA), and subsequently left for 1 min; then the excess solution was removed by blotting with a filter paper. The sample was negatively stained with a 10-μL solution of 2% (w/v) uranylacetate (Electron Microscopy Sciences) for 1.5 min. The grid was blotted and dried in air, and was then stored in a desiccating chamber before use.
STEM analysis
High-resolution STEM images were obtained using JEM-ARM200CF (JEOL, Tokyo, Japan), which was operated with an acceleration voltage of 80 kV at magnification of 400,000. For the grid preparation, 10 μL of a fibril sample, which was collected during 24–72 h of incubation time, was loaded on a 400 mesh copper carbon grid (Electron Microscopy Sciences), for 1 min, and then the excess solution was blotted away with filter paper. The sample was washed twice; each time, 5 μL of DDI water was loaded to the grid and then blotted away after 30 seconds. The sample was then fixed with 10–20 μL of 2% w/v glutaraldehyde (Sigma-Aldrich) solution for 30 min under a fume hood, and then the excess glutaraldehyde solution was blotted away and washed twice again. The fixed sample was negatively stained with 10 μL of 2% (w/v) uranylacetate solution for 2 min, and then blotted and dried in air before being stored in a desiccating chamber.
ThT fluorescence spectroscopy
Fluorescence measurements in the presence of ThT (Sigma-Aldrich) were performed on a Hitachi F-2000 fluorescence spectrometer with an excitation at 446 nm and an emission at 482 nm, as described previously.51 A 10-µL aliquot of an Aβ(1–42) fibril solution was diluted with 0.990 mL of 50 mM glycine buffer (Sigma-Aldrich) (pH 9.0), and the solution was then mixed with 10 µL of a 300 µM ThT solution. The final concentration of ThT was 3 µM. The curve fitting was performed by a χ2 analysis, and the error range for the lag time was estimated at the 90 % confidence level. Fitting of sigmoidal curves was performed using an equation of y(t) = a/[1 + exp(−k(t-t0))], where y(t) denotes the ThT fluorescence at the incubation time t, a and k are fitting parameters, and t0 defines a lag time of tL as tL = t0 −2/k.46 For the Aβ(1–40) seeded data, which showed no lag time, curve fitting was performed using an equation of y(t) = a[1 – exp(−kt)].
SSNMR spectroscopy
All the SSNMR experiments were performed at a 9.4 T magnetic field (1H frequency of 400.2 MHz) with MAS at 10–20 kHz, using Varian Infinity-Plus or Bruker Avance III SSNMR spectrometer with a home-built 1H, 13C, 15N triple-resonance 2.5-mm MAS probe. The sample temperature was ∼15°C at 20 kHz MAS. In 13C cross-polarization (CP) MAS experiments, the13C radio-frequency (RF) amplitude was swept from 49–66 kHz at the average of 57.5 kHz following a tangential shape while the 1H RF amplitude was kept constant at (57.5 + νR) kHz, where νR is the spinning speed. 13C signals were observed under 1H TPPM decoupling at 90 kHz with phase alternation of ±12.5° unless otherwise mentioned. The same 1H TPPM decoupling scheme was also employed during the 15N-13C and 13C-13C dephasing and mixing periods. Recycle delays were 2–3 s, unless otherwise specified. All assignments are listed in s. All the 1D and 2D data were processed by Bruker Topspin and NMRPipe software,52 respectively.
For the 2D 13C–13C correlation data in Fig. 1(c, e, g), a pulse sequence with a 50-ms DARR mixing43 was employed. During the 13C-13C mixing period, 1H RF field was applied with a constant strength matched to νR at 20 kHz. A total of 130 complex t1 points were recorded with a t1 increment of 50 μs. For each t1 point, 64–144 scans were accumulated with an acquisition period of 10.29 ms. The obtained NMR data were processed by NMRPipe software.52 The data were apodized with a Lorenz-to-Gauss window function with an inverse exponential narrowing (IEN) of 10 Hz and a Gaussian broadening (GB) of 130 Hz in the t2 domain, and with a Lorenz-to-Gauss window function with IEN of 50 Hz and GB of 100 Hz in the t1 domain. The overall experimental time was 12–24 hours.
