

Abstract
Rationale: Pulmonary arterial hypertension is characterized by endothelial dysfunction, impaired bone morphogenetic protein receptor 2 (BMPR2) signaling, and increased elastase activity. Synthetic elastase inhibitors reverse experimental pulmonary hypertension but cause hepatotoxicity in clinical studies. The endogenous elastase inhibitor elafin attenuates hypoxic pulmonary hypertension in mice, but its potential to improve endothelial function and BMPR2 signaling, and to reverse severe experimental pulmonary hypertension or vascular pathology in the human disease was unknown.
Objectives: To assess elafin-mediated regression of pulmonary vascular pathology in rats and in lung explants from patients with pulmonary hypertension. To determine if elafin amplifies BMPR2 signaling in pulmonary artery endothelial cells and to elucidate the underlying mechanism.
Methods: Rats with pulmonary hypertension induced by vascular endothelial growth factor receptor blockade and hypoxia (Sugen/hypoxia) as well as lung organ cultures from patients with pulmonary hypertension were used to assess elafin-mediated reversibility of pulmonary vascular disease. Pulmonary arterial endothelial cells from patients and control subjects were used to determine the efficacy and mechanism of elafin-mediated BMPR2 signaling.
Measurements and Main Results: In Sugen/hypoxia rats, elafin reduced elastase activity and reversed pulmonary hypertension, judged by regression of right ventricular systolic pressure and hypertrophy and pulmonary artery occlusive changes. Elafin improved endothelial function by increasing apelin, a BMPR2 target. Elafin induced apoptosis in human pulmonary arterial smooth muscle cells and decreased neointimal lesions in lung organ culture. In normal and patient pulmonary artery endothelial cells, elafin promoted angiogenesis by increasing pSMAD-dependent and -independent BMPR2 signaling. This was linked mechanistically to augmented interaction of BMPR2 with caveolin-1 via elafin-mediated stabilization of endothelial surface caveolin-1.
Conclusions: Elafin reverses obliterative changes in pulmonary arteries via elastase inhibition and caveolin-1–dependent amplification of BMPR2 signaling.
Keywords: neutrophil elastase inhibition, apelin, arterial smooth muscle cells, endothelial cell apoptosis, pulmonary vascular regeneration and angiogenesis
At a Glance Commentary
Scientific Knowledge on the Subject
Pulmonary arterial hypertension (PAH) is a debilitating disease with a poor prognosis. Contributors to the pathogenesis of PAH are impaired bone morphogenetic protein receptor 2 signaling and inflammation associated with increased elastase activity, promising targets of therapeutic intervention.
What This Study Adds to the Field
This report shows the efficacy of elafin, a naturally occurring elastase inhibitor, in reversing severe experimentally induced pulmonary hypertension associated with an obliterative vasculopathy. We show for the first time that a prospective therapy can induce regression of the neointima in cultured explants of human pulmonary arteries from patients with PAH. In addition to elastase inhibition, elafin mediates caveolin-dependent amplification of bone morphogenetic protein receptor 2 signaling to promote restoration of endothelial function and vascular regeneration as well as regression of occlusive lesions.
Pulmonary arterial hypertension (PAH) is characterized by loss of distal pulmonary arteries (DPAs) and by neointimal formation causing narrowing and occlusion of preacinar and intraacinar pulmonary arteries (PA). Altered mitochondrial function and chronic inflammation, increased elastase activity, proliferation of smooth muscle cells (SMCs) and fibroblasts, and endothelial cell (EC) dysregulation have all been implicated in the pathobiology of this adverse remodeling process (for review, see Reference 1). Pulmonary vascular cell function is disturbed by mutations in the bone morphogenetic protein receptor (BMPR) 2, found in more than 70% of patients with familial PAH (FPAH), and in approximately 15% of those with nonfamilial idiopathic PAH (IPAH). BMPR2 mutations are associated with more severe pulmonary vascular pathology (2). Moreover, reduced BMPR2 expression and function is present in patients with PAH without mutations (3). Aberrant BMPR2 signaling is associated with an increase in PA endothelial cell (PAEC) apoptosis (4, 5) and with enhanced pulmonary arterial smooth muscle cell (PASMC) proliferation in response to growth factors (6). The BMPR2 signal is transduced via phosphorylation and nuclear translocation of SMAD1/5, and multiple other signaling pathways (for review, see Reference 7). Restoring BMPR2 signaling improves PAEC survival and normal angiogenesis, and also represses proliferation and induces apoptosis of SMCs, at least in part via apelin, a secreted product of BMPR2-mediated gene regulation (8, 9).
The interaction of BMP receptors with caveolin-1 (Cav1) has important implications for downstream signaling of BMPR2. Cav1 is highly expressed in ECs and is the major constitutive protein of caveolae, the flask-shaped plasma membrane invaginations. Cav1 also interacts with and regulates numerous ion channels and membrane-bound enzymes, notably endothelial nitric oxide synthase (eNOS) (for review, see Reference 10). Progressive loss of Cav1 is observed with PAEC damage in rats that acquire pulmonary hypertension (PH) after injection of monocrotaline (11) and administration of Cav1 peptides reverses PH (12). Cav1 knockout mice have PH associated with pulmonary vascular abnormalities, including loss of DPAs (13). In patients with PAH, Cav1 is reduced in remodeled PAs and is barely detectable in plexiform lesions (14). Mutations in Cav1 have been reported in patients with FPAH and IPAH (15).
Synthetic elastase inhibitors were used to prevent and reverse PH in the monocrotaline rat model (16). Clinical use of these compounds was not pursued because of hepatotoxicity, but studies using an endogenous elastase inhibitor, elafin, prevented experimentally induced PH (17) and other cardiovascular pathologies (18, 19). Here we report the efficacy of daily subcutaneous injections of elafin in reversing severe PH in the Sugen/hypoxia (Su/Hx) rat model (20). Elafin-induced regression of occlusive lesions and increase in DPA number was associated with heightened production of apelin (8). In lung explants from patients with PAH elafin caused regression of the PA neointima by inducing apoptosis. Elafin improved survival and angiogenesis in cultured human PAH-PAECs. The mechanism was linked to enhanced BMPR2 signaling by promoting the interaction of BMPR2 with Cav1.
Methods
An expanded Methods section is available in the online supplement.
Studies in Su/Hx Rat PH Model
We used the Su/Hx rat model of PH (20). Recombinant human elafin, 0.2 mg/kg, dissolved in 0.9% saline vehicle, or an equivalent volume of vehicle was administered for 2 weeks by daily subcutaneous injections in the dorsal cervical area. Echocardiographic measurements of cardiac function and closed chest assessments of right ventricular systolic pressure (RVSP) and left ventricular end diastolic pressure were obtained as previously described (21). We assessed RV hypertrophy by the ratio of the weight of the RV to that of the left ventricle (LV) and septum (S). The number of DPAs at alveolar wall and duct level per 100 alveoli, the number of alveoli per square millimeter, and the percentage of obliterated to total DPAs were evaluated using morphometric techniques (21).
