

Renal Endothelium
The kidney has one of the richest and most diversified endothelial cell (EC) populations found within any organ. This extensive diversity can be explained on two counts. First, renal endothelium contributes to differential transport capabilities across the various segments of the nephron. Second, the endothelium must withstand unparalleled environment extremes in oxygenation and osmolality. Endothelial cells (ECs) in the outer cortex are exposed to normal osmolality and oxygen tension, whereas those in the inner medullary region are exposed to an osmolality of up to 1,200 mOSM and O2 content as low as 20 mm Hg.
Though significant progress has been done in the field of endothelial biology, still we know little about the different endothelial populations within kidney and the molecular mechanism that govern their structure, functions and potential injury. Therefore the goal of the review is to summarize the current knowledge concerning ECs in kidney and delineate the role of ECs under physiological and pathophysiological state especially in sepsis.
Structure and function
The unique structure of the vascular tree within the kidney is shown in the Figure 1. Anatomically, the kidney is divided into four zones. The cortex, occupies the outermost aspect of the kidney. As one moves toward the renal pelvis from the cortex, one encounters the outer medullary region, consisting of outer and inner stripes, and finally the inner medullary region. Although the renal arteries enter the kidney via the inner renal pelvis, a series of large branching arteries deliver arteriolar blood into the outer cortex via the interlobular arteries terminating in a network of glomerular afferent arterioles. Although the overall blood flow to the kidney averages 4mL/g tissue/min, there is great heterogeneity within the kidney. The outer cortex receives 5–6mL/g/min with consecutively deeper areas receiving less flow per unit weight. The inner aspect of the cortex, outer medulla and inner medulla receive 2–3, 1, and 0.5 mL/g/min, respectively. Thus 90% of the blood flow is directed to the cortical area. The vast majority of blood flow reaches the medulla through juxtamedullary efferent arterioles; however, some may also be from periglomerular shunt pathways [1]. Afferent arterioles have a continuous smooth muscle layer and are the major resistance vessels controlling the glomerular filtration rate (GFR). They terminate to form the glomerular capillary network where formation of the primary urine takes place via filtration. Each glomerular capillary network gives rise to a single efferent arteriole. In contrast to their afferent counterpart, efferent arterioles have a limited discontinuous layer of smooth muscle cells.
Figure 1.

Schematic representation of the visional difference in the microvasculature within the kidney. OSOM, outer stripe of the outer medulla; ISOM, inner stripe of the outer medulla; IM, inner medulla; PCT, proximal convoluted tubule; PST, proximal straight tubule; DLT, descending thin limb; DCT, distal convoluted tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct; AA, arctuate artery; IA, interlobular artery. (Reprinted with permission from Endothelial Biomedicine, Ed. William C. Aird, 2007, Cambridge University Press, Chapter 138, Pg. 1271).
Efferent arterioles follow one of the two paths. First, those that arise from glomeruli within the mid and outer cortex give rise to an O2-rich, dense network of capillaries (peritubular plexus) that run alongside the proximal and distal convoluted tubules in the kidney. This arrangement forms a unique vasculature where two capillary beds (glomerular and peritubular capillaries) are connected by an arteriole. Second, those efferent arterioles that arise from the innermost or juxtamedullary glomeruli give rise to the vasa recta, which run parallel to the loops of Henle and collecting tubules in the medulla. Descending arteriolar vasa recta branch into several vessels that penetrate the inner depths of the medulla, give rise to a dense capillary network within the inner medulla, and return to the cortex via venous ascending vasa recta (AVR). Descending vasa recta (DVR) and AVR, pack together in vascular bundles in the outer medulla, are the sole blood supply to the medulla.