For the long-range 2D 13C–13C correlation data in Fig. 2(a–c) and Fig. S2, the same pulse sequence was employed at a varied spinning speed of 12–14.5 kHz with a 200-ms DARR mixing, where a 1H RF field was matched to νR. A total of 120 complex t1 points were recorded with a t1 increment of 50 μs. For each t1 point, 64–144 scans were accumulated with an acquisition period of 10.29 ms. The data were apodized with a Lorenz-to-Gauss window function with IEN of 80 Hz and GB of 150 Hz in the t1 and t2 time domains. An overall experimental time was 24–48 hours. The contour levels in Fig. 2 for 200-ms mixing were set to (a) 11%, (b) 10%, and (c) 12% of the diagonal signals of (a) 13Cα of Ala21 or (b, c) Ala30, while those were set to (a) 5%, (b) 7%, and (c) 10% of the diagonal signals of (a) 13Cα of Ala21 or (b, c) Ala30 for 50-ms mixing.
To collect the 2D 13C–15N correlation data in Fig. 1(b, d, f), we monitored 15N chemical-shift evolution during the t1 period and detected 13C signals after CP from 15N to 13C spins at νR of 20 kHz. During the initial CP from 1H to 15N spins in a period of 1.5 ms, the 15N RF field strength was swept from 30 to 40 kHz while the 1H RF strength was kept constant at 55 kHz. During the 15N-13C CP period of 2.5 ms, an 15N RF-field strength was fixed at 15 kHz while a 13C RF strength was swept from 34.3 kHz to 45.7 kHz using adiabatic CP. A total of 80 complex t1 points were recorded with a t1 increment of 100 μs. For each t1 point, 64–144 scans were accumulated with an acquisition period of 5.17 ms. The data were apodized with a Lorenz-to-Gauss window function with IEN of 20 Hz and GB of 100 Hz in the t1 and t2 time domains. The overall experimental time was 24–48 hours each.
The frequency-selective 13C-15N REDOR experiments in Fig 2d were carried out at νR of 8,000 Hz ± 3 Hz using the pulse sequence in ref. 45 with minor modifications. A 15N π-pulse train with a XY-16 phase cycle53 was rotor-synchronously applied for a REDOR mixing with two 15N π-pulses in each rotor cycle; the 15N π-pulse width was 16.66 μs. For selective 13C-15N dipolar dephasing, selective inversion Gaussian pulses for 13CO2- and 15NH3 groups centered in the 1500-μs period were sandwiched by the two identical REDOR mixing sequences. The total time of the REDOR mixing was up to 18 ms. The pulse widths of the Gaussian π-pulses were 1250 µs and 500 µs for 15N and 13C, respectively. 1H TPPM decoupling with an RF field strength of 90–100 kHz was applied during the acquisition, REDOR mixing, and selective pulse periods. The details of 13CO-13CO inter-strand distance measurements by SSNMR are included in the supplementary information. Fitting of the NMR data for the 13C-15N or 13CO-13CO distance measurements to the best-fit simulated curve was confirmed by a χ2 analysis. The ranges of the uncertainty in the site-specific distance measurements were found to be within ±0.1 Å at the 90 % confidence level.
Structure Calculation and Analysis
In our preliminary MD-assisted structural modeling efforts, the peptide torsion angles were systematically changed to minimize the deviation of experimental chemical shifts and those calculated from the SHIFTX244 program. The stable structural models that meet NMR constransts have two unique features: (1) Phe19, Phe20, and Val24 are buried inside turn, and all charged residues from Glu22 to Lys28 are exposed to solvation; and (2) Lys28 forms salt bridge with the C-terminal of Ala42, which was confirmed by the subsequent REDOR measurement in Fig. 2d.