Elastase Assay
Lung tissue was snap frozen in liquid N2 and stored at −80°C for measurement of serine elastase activity by the DQ-elastin substrate assay (22).
Reverse Transcriptase Quantitative Polymerase Chain Reaction
Total RNA was extracted and purified from whole-lung lysates using the RNeasy Mini kit (Qiagen, Valencia, CA). Primer sequences were designed using the National Center for Biotechnology Information’s Primer-BLAST function (see Table E1 in the online supplement).
Lung Organ Culture
Tissue sections, containing PAs, were isolated from the explanted lungs of patients with PAH who had undergone lung transplantation. Mirror image sections were placed in organ culture and treated with either elafin in H2O or an equal volume of H2O vehicle once daily for 8 days. Movat staining was used to measure lumen diameter/external diameter, lumen area/vessel area, and media area/vessel area (23). Apoptosis (% apoptotic cells per artery) was assessed by the TUNEL assay.
PAEC and SMC Cultures
Primary human PAECs were obtained commercially or harvested from explanted lungs of patients undergoing transplantation for IPAH or FPAH, or from unused donor control subjects, obtained through the Pulmonary Hypertension Breakthrough Initiative. Cells were used at passages 3–8. PASMCs were obtained commercially. siRNA oligonucleotides are listed in Table E1.
PAEC Function Assays
Tube formation
PAECs were cultured in 5% fetal bovine serum and seeded in growth factor reduced Matrigel (Cultrex BME; Trevigen Inc., Gaithersburg, MD) (21). Vehicle, BMP9, and/or elafin were added to media containing no fetal bovine serum. The number of tubes formed was assessed after 8 hours.
Survival and proliferation
For caspase 3/7 apoptosis assays, cells were grown to 80–90% confluence. Then under serum-free conditions, BMP9 and/or elafin were added, and after 12 hours luminescence was measured in a plate reader. To assess cell proliferation, cell counts and formazan assays were used.
Western Immunoblotting
Western immunoblotting was performed as previously described (8, 24). Antibodies are listed in Table E2.
Immunoprecipitation
PAEC lysates were incubated with the IP antibody (see Table E2). The final eluate was analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis followed by Western immunoblotting.
Confocal Microscopy
One hour after adding vehicle, BMP9, and/or elafin, PAECs were fixed with 2% paraformaldehyde and treated with an antibody against Cav1, followed by Alexa Fluor 488–conjugated secondary antibody. Sections were mounted and stained with 4′,6-diamidino-2-phenylindole to visualize nuclei and Cav1 distribution.
Results
Elafin Reverses PH and Inhibits Elastase Activity
We used 8-week-old male Sprague Dawley rats (180–220 g) injected by subcutaneous route with a single 20 mg/kg dose of a vascular endothelial growth factor receptor 2 blocker (Sugen 5416), followed by hypoxia (10% O2) for 3 weeks, and normoxia for an additional 3 weeks, to elicit severe PH (Su/Hx) (25). At the 6-week time point, Su/Hx rats were divided into three groups: one had no intervention, and two had 2 weeks of daily subcutaneous injections of either saline (vehicle) or elafin, 0.2 mg/kg. A preliminary dose–response test showed that an elafin dose of 0.2 mg/kg afforded optimal suppression of elevated lung elastase activity in Su/Hx rats with PH. Untreated age-matched control animals were kept in room air throughout the study period. Figure 1A shows a schematic overview of the experimental protocol.
Figure 1.

Elafin improves right ventricular hemodynamic function in Sugen/hypoxia (Su/Hx) rats. (A) Protocol of animal experiments. (B) Pulmonary artery acceleration time (PAAT) and (C) cardiac output (CO) measured by echocardiography before and after treatment. Bars represent mean ± SEM for n = 6 for normoxia-treated (Nx), untreated (Untr), and 0.9% saline vehicle–treated (Veh) rats; n = 9 for elafin-treated rats (E). **P < 0.01 versus Veh or Untr (Day 14), ##P < 0.01 and ####P < 0.0001 versus Nx, §P < 0.05 and §§P < 0.01 versus elafin pretreatment, by one- or two-way analysis of variance and Bonferroni post hoc test. s.c = subcutaneously.
Before initiation of treatment at Week 6, all rats exposed to the Su/Hx protocol showed signs of severe PH compared with control rats maintained in room air, as affirmed by echocardiographic evidence of reduced PA acceleration time and cardiac output (Figures 1B and 1C). Repeat measurements after 2 weeks showed sustained reduction of PA acceleration time and cardiac output in the untreated or vehicle-treated Su/Hx rats, whereas those that received elafin displayed near normal cardiac output and improved PA acceleration time. Consistent with the echocardiographic measurements, elafin-treated Su/Hx rats had a marked reduction in RVSP and RV hypertrophy compared with untreated or vehicle-treated rats (Figures 2A and 2B). We found no evidence of elevated systemic (carotid artery) blood pressure or left ventricular end diastolic pressure in any of the treatment groups (see Figures E1A and E1B). As expected, the increased lung elastase activity seen in untreated and vehicle-treated Su/Hx rats relative to room air control animals was inhibited in the elafin-treated Su/Hx group (Figure 2C). The room air elafin rats were indistinguishable from room air control animals, as assessed by RVSP, RV/(LV + S), echocardiographic determination of cardiac function (see Figures E1C–E1F), or hematologic and blood chemistry results (see Table E3).
Figure 2.

Elafin reverses pulmonary hypertension, heightened elastase activity, and obstructed distal pulmonary arteries, and increases vessel number and lung expression of endothelial nitric oxide synthase (eNOS) and apelin in Sugen/hypoxia (Su/Hx) rats. Rats were exposed to room air only (normoxia [Nx]; n = 6) or to the Su/Hx protocol. The Su/Hx rats were then divided into three groups: no intervention (Untr; n = 6), 0.9% saline vehicle treatment (Veh; n = 6), or elafin treatment (E; n = 9). (A) Right ventricular systolic pressure (RVSP). (B) Right ventricular hypertrophy, defined by the Fulton index, weight of right ventricle/(left ventricle + septum). (C) Elastase activity was measured in whole-lung lysate by the DQ elastin assay. (D) Representative histology of distal pulmonary arteries (DPA) from Nx, Su/Hx (Untr, Veh, and E) rats; scale bars = 50 μm. (E) Number of occluded alveolar duct and wall DPA. (F) Representative immunofluorescence photomicrographs of DPA from rats treated as described previously and stained for nuclei (4′,6-diamidino-2-phenylindole [DAPI]; blue), von Willebrand factor (vWF; green), and α-smooth muscle actin (SMA; red). Scale bars = 50 μm. Note thinning of arterial wall in elafin-treated versus -untreated Su/Hx and vehicle-treated. Room air control is used for comparison. (G) The number of DPA (vessels) per 100 alveoli. Whole-lung mRNA levels of (H) apelin and (I) eNOS were assessed in Nx and Su/Hx (Untr, Veh, and E) rats. Bars represent mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus Veh, #P < 0.05 and ####P < 0.0001 versus Nx, by one-way analysis of variance and Bonferroni post hoc test. LV = left ventricle; RLU = relative light units; RV = right ventricle; S = septum.