The unique vascular architecture of the kidney results in a gradient of decreasing oxygenation from the cortex to the medulla. This arrangement is essential for development of the countercurrent mechanism necessary for the recycling of osmotic compounds to maintain the marked osmotic gradient that increases as one goes from the other cortex to the inner medullary region [2]. The density of the vascular space within the kidney also varies between the cortex and medullary regions. Traditionally, microvascular space has been considered as part of renal interstitium as H&E stain don’t identify ECs and thus one can’t differentiate the microvascular area from the true interstitial space. ECs within this heterogeneous microvasculature system also vary tremendously structurally and functionally depending upon location (Table 1). Afferent arterioles, with a mean pressure of 90mm Hg, give rise to glomerular capillaries that are lined by fine fenestrated endothelium and have a mean pressure of 60mm Hg. Diaphragms don’t not extend across these fenestrae and the fenestrae are surrounded by microfilaments [3]. Glomerular ECs are known to synthesize both nitric oxide (NO) and endothelin-1 and express vascular endothelial growth factor (VEGF) receptors that have been shown to enhance EC permeability and induce endothelial fenestrations [4]. VEGF was first shown to play major role in renal development by blocking VEGF activity utilizing an antibody in newborn mice. This resulted in disrupted and reduced renal cortical capillary tufts, and an overall reduction in number of nephrons [5]. Additional evidence indicates that VGEF is important for EC survival and repair in glomerular disease characterized by EC damage [5–7]. Glomerular endothelial fenestrae can also be lost within hours following VEGF removal. This results in glomerular endotheliosis, reduction in GFR, and proteinuria [8, 9]. Overexpressoin of VEGF leads to collapsing glomerulopathy, further demonstrating that tight regulation of VEGF is critical to glomerular development and function [10]. VGEF produced by epithelial cells appears to be an important regulator of glomerular EC functionality and permeability. Within the cortex, microvascular ECs are fenestrated on those portions of the cell that border tubule epithelial cells. Erythropoietin-producing cells are localized in the renal cortical interstitial space adjacent to capillary endothelial and tubular epithelial cells. As efferent arterioles from juxtamedullary glomeruli penetrate into the outer stripe of the other medulla, the outer smooth muscle layer is replaced by a discontinuous layer of pericytes. This marks the beginning of the DVR and upon further penetration in to the inner medulla pericytes becomes less numerous. In both the inner and outer medullary regions the ascending arterial vasa recta outnumber the descending arterial vasa recta. The DVR have a continuous nonfenestrated endothelium with a zona occludens limiting their permeability. They express both water channels and a facilitated urea transporter[11]. In the inner medullary region, the DVR terminate and form a sparse capillary plexus that coalesces to form the AVR. Within the AVR EC fenestrations again occur with approximately 50% of the endothelial wall covered by fenestrae in the inner medullary region and 15% to 30% in the outer medullary region. ECs within the AVR are extremely thin and have remarkably high hydraulic conductivity.
Table 1.
Function of Renal endothelium (Modified from Endothelial Biomedicine, Ed. William C. Aird, 2007, Cambridge University Press, Chapter 138, Pg. 1271).
| Blood Vessel | Smooth Muscle Cells | Endothelium | Major function |
|---|---|---|---|
| Afferent arteriole | Continuous | Regulate single nephron blood flow | |
| Glomerular capillary | Continuous, fenestrated, no diaphragm | Selective filtration of macromolecules based on size and charge | |
| Efferent arteriole | Discontinuous | Regulate filtration fraction | |
| Peritubular plexus | Fenestrated facing epithelial cells | Handle bulk of reabsorbed H2O and ions | |
| DVR | Discontinuous pericyte | Continuous, nonfenestrated | Countercurrent exchanger serving to prevent washout of osmotic gradient created by loops of Henle: enters increasingly hyperosmolar medulla, thus gains solutes and loses water |
| AVR | Discontinuous pericyte | Continuous, highly fenestrated, diaphragm | Countercurrent exchanger serving to prevent washout of osmotic gradient created by loops of Henle: leaves high osmolar environment as it moves towards cortex, thus loses in DVR and gains back water; net effect is the osmolality of blood at end of AVR approaches that of blood at beginning of DVR (slightly higher NaCl) |
(Modified from Endothelial Biomedicine, Ed. William C. Aird, 2007, Cambridge University Press, Chapter 138, Pg. 1271).
Endothelial markers have been determined to differentiate the tremendous heterogeneous population of ECs. In human kidney, peritubular capillaries ECs exhibited immunohistochemical characteristics similar to those of peripheral macrophages, including OKM5 (medullary only), von Willebrand factor (vWF), and interleukin (IL)-1. Glomerular capillary ECs expressed vWF but not OKM5. Additional studies have identified RECa-1 and JG-12 as pan-EC specific monoclonal antibodies for all the vascular beds in the rat.