Using the preliminary models as a guide, a further two-step structural optimization was performed so that the final atomic model satisfies all the distance, dihedral-angle, and chemical-shift constraints from our SSNMR experiments. At the first stage, an ensemble of 1000 structures were generated with CYANA 2.1 program by adopting a similar approach used for Het-s prion fibril.35 The initial model of a 12-mer for the residues 11–42 of Aβ(1–42) was built as the first 10 residues were found to be flexible and likely disordered. Neighboring strands of the Aβ molecules were connected by virtual atom linkers, each of which was comprised of 210 residues in length. A list of upper limit restraints set to 6.5 Å were created from the long-range cross-peaks summarized in Table S2. Additionally, a list of lower limit restraints were generated for a pair of well structured residues (residues 17–42) for which no cross-peaks were identified in DARR experiments with a 200-ms mixing period (s). The lower-limit restraints were implemented at the Cβ atoms of non-glycine residues and set to 6.5 Å. Our REDOR experiment identified a unique contact between 13CO2− terminus of Ala42 and 15NH3+ side chain of Lys28; therefore, the distance constraints with the lower and upper limits were set to 3.9 and 4.1 Å, respectively. To elucidate likely dihedral angles (ϕ, ψ) from the obtained 13C and 15N chemial shifts, TALOS-N software41 was employed, and these dihedral angles were used as restraints only when the program determined the prediction as a consistent match to the data base (i.e. “Strong” or “Generous”). From intermolecular 13CO-13CO distance measurement results, we concluded that neighboring strands form in-register parallel β-strand throughout well structured residues 17–42. Thus, internuclear 13CO-13CO distance restraints were included at residues 20, 24, 30, and 34 as lower and upper restraints of 4.6 and 5.0 Å, respectively. The SSNMR spectra consistently showed a single set of resonances for each correlation observed. This indicates that the molecules are nearly identical between strands and are semi-crystalline in nature. In fact, fibrils are known to arrange with quasi-one-dimensional arrays along the fiber axis. We exploit this nature by imposing symmetry in terms of distance restraints between neighboring atoms (heavy-atom only) with lower-upper distance bounds of 4.7–5.1 Å. A list of the distance and dihedral structural restraints used in this study are given in Tables S1 and S2 and are weighted according to CYANA2.1 default values except for the distance restraint Lys28(Nζ)-Ala42(CO), which was increased by a factor of 5 to compensate for the lack of Coulombic interactions in CYANA. Moreover, all distances are considered ambiguous except where obvious such as non-Glycine CA positions. A total of 1000 structures were calculated within CYANA using the standard anneal.cya method included in the program with a slight modification. We modified the the annealing procedure by CYANA to include three rounds of high temperature annealing instead of one for each molecule as this provided structures that overall better satisfy experimental restraints. Of the 1000 structures, the set with 100 lowest target energies were retained for optimization at the next stage.
At the second stage, refinement by thermal annealing with AMBER12 was performed, as previously reported for globular proteins.54 All the distance and torsional restraints used in CYANA were transferred to AMBER 12. The CYANA structures were first energy minimized for a 1000 steps without experimental restraints. Then, thermal annealing was carried out with the structural restraints. For the distance, torsitional, chirality, angular (bond), and symmetry restraints, force constants were set to 10 kcal/(mole Å2), 540 kcal/(mole rad2), 100 kcal/(mole rad2), 40 kcal/(mole rad2), and 1 kcal/(mole Å2), respectively. The refinement process involved a total of three rounds of simulated annealing from 0 to 1000 K and then back to 0 K, regulated by a Berendsen thermostat; each round of the annealing process was implemented for a 20-ps period. The temperature ramping and restraint weighting were the same as those previously reported.54 A standard pairwise Generalized Born solvation55 was used with a cutoff of 12 angstroms. The time step of the MD simulation was set to 1 fs with a total of 60,000 steps or 60 ps for the three rounds of annealing. The last step involved a final energy minimization, including NMR restraints and implicit solvation, for 2,000 steps.