Elafin Reverses Adverse Structural Remodeling in DPAs
The percentage of occluded vessels was reduced from more than 60% in lungs of untreated and vehicle-treated Su/Hx groups to less than 20% of vessels in the rats given elafin (Figures 2D and 2E). Healthy control rats exhibit little or no muscularization of DPAs, as assessed by α-smooth muscle actin staining; in contrast, there was marked medial hypertrophy of DPAs in the untreated and vehicle-treated Su/Hx rats, which elafin treatment mitigated (Figure 2F). Elafin treatment also increased the number of DPAs relative to those of both untreated and vehicle-treated Su/Hx groups (Figure 2G).
PH in the Su/Hx rat model is initiated by disrupting EC homeostasis in the pulmonary circulation after vascular endothelial growth factor receptor blockade. This perturbation increases susceptibility of PAECs to apoptosis during hypoxia (20). Regression of vascular obliterative changes and the increase in DPA number in elafin-treated rats was associated with an elevation in whole-lung mRNA expression of apelin and its target eNOS (26). These features seem to reflect new or regenerating arteries (Figures 2G–2I) (27).
Elafin Induces Regression of Neointima in PAH Lung Organ Cultures
Although the Su/Hx rat model shows obliterative changes in DPAs, it does not recapitulate the extensive occlusive neointima formation observed in larger PAs in patients with PAH (2). Our previous studies have shown that rat PAs can be maintained in organ culture as explants with preserved viability for 8 days, and that administration of synthetic elastase inhibitors (23) or epidermal growth factor receptor blockers (28) can result in regression of PA hypertrophy. We therefore adapted this technique to assess lung explants using tissue removed at the time of lung transplantation from six consecutive patients: two with IPAH, one with PAH associated with pulmonary capillary hemangiomatosis, two with PAH related to a congenital heart defect, and one with PAH related to drug and toxin ingestion (Table 1).
Table 1.
Characteristics of Patients with PAH: Organ Culture Experiments
| ID | Age (yr) | Sex | PAH Diagnosis | PAP (s/d/m) (mm Hg) | PVR (WU) | 6MWD (m) | PAH Medications |
|---|---|---|---|---|---|---|---|
| OC-1 | 28 | F | IPAH | NA/NA/50 | 11.8 | NA | Epoprostenol, bosentan |
| OC-2 | 35 | F | CHD | NA | NA | 164 | NA |
| OC-3 | 26 | M | CHD | NA/NA/105 | 18.5 | 185 | Epoprostenol |
| OC-4 | 39 | F | IPAH | NA/NA/55 | 15.4 | NA | Bosentan |
| OC-5 | 26 | F | PCH | NA/NA/73 | NA | 513 | Epoprostenol, bosentan, sildenafil |
| OC-6 | 63 | F | Drug and toxin | NA/NA/62 | 17.1 | 212 | Epoprostenol, bosentan, sildenafil |
Definition of abbreviations: 6MWD = distance walked in 6 minutes; CHD = congenital heart disease; ID = identification; IPAH = idiopathic pulmonary arterial hypertension; NA = data not available; OC = organ culture; PAH = pulmonary arterial hypertension; PAP (s/d/m) = pulmonary artery pressure (systolic/diastolic/mean); PCH = pulmonary capillary hemangiomatosis; PVR = pulmonary vascular resistance (baseline Fick PVR); WU = Wood units.
Hemodynamic data were obtained from catheterization study performed closest to transplantation.
In some of the organ culture studies there is only limited clinical information in our records because these patients were referred before 2006 and not in our database. For example, patient OC-2, a patient with Eisenmenger syndrome, was referred from Mexico.
PAH medications are listed according to total drug exposure during treatment period of follow-up, not necessarily in combination.
Daily administration of elafin for 8 days resulted in a reduction in PA neointima size as judged by an increase in lumen-to-vessel area and lumen-to-vessel diameter (Figures 3A–3D). Regression of the neointima was attributed to apoptosis of cells as assessed by TUNEL assay (Figure 3E). Most cells in the neointima express α-smooth muscle actin, a marker of SMC. However, inflammatory cells, myofibroblasts, or other stromal cells also could contribute to the neointimal apoptotic population. The mechanism of elafin-mediated neointimal cell apoptosis could be related to induction of apelin in PAECs. Apelin can, by a paracrine effect, cause apoptosis of PASMCs, attributed in part to miR-424 and miR-503 repression of fibroblast growth factor 2 (8, 9). However, elafin can also directly induce apoptosis of PASMC and suppress their proliferation in response to growth factors (Figure 3F).
Figure 3.

Elafin induces regression of neointima in cultured lung explants of patients with pulmonary arterial hypertension (PAH), induces apoptosis, and inhibits platelet-derived growth factor (PDGF)–induced smooth muscle cell proliferation. Sections from cultured lung explants taken from six patients with PAH were treated with either vehicle (H2O) or elafin (E; 0.5 μg/ml) for 8 days. (A) Representative photomicrographs of Movat-stained sections of pulmonary arteries (PAs) from two patients. (B) Ratios of lumen to vessel area, (C) lumen to vessel diameter, and (D) media to vessel area based on analysis of 15 vessels per lung section for each treatment (scale bar = 100 μm). (E) On the left, representative photomicrographs of PA immunohistochemistry for TUNEL-stained cell nuclei from vehicle- and elafin-treated PAs (note the abundant immunoperoxidase-positive nuclei stained in the neointima [I] and in the media [M] and adventitia [A] of the PAH vessel on the right). On the right, quantitative assessment of percentage of TUNEL-positive cells per total cells per artery (n = 6 for each condition). +P < 0.05 by unpaired t test. Bars represent mean ± SEM. Scale bar = 50 μm. (F) Proliferation assay by cell count of cultured control PA smooth muscle cells (PASMCs) 24 hours after treatment with saline vehicle (Veh), PDGF (20 ng/ml), E (0.5 μg/ml), or a combination of PDGF and E. Bars represent mean ± SEM for n = 6, *P < 0.05, and ****P < 0.0001 versus Veh, ####P < 0.001 versus PDGF by one-way analysis of variance and Bonferroni post hoc test.
Elafin Improves Angiogenesis and Enhances BMP Function in PAH-PAECs
We next used tube formation assays to investigate the mechanism by which elafin improved PAEC function in Su/Hx rats, as inferred from increased levels of lung apelin, a target of BMPR2 signaling in PAECs and a proangiogenic factor (8). Although elafin did not increase tube formation in control PAECs, elafin mitigated the defective angiogenesis seen in IPAH-PAECs (Figures 4A and 4B). We attribute this response to improved survival of IPAH-PAECs because elafin did not increase their proliferation (see Figures E2A and E2B).
Figure 4.

Elafin improves angiogenic properties and survival in idiopathic pulmonary arterial hypertension (IPAH) pulmonary artery endothelial cells (PAECs). (A) Representative inverted (black on white) photomicrographs of tube formation assay in donor control and IPAH patient PAECs. Scale bars = 150 μm. (B) Tube number was assessed 8 hours after treatment with 0.9% saline vehicle (Veh) or elafin (E) (1 μg/ml). Bars represent mean ± SEM for n = 4; +P < 0.05 by unpaired t test. (C and D) Survival was assessed by a decrease in apoptosis evaluated by the caspase-3/7 assay 12 hours after serum starvation and treatment with Veh, bone morphogenetic protein 9 (B9; 10 ng/ml), E (1 μg/ml), or a combination of B9 + E in (C) control PAECs and (D) IPAH-PAECs. Bars represent mean ± SEM of n = 4; *P < 0.05, **P < 0.01, ****P < 0.0001 versus Veh, and #P < 0.05 versus B9, by one-way analysis of variance and Bonferroni post hoc test. RLU = relative light units.
To investigate whether elafin improves PAEC survival via BMPR2 signaling, we compared the survival of PAECs from control subjects and patients with IPAH (Tables 2 and 3) after serum withdrawal in the presence of the BMPR2 ligand BMP9, elafin, or both. In control PAECs, BMP9 enhanced PAEC survival, judged by reduced caspase 3/7 activity. Elafin alone did not enhance survival in control PAECs, but when combined with BMP9, there was a synergistic prosurvival effect (Figure 4C). In IPAH-PAECs, elafin and BMP9 independently increased survival, and the effect of the combination was additive (Figure 4D).
Table 2.
Characteristics of Patients with PAH: Cell Culture Experiments
| ID | Assay | Age (yr) | Sex | Race/Ethnicity | Diagnosis | BMPR2 Mutation | PAP (s/d/m) (mm Hg) | PVR (WU) | 6MWD (m) | PAH Medications |
|---|---|---|---|---|---|---|---|---|---|---|
| PAH-01 | Caspase WB | 27 | F | White non-Hispanic | IPAH | c.76 + 5G>GA probable mutation, not in dbSNP; may disrupt splicing | 110/49/69 | 12.11 | 420.6 | Sildenafil, tresprostinil, bosentan, iloprost |
| PAH-02 | Caspase WB | 33 | F | White non-Hispanic | FPAH | c.961C>T nonsense, exon 7 (kinase domain) | 87/29/48 | 9.74 | 288 | Bosentan, treprostinil, sildenafil, epoprostenol |
| PAH-03 | Caspase WB | 33 | F | Black/AA non-Hispanic | FPAH | c.1471C>T missense, exon 11 (kinase domain) | 75/33/48 | 15.57 | 326.1 | Epoprostenol, bosentan, sildenafil, treprostinil |
| PAH-04 | WB | 37 | M | White non-Hispanic | FPAH | c.1471C>T missense, exon 11 (kinase domain) | 119/51/77 | 14.22 | 309 | Sildenafil, sitaxsentan, ambrisentan, epoprostenol, imatinib (investigational drug) |
| PAH-05 | Caspase WB | 54 | F | White non-Hispanic | IPAH | NA | 100/45/60 | 11.96 | 296.3 | Sildenafil, epoprostenol, ambrisentan, bosentan |
| PAH-06 | Caspase WB | 27 | M | White non-Hispanic | IPAH | No | 90/51/68 | 11.38 | 423.7 | Bosentan, sildenafil, epoprostenol |
| PAH-07 | Caspase WB | 56 | M | White non-Hispanic | IPAH | No | 125/50/75 | 9.58 | 234.7 | Tadalafil, bosentan |
| PAH-08 | Caspase Matrigel WB | 32 | F | White non-Hispanic | IPAH | No | 68/38/49 | 15.34 | 238 | Bosentan, epoprostenol |
| PAH-09 | Matrigel WB | 40 | F | White non-Hispanic | IPAH | NA | 105/63/55 | 9.84 | 472.4 | Sildenafil, bosentan |
| PAH-10 | Matrigel WB | 56 | F | White non-Hispanic | FPAH | No | 110/55/75 | NA | 372.2 | Epoprostenol, bosentan, ambrisentan, sildenafil |
| PAH-11 | Matrigel | 55 | F | Black/AA non-Hispanic | IPAH | No | 89/41/53 | 12.29 | 273.4 | Sildenafil, bosentan, epoprostenol |
Definition of abbreviations: 6MWD = distance walked in 6 minutes; AA = African American; BMPR2 = bone morphogenetic protein receptor 2; dbSNP = Single Nucleotide Polymorphism database; FPAH = familial pulmonary arterial hypertension; ID = identification; IPAH = idiopathic pulmonary arterial hypertension; NA = data not available; PAH = pulmonary arterial hypertension; PAP (s/d/m) = pulmonary artery pressure (systolic/diastolic/mean); PVR = pulmonary vascular resistance (baseline Fick PVR); WB = Western immunoblot; WU = Wood units.
Hemodynamic data were obtained from catheterization study performed closest to transplantation.
PAH medications are listed according to total drug exposure during treatment period of follow-up, not necessarily in combination.
Table 3.
Characteristics of Control Subjects (Unused Donor Lungs): Cell Culture Experiments
| ID | Assay | Age (yr) | Sex | Race/Ethnicity | Cause of Death |
|---|---|---|---|---|---|
| CON-01 | Caspase | 11 | M | White non-Hispanic | Hypoxic brain injury |
| CON-02 | Matrigel caspase | 45 | F | White non-Hispanic | CVA/subarachnoid hemorrhage |
| CON-03 | Matrigel caspase | 52 | F | White unknown | Hypoxic brain injury |
| CON-04 | Matrigel | 24 | M | White non-Hispanic | Intracerebral hemorrhage |
| CON-05 | WB | 57 | F | White non-Hispanic | Anoxia |
Definition of abbreviations: CON = control subject; CVA = cerebrovascular accident; ID = identification; WB = Western immunoblot.