The structures from AMBER with the 20 lowest non-restraint energies were kept for structural analysis. Lastly, the SHIFTX244 program was used to further assess structural quality by back calculating the 13Cα, 13Cβ, 13CO, and amide 15N, chemical shifts from the determined structures and comparing these results to the experimentally measured shifts. The shift prediction was performed for each Aβ molecule and the ensemble average of the 13C or 15N was obtained for each site. The top 10 models that show the lowest root-mean-square deviations (RMSD) between the predicted and experimental shifts were selected as representative structural models (Table S5). For the best-fit model in Fig. 3, the average RMSD shift of 13Cα, 13Cβ, 13CO, and amide 15N is 1.29 ppm (Table S3), indicating reasonable fitting. The average RMSD value obtained for our model in Fig. 3 is comparable to the RMSD value of 1.38 ppm that was obtained from the amyloid-fibril structure for Het-s prion protein (pdb ID: 2RNM) and its experimental shifts (Table S6).35 Further discussion about the comparisons is given in supplementary material. Analysis by PROCKECK-NMR56 shows nearly all the residues for the fibril model to reside in allowed ϕ/ψ-space (Table S7). Distance and dihedral restraint violations were performed within PSVS and AMBER for the final structures (Table 1). The minimal number of violtations show that the structures are consistent with all the SSNMR structural constraints. Overlaid ensemble structures (Fig. S3) present that all the 10 models show very similar tertiary folds except for a few sidechains near the loop regions and the dynamic N-terminus residues 11–16. The obtained structures were diplayed by VMD 1.9.1 software using the secondary structures elucidated by STRIDE program.57
Supplementary Material
Acknowledgments
This work was supported primarily by the National Institutes of Heath RO1 program (GM 098033) and Alzheimer’s Association IIRG grant (08-91256) for YI. This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E for BM and RN. This research was supported in part by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research for BM and RN. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. MD simualtions to generate initial structural models were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the NIH, Bethesda, MD (http://biowulf.nih.gov). YI is grateful to Drs. S. Chimon, C. Jones, and N. Wickramasinghe for their initial efforts on the preparation of Aβ(1–42) fibrils at UIC.
Footnotes
Database accession numbers
The coordinate data is available at the Protein Data Bank (PDB ID: 2MXU).
Author contribution
Y.X and Y.I. designed the overall study and analyzed the data extensively. Y.X., S.P, F.L, M.H., and Y.I contributed to establishing sample preparation procedures. Y.X prepared the Aβ(1–42) fibril samples for the study with the help of S.P. and F.L. and with the advice of Y.I. Y.X and Y.I. performed SSNMR experiments. Y.X. performed electron microscopy experiments with staff assistance from the UIC RRC. Y.X. and F.L designed and performed kinetics experiments with ThT fluorescence spectroscopy. B.M., D.M, R.N., and Y.I. contributed to structural modeling and its design. Y.X, D.M., B.M., R.N., and Y.I wrote the paper. All the authors are involved in the editing of the manuscript.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 3.Stoehr J, et al. Purified and synthetic Alzheimer’s amyloid beta (A beta) prions. Proc Natl Acad Sci U S A. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu JX, et al. Molecular Structure of beta-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luhrs T, et al. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olofsson A, Sauer-Eriksson AE, Ohman A. The solvent protection of Alzheimer amyloid-beta-(1–42) fibrils as determined by solution NMR spectroscopy. J Biol Chem. 2006;281:477–483. doi: 10.1074/jbc.M508962200. [DOI] [PubMed] [Google Scholar]
- 7.Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A. A New Structural Model of A beta(40) Fibrils. J Am Chem Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 10.Masuda Y, et al. Identification of Physiological and Toxic Conformations in A beta 42 Aggregates. Chembiochem. 2009;10:287–295. doi: 10.1002/cbic.200800411. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt M, et al. Comparison of Alzheimer A beta(1–40) and A beta(1–42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez del Amo JM, et al. An Asymmetric Dimer as the Basic Subunit in Alzheimer’s Disease Amyloid beta Fibrils. Angew Chem Int Edit. 2012;51:6136–6139. doi: 10.1002/anie.201200965. [DOI] [PubMed] [Google Scholar]
- 13.Chimon S, et al. Evidence of fibril-like ß-sheet structures in neurotoxic amyloid intermediate for Alzheimer’s ß-amyloid. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed M, et al. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyung SJ, et al. Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-beta species. Proc Natl Acad Sci U S A. 2013;110:3743–3748. doi: 10.1073/pnas.1220326110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM. Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature. 2011;480:268–U161. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar B, et al. Significant Structural Differences between Transient Amyloid-beta Oligomers and Less-Toxic Fibrils in Regions Known To Harbor Familial Alzheimer’s Mutations. Angew Chem Int Edit. 2014;53:6888–6892. doi: 10.1002/anie.201402636. [DOI] [PubMed] [Google Scholar]