Hemodynamic data were obtained from catheterization study performed closest to transplantation.
Elafin Enhances BMP Signaling in Healthy and PAH-PAECs via BMPR2
To further investigate how elafin augments the signaling properties of BMP ligands, we performed Western immunoblotting to assess a canonical downstream molecule, pSMAD1/5, and the factor it activates, inhibitor of DNA binding 1(ID1). Elafin alone did not increase pSMAD1/5 or ID1 in control PAECs but augmented induction of this signaling pathway by BMP9 (Figure 5A). A similar result was apparent in IPAH-PAECs (Figure 5B), including those with known BMPR2 mutations (Figure 5C), although the magnitude of enhanced signaling was reduced. We also stimulated PAECs from control subjects, IPAH, and BMPR2 mutant patients with PAH with BMP7 or BMP4. These ligands, when compared with BMP9, produced more intense activation of pERK42/44 (not shown). We found that elafin augmented pERK42/44 in PAECs from control subjects and patients with IPAH (Figures 5D and 5E), including those with a BMPR2 mutation (Figure 5F), albeit to a lesser extent in the latter group. Elafin did not increase levels of BMPR2 in any group (see Figure E3).
Figure 5.

Elafin enhances bone morphogenetic protein (BMP) signaling in pulmonary artery endothelial cells (PAECs). (A–C) Representative Western immunoblots with relative densitometric analyses showing pSMAD1/5 relative to SMAD1/5 and ID1 relative to β-actin, after stimulation with 0.9% saline vehicle (Veh), BMP-9 (B9; 10 ng/ml), elafin (E; 1 μg/ml), or a combination of B9 + E in (A) control PAECs, (B) idiopathic pulmonary arterial hypertension (IPAH)-PAECs, and (C) PAH-PAECs from patients with a bone morphogenetic protein receptor 2 (BMPR2) mutation. (D–F) Representative Western immunoblots and relative densitometry of pERK42/44 relative to β-actin after stimulation of PAECs from (D) control PAECs, (E) IPAH-PAECs, (F) PAH-PAECs from patients with a BMPR2 mutation, with Veh, B4 or B7 (10 ng/ml), E (1 μg/ml), or B4 or B7 + E. Bars represent mean ± SEM of n = 3. +P < 0.01, ++P < 0.01, ++++P < 0.0001 by unpaired t test where only signals from two conditions were detectable; *P < 0.05, **P < 0.01, ****P < 0.0001 versus Veh, #P < 0.05 versus B9, B4, or B7, by one-way analysis of variance and Bonferroni post hoc test. ERK = extracellular signal–regulated kinase; ID1 = inhibitor of DNA binding 1; pSMAD = phosphorylated mothers against decapentaplegic homolog 7.
To determine if reduced BMP signaling in the BMPR2 mutant cells might relate to lower levels of BMPR2, we repeated the pSMAD1/5-ID1 signaling experiments and measured BMPR2 protein levels (see Figure E3). PAECs from donor control subjects and patients with IPAH showed a similar increase in pSMAD1/5 when elafin was added to BMP9 compared with BMP9 alone, despite lower expression of BMPR2 in the IPAH-PAECs. However, in the PAECs from patients with IPAH bearing the BMPR2 mutation, BMPR2 levels were even lower than in the IPAH-PAECs, and in these cells, the combination of BMP9 and elafin failed to elicit a significant increase in pSMAD1/5 over BMP9 alone (see Figure E3). IPAH-PAECS also showed an increase in ID1 in response to BMP9 that was greater when elafin was added but this additive effect was also absent in the patients with a BMPR2 mutation. Because later passage cells were used in these experiments compared with those in Figure 5, it is possible that BMPR2 levels progressively decline in culture accounting for the absence of response in those with a BMPR2 mutation.
To determine whether there might be a threshold below which elafin would not yield a synergistic effect when combined with BMP ligands, and to determine the specificity of the response, we reduced levels of BMPR2 by more than 80% using siRNA (see Figures E4A and E4B). PAECs transfected with BMPR2 siRNA showed increased pSMAD1/5 and ID1 when exposed to BMP9 alone, although the response was considerably less than in those transfected with control siRNA. Elafin amplified pSMAD1/5 and ID1 in response to BMP9 in the control siRNA transfected cells but not in the BMPR2 siRNA transfected cells (see Figure E4A). Similarly, we found a synergistic amplification of pERK42/44 by BMP7 and elafin in control PAECs, but not in BMPR2 siRNA transfected cells (see Figure E4B). The failure of elafin to amplify BMPR2 signaling in PAECs with reduced BMPR2 by siRNA was also reflected in failure to improve survival of these cells in response to BMP9 (see Figure E4C). Taken together, our data indicate that elafin’s amplifying effect on BMP signaling and survival of PAECs is specific for BMPR2 and may require a threshold amount of this receptor.
To determine whether another elastase inhibitor could mimic elafin-mediated augmentation of BMPR2 signaling, we assessed the response to α1-antitrypsin. We found no increase in pSMAD1/5 or in ID1, nor was there improved cell survival compared with BMP9 alone with three different doses of α1-antitrypsin (see Figure E5).
Enhanced BMP Signaling by Elafin Is Cav1 Dependent
Cav1 directly interacts with BMPR2, and increases BMP9-dependent SMAD1/5 phosphorylation and induction of ID1 (29). To determine whether elafin might be enhancing BMPR2 signaling and PAEC survival via Cav1, we transfected cultured PAECs with either Cav1 siRNA or nontargeting control siRNA. We induced a reduction of more than 50% in Cav1 protein with the Cav1 siRNA (Figure 6A), and under these conditions elafin did not enhance BMPR2 signaling, as assessed by Western immunoblotting for pSMAD/1/5, ID1, and pERK42/44 (Figures 6A and 6B). Consistent with these findings, loss of Cav1 prevented the additive effect of elafin on BMP9-mediated PAEC survival (Figure 6C).
Figure 6.