- 18.Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiological Reviews. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 19.Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 20.Davis J, VanNostrand WE. Enhanced pathologic properties of Dutch-type mutant amyloid beta-protein. Proc Natl Acad Sci U S A. 1996;93:2996–3000. doi: 10.1073/pnas.93.7.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murakami K, et al. Neurotoxicity and physicochemical properties of A beta mutant peptides from cerebral amyloid angiopathy – Implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J Biol Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 22.Luheshi LM, et al. Systematic in vivo analysis of the intrinsic determinants of amyloid beta pathogenicity. Plos Biology. 2007;5:2493–2500. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gravina SA, et al. Amyloid β protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43) J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 24.Roher AE, et al. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwatsubo T, et al. Visualization of A-beta-42(43) and A-beta-40 in senile plaques with end-specific A-beta monoclonals – evidence that an initially deposited species is A-beta-42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 26.De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease – Talking Point on the role of presenilin mutations in Alzheimer disease. Embo Reports. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajava AV, Baxa U, Steven AC. beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. Faseb Journal. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma BY, Nussinov R. Stabilities and conformations of Alzheimer’s beta-amyloid peptide oligomers (A beta(16–22′) A beta(16–35′) and A beta(10–35)): Sequence effects. Proc Natl Acad Sci U S A. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma B, Nussinov R. Polymorphic Triple beta-Sheet Structures Contribute to Amide Hydrogen/Deuterium (H/D) Exchange Protection in the Alzheimer Amyloid beta 42 Peptide. J Biol Chem. 2011;286:34244–34253. doi: 10.1074/jbc.M111.241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MMB. Plasma A beta(1–40) and A beta(1–42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–660. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 31.Graff-Radford NR, et al. Association of low plasma A beta 42/A beta 40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Archives of Neurology. 2007;64:354–362. doi: 10.1001/archneur.64.3.354. [DOI] [PubMed] [Google Scholar]
- 32.Pauwels K, et al. Structural Basis for Increased Toxicity of Pathological Aβ42:Aβ40 Ratios in Alzheimer Disease. J Biol Chem. 2012;287:5650–5660. doi: 10.1074/jbc.M111.264473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoehr J, et al. Distinct synthetic A beta prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci U S A. 2014;111:10329–10334. doi: 10.1073/pnas.1408968111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lange A, et al. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 35.Wasmer C, et al. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 36.Cady SD, et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–U127. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loquet A, et al. Atomic model of the type III secretion system needle. Nature. 2012;486:276–+. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spera S, Bax A. Empirical Correlation between Protein Backbone Conformation and C-Alpha and C-Beta C-13 Nuclear-Magnetic-Resonance Chemical-Shifts. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 39.Petkova A, et al. A structural model for Alzheimer’s b-amyloid peptide fibrils based on experimental constraints from solid-state NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Supramolecular structural constraints on Alzheimer’s beta-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 43.Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-driven C-13-C-13 recoupling without C-13 rf irradiation in nuclear magnetic resonance of rotating solids. J Chem Phys. 2003;118:2325–2341. [Google Scholar]
- 44.Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaroniec CP, Tounge BA, Herzfeld J, Griffin RG. Frequency selective heteronuclear dipolar recoupling in rotating solids: Accurate C-13-N-15 distance measurements in uniformly C-13,N-15-labeled peptides. J Am Chem Soc. 2001;123:3507–3519. doi: 10.1021/ja003266e. [DOI] [PubMed] [Google Scholar]
- 46.Nielsen L, et al. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 47.O’Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R. Thermodynamics of A beta(1–40) amyloid fibril elongation. Biochemistry. 2005;44:12709–12718. doi: 10.1021/bi050927h. [DOI] [PubMed] [Google Scholar]
- 48.Bernstein SL, et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nature Chemistry. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
Online Method References
- 50.Noguchi A, et al. Isolation and Characterization of Patient-derived, Toxic, High Mass Amyloid beta-Protein (A beta) Assembly from Alzheimer Disease Brains. J Biol Chem. 2009;284:32895–32905. doi: 10.1074/jbc.M109.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chimon S, Ishii Y. Capturing intermediate structures of Alzheimer’s b-amyloid, Ab(1–40), by solid-state NMR spectroscopy. J Am Chem Soc. 2005;127:13472–13473. doi: 10.1021/ja054039l. [DOI] [PubMed] [Google Scholar]
- 52.Delaglio F, et al. Nmrpipe – a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 53.Gullion T, Baker DB, Conradi MS. New, Compensated Carr-Purcell Sequences. J Magn Reson. 1990;89:479–484. [Google Scholar]
- 54.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci U S A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsui V, Case DA. Molecular dynamics simulations of nucleic acids with a generalized born solvation model. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- 56.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 57.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.