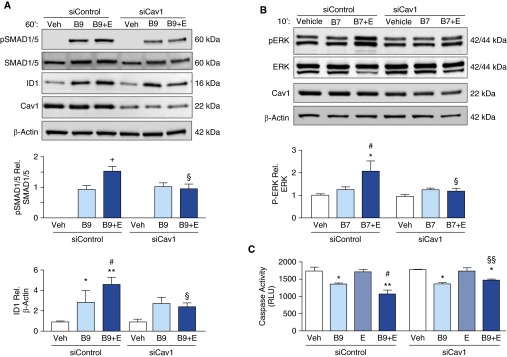
Elafin amplifies bone morphogenetic protein (BMP) signaling and enhances pulmonary artery endothelial cell (PAEC) survival via caveolin-1 (Cav1). (A and B) Representative Western immunoblot of commercially available PAECs treated with nontargeting siRNA (siControl) or siRNA specific for Cav1 (siCav1) with densitometric analysis showing (A) pSMAD1/5 relative to SMAD1/5, and ID1 relative to β-actin and (B) pERK42/44 relative to ERK42/44 after stimulation with 0.9% saline vehicle (Veh), BMP9 (B9; 10 ng/ml), or BMP7 (B7; 10 ng/ml), ±elafin (E; 1 μg/ml). (C) Representative caspase-3/7 assays in PAECs treated with siControl or siCav1, 12 hours after serum starvation and stimulation with Veh, B9, E, or B9 + E at the concentrations indicated previously. Bars represent mean ± SEM of n = 4. +P < 0.05 by unpaired t test; *P < 0.05, **P < 0.01 versus Veh and #P < 0.05 versus B9 or B7 by one-way analysis of variance and Bonferroni post hoc test; §P < 0.05, §§P < 0.01 versus siControl, B9 + E or B7 + E, by two-way analysis of variance and Bonferroni post hoc test. ERK = extracellular signal–regulated kinase; ID1 = inhibitor of DNA binding 1; pSMAD = phosphorylated mothers against decapentaplegic homolog 7; RLU = relative light units.
Caveolae affect multiple cellular signaling pathways by redistribution and endocytosis of transmembrane receptors and receptor–ligand complexes (29–31). BMPR2 undergoes caveolae-dependent internalization, and a disrupted interaction between Cav1 and BMPR2 resulted in receptor mislocalization and aberrant signaling (29). We therefore assessed, by confocal immunofluorescence microscopy, the effect of BMP9 and BMP9 plus elafin on Cav1 distribution in PAECs. Under nonstimulated conditions, Cav1 was localized predominantly in a linear pattern along the plasma membrane, but BMP9 stimulation induced a redistribution of Cav1 to an intracytoplasmic vesicular pattern consistent with internalization. Elafin treatment alone had no effect on Cav1 distribution. However, when elafin was combined with BMP9 there was enhanced localization of Cav1 on PAEC surfaces (Figures 7A and 7B).
Figure 7.

Elafin enhances the caveolin-1 (Cav1)–bone morphogenetic protein receptor 2 (BMPR2) interaction and stabilizes Cav1 at the cell surface. (A and B) Representative confocal immunofluorescence images and quantification of Cav1 organization in pulmonary artery endothelial cells stained for Cav1 (green) and nuclei (4′,6-diamidino-2-phenylindole, blue) treated with 0.9% saline vehicle (Veh), BMP-9 (B9; 10 ng/ml), elafin (E; 1 μg/ml), or a combination of B9 + E for 60 minutes. Scale bar = 50 μm. Solid arrows, cell surface Cav1; dashed arrows, cytoplasmic Cav1. Bars represent mean ± SEM for n = 100 nuclei per condition. *P < 0.05 versus Veh, and #P < 0.05 versus B9 by one-way analysis of variance and Bonferroni post hoc test. (C and D) Representative immunoprecipitation with an antibody to (C) Cav1 or to (D) BMPR2 and immunoblots for BMPR2, Cav1, and β-actin from pulmonary artery endothelial cell lysates treated with Veh or E for 30 minutes. Bars represent mean ± SEM for n = 3 for Cav1 IP. ++P < 0.01 versus vehicle by unpaired t test. Immunoprecipitation for BMPR2 in D is representative of two experiments with similar results.
To investigate whether elafin enhances BMP signaling by increasing the interaction of Cav1 and BMPR2, we performed coimmunoprecipitation studies with both BMPR2 and Cav1 antibodies. Elafin stimulation had no effect on the total amount of Cav1 or BMPR2 proteins in the cell lysates, but it enhanced the interaction between BMPR2 and Cav1 (Figures 7C and 7D).
Discussion
This study demonstrates that elafin can reverse PH by a dual mechanism of action. In addition to elastase inhibition, we describe a novel property whereby elafin facilitates an interaction between BMPR2 and Cav1 that reduces internalization of the complex and intensifies BMPR2 signaling. This is associated with improved PAEC survival and angiogenesis, evident in cultured human PAH-PAECs and in the increase in DPAs in the Su/Hx rat model. Improved BMPR2 signaling by elafin can also be related to regression of the obliterative pulmonary vasculopathy in the Su/Hx rats and of neointima formation in human PAH lung organ cultures (see schema in Figures 8A and 8B).
Figure 8.
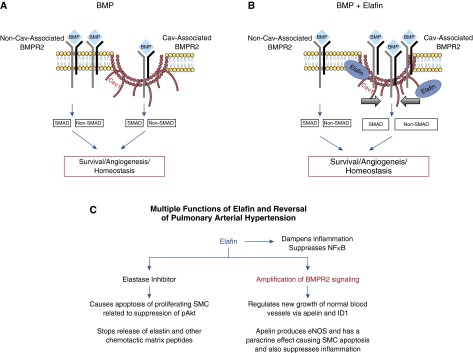
Proposed model of the effect of elafin on bone morphogenetic protein receptor (BMPR) 2 signaling and multiple functions of elafin. (A) BMP signaling can be initiated in caveolin-1 (Cav1)-dependent and Cav1-independent compartments, resulting in SMAD1/5 (canonical) and non-SMAD1/5 (noncanonical) downstream signaling. (B) Elafin leads to stabilization of Cav1 on the cell surface (thick arrows), and this causes increased BMPR2 recruitment into the Cav1 compartment, resulting in enhanced BMP signaling and improved cell survival and angiogenesis. (C) A schema of the multiple functions of elafin related to reversal of pulmonary arterial hypertension, with the novel function of amplification of BMPR2 signaling in red. eNOS = endothelial nitric oxide synthase; ID1 = inhibitor of DNA binding 1; NFκB = nuclear factor-κB; SMAD = mothers against decapentaplegic homolog 7; SMC = smooth muscle cell.
Increased elastase activity and fragmented PA elastin are early features of PAH in humans and in animal models of PH (32, 33). Elastase activity is related to progressive pulmonary vascular pathology by degradation and remodeling of the extracellular matrix, resulting in release of SMC mitogens (34) and activation of their receptors (35). Elastin peptides and other products of elastase-mediated matrix breakdown are potent chemoattractant molecules associated with inflammation and SMC proliferation (36).
PAEC dysfunction related to leak of serum and release of endothelial factors into the subendothelium can stimulate PASMC elastase activity and proliferation (37). Elastase inhibition reverses muscularization of DPAs in the monocrotaline rat model (16) and attenuates neointima formation in S100A4 overexpressing mice infected with murine gamma herpes virus (22). We speculate that in the Su/Hx rat the initial PAEC injury triggers PASMC activation and subsequent release of elastase identified as neutrophil elastase (22). The early inflammatory component of the Su/Hx model (38) may be important in perpetuating activity of elastase and perhaps other proteases, notably proteinase-3, which is also inhibited by elafin (39). Because there is no significant inflammatory infiltrate in the lungs of the Su/Hx rats after 6 weeks (20), SMCs could be the source of the sustained elevation in elastase activity. Because elafin represses nuclear factor-κB (40) and modulates innate immunity (41), it might be of particular benefit to patients with PAH in whom chronic inflammation contributes to adverse pulmonary vascular remodeling (for review, see Reference 42).
Because we observed increased DPAs linked to elevated levels of apelin and its target eNOS (26), elafin seemed to promote regeneration of DPAs by augmenting endothelial function. Apelin-null mice show increased PA pressures compared with wild-type mice (26) and apelin administration reversed PH in mice with conditional deletion of PPARγ in ECs (8). Patients with PAH compared with control subjects have decreased plasma levels of apelin, and apelin is reduced in cultured PAECs from their lungs. Because apelin is an EC target downstream of BMPR2, we investigated the role of elafin in enhancing BMPR2 signaling as a mechanism to promote PAEC survival and PASMC apoptosis.
BMPR2 heterodimeric complexes are localized in clathrin-coated pits (43), whereas ligand-inducible BMPR complexes are located in caveolae (30) and bind BMP ligands with high affinity. Cav1-BMPR2 interaction facilitates pSMAD signaling (29, 30). Our finding that elafin stabilizes Cav1 on PAEC surfaces suggests that it may enhance BMP signaling by delaying internalization and degradation of the BMPR2-coreceptor complexes, as observed with other receptors (44). This would be consistent with studies indicating that elafin is a proteasome inhibitor (40). It is therefore also possible that elafin increases ID1 (45) by preventing its degradation by the ubiquitin/26S proteasome pathway (46).
As a cholesterol binding protein, Cav1 is preferentially localized in membrane microdomains enriched with cholesterol and sphingolipids. The polycationic nature of elafin allows it to permeate, and accumulate within the phospholipid bilayer (47). In this hydrophobic environment the N-terminus of elafin forms an α-helix that can serve as a membrane anchor (48). These biochemical properties of elafin may enable it to interact with Cav1 directly or through an intermediary molecule. Development of PH in the Cav1 knock-out mouse was attributed to uncoupling of eNOS in pulmonary ECs (13). Oxidative stress in Su/Hx rats has also been linked to eNOS uncoupling (49). Elafin might facilitate interaction of Cav1 with eNOS, and in so doing reduce oxidative stress.
Amplifying BMPR2 function is a desirable treatment strategy for patients with PAH carrying a BMPR2 mutation and also for patients without a known mutation in whom BMPR2 expression or function is reduced (3). Activation of the BMPR2 pathway by either gene delivery (50) or pharmacologic compounds (23) can prevent or induce regression of pulmonary vascular changes in animal models of PH and can reverse abnormalities in cultured vascular cells from patients with PAH (23).
Elafin given by single intravenous dose is in clinical trial abroad as a preventive strategy for other indications, and our study indicates that it could reverse established disease when administered daily by the subcutaneous route. We demonstrate the impact of elafin in reversing obliterative pulmonary vascular remodeling in rats and for the first time, the efficacy of any agent in the regression of human PA neointimal vascular lesions as assessed in lung organ culture. In so doing, we provide a compelling rationale to purse elafin as a PAH treatment.
Acknowledgments
Acknowledgment
The authors greatly appreciate the editorial and technical assistance of Dr. Michal Bental Roof in preparing the manuscript and figures, and the administrative help of Ms. Michelle Fox. Lung tissues from pulmonary arterial hypertension and control patients were provided by the Pulmonary Hypertension Breakthrough Initiative, which is funded by the NIH/NHLBI and the Cardiovascular Medical Research and Education Fund. The tissues were procured at the Transplant Procurement Centers at Baylor, Stanford, Cleveland Clinic, and Vanderbilt, and deidentified patient data were obtained via the Data Coordinating Center at the University of Michigan. Elafin was a kind gift from Prof. Oliver Wiedow of the University of Kiel and Proteo Biotech, facilitated by Birge Bargmann and Dr. Barbara Kahlke of Proteo.
Footnotes
Supported by an NIH-NHLBI Translational Program Project Grant 5P01HL108797 (M.R., M.R.N., R.D.B., and R.T.Z.). N.P.N. (Ni 1456/1-1) and J.K.H. (He 6855/1-1) were supported by grants from Deutsche Forschungsgemeinschaft, and I.D. by Deutsche Herzstiftung e.V. (S/06/11). E.S. is currently supported by the NHLBI (1K08HL107450-01), M.G. by an AHA postdoctoral fellowship, and K.M. by a fellowship from the Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad.
Author Contributions: N.P.N. performed and designed rat and pulmonary artery endothelial cell experiments, data analysis, and write-up. E.S. and R.W.S. performed organ culture. M.G. performed all experiments with bone morphogenetic protein receptor 2 siRNA and bone morphogenetic protein receptor 2 levels in patients related to signaling. K.-Y.K. helped with rat and pulmonary artery endothelial cells. K.M., L.W., and A.C. helped with hemodynamics, pulmonary artery endothelial cell harvest, and culture. M.K. helped with morphometry. S.S. helped with cell culture and proliferation assays. C.G.L. harvested animal tissues, designed IP experiments including antibody validation, and helped with confocal imaging. H.L. helped with IP. I.D. helped with morphometry and echocardiography. J.K.H. helped with morphometry and IP. X.J., M.R.N., R.T.Z., and R.D.B. provided conceptual and editing input. M.R. oversaw design and analysis of all experiments, and revised and edited manuscript.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201412-2291OC on April 8, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 4.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184:83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98:209–217. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 6.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 8.Alastalo TP, Li M, Perez VdeJ, Pham D, Sawada H, Wang JK, Koskenvuo M, Wang L, Freeman BA, Chang HY, et al. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. 2011;121:3735–3746. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. 2013;19:74–82. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012;2:120. doi: 10.3389/fphys.2011.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Wolk JH, Gewitz MH, Mathew R. Caveolin-1 expression during the progression of pulmonary hypertension. Exp Biol Med (Maywood) 2012;237:956–965. doi: 10.1258/ebm.2012.011382. [DOI] [PubMed] [Google Scholar]
- 12.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–920. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- 13.Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, Minshall RD. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1-/- mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–L873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129:696–705. doi: 10.1378/chest.129.3.696. [DOI] [PubMed] [Google Scholar]
- 15.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, III, Palomero T, Sumazin P, Kim HR, Talati MH, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 17.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105:516–521. doi: 10.1161/hc0402.102866. [DOI] [PubMed] [Google Scholar]
- 18.Zaidi SHE, Hui C-C, Cheah AYL, You X-M, Husain M, Rabinovitch M. Targeted overexpression of elafin protects mice against cardiac dysfunction and mortality following viral myocarditis. J Clin Invest. 1999;103:1211–1219. doi: 10.1172/JCI5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowan B, Baron O, Crack J, Coulber C, Wilson GJ, Rabinovitch M. Elafin, a serine elastase inhibitor, attenuates post-cardiac transplant coronary arteriopathy and reduces myocardial necrosis in rabbits afer heterotopic cardiac transplantation. J Clin Invest. 1996;97:2452–2468. doi: 10.1172/JCI118692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 21.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. doi: 10.1172/JCI65592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, Acharya S, Rodriguez-Colon G, Orton A, Zhao M, et al. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol. 2011;179:1560–1572. doi: 10.1016/j.ajpath.2011.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowan KN, Jones PL, Rabinovitch M. Regression of hypertrophied rat pulmonary arteries in organ culture is associated with suppression of proteolytic activity, inhibition of tenascin-C, and smooth muscle cell apoptosis. Circ Res. 1999;84:1223–1233. doi: 10.1161/01.res.84.10.1223. [DOI] [PubMed] [Google Scholar]
- 24.Sawada H, Saito T, Nickel NP, Alastalo TP, Glotzbach JP, Chan R, Haghighat L, Fuchs G, Januszyk M, Cao A, et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med. 2014;211:263–280. doi: 10.1084/jem.20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toba M, Alzoubi A, O’Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306:H243–H250. doi: 10.1152/ajpheart.00728.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2011;31:814–820. doi: 10.1161/ATVBAHA.110.219980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Hu T, He L, Huang X, Tian X, Zhang H, He L, Pu W, Zhang L, Sun H, et al. Genetic targeting of sprouting angiogenesis using Apln-CreER. Nat Commun. 2015;6:6020. doi: 10.1038/ncomms7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 29.Wertz JW, Bauer PM. Caveolin-1 regulates BMPRII localization and signaling in vascular smooth muscle cells. Biochem Biophys Res Commun. 2008;375:557–561. doi: 10.1016/j.bbrc.2008.08.066. [DOI] [PubMed] [Google Scholar]
- 30.Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol. 2006;26:7791–7805. doi: 10.1128/MCB.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 32.Todorovich-Hunter L, Johnson DJ, Ranger P, Keeley FW, Rabinovitch M. Altered elastin and collagen synthesis associated with progressive pulmonary hypertension induced by monocrotaline: a biochemical and ultrastructural study. Lab Invest. 1988;58:184–195. [PubMed] [Google Scholar]
- 33.Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, Olley PM, Cutz E. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension: a correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. 1986;55:632–653. [PubMed] [Google Scholar]
- 34.Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular-matrix bound basic fibroblast growth factor. J Cell Physiol. 1996;166:495–505. doi: 10.1002/(SICI)1097-4652(199603)166:3<495::AID-JCP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 35.Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the α v β 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139:279–293. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senior RM, Griffin GL, Mecham RP. Chemotactic responses of fibroblasts to tropoelastin and elastin-derived peptides. J Clin Invest. 1982;70:614–618. doi: 10.1172/JCI110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi J, Wigle D, Childs T, Zhu L, Keeley FW, Rabinovitch M.Serum-induced vascular smooth muscle cell elastolytic activity through tyrosine kinase intracellular signaling J Cell Physiol 1994160121–131 [DOI] [PubMed] [Google Scholar]
- 38.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175:1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiedow O, Lüademann J, Utecht B. Elafin is a potent inhibitor of proteinase 3. Biochem Biophys Res Commun. 1991;174:6–10. doi: 10.1016/0006-291x(91)90476-n. [DOI] [PubMed] [Google Scholar]
- 40.Butler MW, Robertson I, Greene CM, O’Neill SJ, Taggart CC, McElvaney NG. Elafin prevents lipopolysaccharide-induced AP-1 and NF-kappaB activation via an effect on the ubiquitin-proteasome pathway. J Biol Chem. 2006;281:34730–34735. doi: 10.1074/jbc.M604844200. [DOI] [PubMed] [Google Scholar]
- 41.Roghanian A, Williams SE, Sheldrake TA, Brown TI, Oberheim K, Xing Z, Howie SE, Sallenave JM. The antimicrobial/elastase inhibitor elafin regulates lung dendritic cells and adaptive immunity. Am J Respir Cell Mol Biol. 2006;34:634–642. doi: 10.1165/rcmb.2005-0405OC. [DOI] [PubMed] [Google Scholar]
- 42.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, Knaus P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002;277:5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- 44.Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2008;20:713–719. doi: 10.1097/bor.0b013e3283103d27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdimarsdottir G, Goumans MJ, Rosendahl A, Brugman M, Itoh S, Lebrin F, Sideras P, ten Dijke P. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106:2263–2270. doi: 10.1161/01.cir.0000033830.36431.46. [DOI] [PubMed] [Google Scholar]
- 46.Berse M, Bounpheng M, Huang X, Christy B, Pollmann C, Dubiel W. Ubiquitin-dependent degradation of Id1 and Id3 is mediated by the COP9 signalosome. J Mol Biol. 2004;343:361–370. doi: 10.1016/j.jmb.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 47.Bellemare A, Vernoux N, Morin S, Gagné SM, Bourbonnais Y. Structural and antimicrobial properties of human pre-elafin/trappin-2 and derived peptides against Pseudomonas aeruginosa. BMC Microbiol. 2010;10:253. doi: 10.1186/1471-2180-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinert PM, Marekov LN. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem. 1995;270:17702–17711. doi: 10.1074/jbc.270.30.17702. [DOI] [PubMed] [Google Scholar]
- 49.Rafikova O, Rafikov R, Kumar S, Sharma S, Aggarwal S, Schneider F, Jonigk D, Black SM, Tofovic SP. Bosentan inhibits oxidative and nitrosative stress and rescues occlusive pulmonary hypertension. Free Radic Biol Med. 2013;56:28–43. doi: 10.1016/j.freeradbiomed.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reynolds AM, Xia W, Holmes MD, Hodge SJ, Danilov S, Curiel DT, Morrell NW, Reynolds PN. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]