Cross talk with pericytes and Peritubular capillaries (PTC)
Pericytes are a type of mural smooth muscle like cell, located to the non-albuminal side of EC, which discontinuously encircles the endothelial cells and are embedded in the basement membranes of the capillaries (Figure 1). As name indicates, pericytes are perivascular cells. Numerous claw like invagination emanating from the cell body of the pericyte encircles capillaries, arterioles and venules. They have been identified in many organ and tissues of many species [12]. Of the many implicated roles of pericytes vessel stabilization, fibrosis, endothelial cell regulation and angiogenesis are the principal ones. Pericytes have been molecularly characterized by the presence of an extracellular chondroitin sulfate proteoglycan called NG2. Renal pericytes have been demonstrated to express α-smooth muscle actin (α-SMA) which is necessary for the regulation of vessels diameter and thus renal blood flow [13]. The density of pericytes in kidney is quite heterogeneous. The outer medulla has far more pericytes compared to inner medulla [14]. Pericytes and ECs are connected bi-directionally via peg and socket junctions. Membrane invaginations from pericytes and ECs form tight, gap and adherence junctions. These connections are not mere physical contacts but they actively participate to facilitate communication between the two cell types [15, 16]. Pericytes have been proposed to acts as a signaling bridge between endothelial and peritubular capillaries [17]. The glomerular endothelium essentially lacks stabilization by the loss of crosstalk between ECs and pericytes. Pallone and colleagues have demonstrated the role of pericyte in regulating the medullary blood flow by controlling the vasa recta capillary diameter [18]. Pericytes detect the vasoactive signals from endothelial cells and respond by regulating medullary blood flow via the vasa recta [14, 19]. Various endogenous stimuli, acetylcholine [20], angiotensin-II, adenosine triphosphate (ATP), endothelin-1, noradrenaline, UTP and vasopressin, have been shown to evoke pericyte mediated vasoconstriction of vasa recta. Besides peptides and hormones, free radicals and reactive oxygen species (ROS) have also been implicated in the regulation of medullary blood flow [21, 22]. All three forms of nitric oxide synthase (endothelial, inducible and neuronal) have been identified in tubular and vascular capillaries of kidney [23, 24]. Schanackenberg et al. have demonstrated the association of ROS mediated vasoconstriction to hypertension and diabetic nephropathy [23, 24]. Similarly, regulation of pericyte mediated vasodilation of vasa recta have been demonstrated by acetylcholine (NO mediated), adenosine, angiotensin-2 (Ang-2), nitric oxide, PGE2, and vasopressin. In the medulla, adenosine acts as vasodilator resulting in increase of medullary blood flow [25, 26]. Adenosine, which is released from medullary thick ascending limb (mTAL) after oxidative stress also dilates vasa recta by acting on pericytes [14]. Dilation of mTAL, that, results in increased oxygen supply and simultaneously oxygen consumption, is reduced by inhibition of mTAL by NaCl reabsorption [2, 26, 27]. Endothelin-1 (ET-1) synthesized by collecting duct epithelial and ECs, is known to constrict DVR by acting on pericytes [14, 28]. Other vasoactive agents, Ang-II and NO, also act via pericytes [19]. Pericytes along the vasa recta have been shown to contract and relax when acted upon by nucleotides released from endothelial cells, ATP and UTP, respectively [14, 29, 30]. Kidney slices exposed to hypotonic solution release ATP resulted in vasodilation of vasa recta providing direct evidence of tubulo-vascular cross talk in the medulla [19].
Pallon et al. have hypothesized that “feedback of vasoactive stimuli to juxtamedullary resistance vessels may provide the medulla with an intrinsic feedback loop, which could allow the medulla to control its own perfusion” [17, 18]. To summarize, pericytes acts as sensor of the extracellular environment and respond accordingly either by constricting and/or dilating the microvasculature thus fine tuning the medullary blood flow.
Role of pericyte in endothelium stability
Although the role of ECs in regulation of blood flow and vessel fate has been well studied, only recently did the role of pericytes in relation to ECs start to emerge. Pericytes have been demonstrated to regulate microvascular stability in central nervous system. In the absence of pericytes the brain forms micro-aneurysms and undergoes spontaneous hemorrhage [31]. They are also vital in the formation of blood-brain and blood-retina barriers. Diabetic retinopathy is characterized by loss of pericytes [32]. Under physiological conditions, two signaling pathways have been shown to play key roles in stability and vessel development. First, platelet derived growth factor-B/PDGF receptor-β (PDGFR-β) and angiopoietin-Tie2 are important for pericyte differentiation, recruitment and expansion. Second, transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), Notch and sphingosine-1-phosphate (S1P) are known to regulate stabilization of vessels [33–35]. In pathophysiological conditions, like AKI-induced sepsis or ischemia-reperfusion (IR) injury pericytes are activated, detach from peritubular capillaries [36], migrates to the interstitium and differentiate into myofibroblasts [37]. Detachment of pericytes from the endothelium results in unstable, proliferating and non-specific permeability of the endothelium. Injury to the endothelium ultimately results in loss of microvasculature – also known as rarefaction. Thus, pericyte detachment form endothelium has a two prong effect on renal disease progression. First, conversion of pericyte into microfibroblasts leads to fibrosis and, second, leaky endothelium contribute to the inflammation cascade and oxidative stress [38]. Ischemia and oxidative insults to the ECs acts as stimuli for apoptosis as well. In-vitro studies have shown that ECs derived from injured kidney show limited growth potential. Basile et al. have shown that, even in the presence of excessive VEGF and functional VEGF receptor, ECs from injured kidney failed to respond [39]. Lin et al have shown that blockade of either PDGFR-β on pericytes or VEGFR2 on ECs prevents pericyte detachment, amelioration of fibrosis, stabilization of capillary rarefaction and attenuation of inflammatory response [40]. Similarly Greenberg et al. observed that VEGF-A bolstered pericytes detachment and microvasculature destabilization via PDGFR-β [41]. Switching between three isoforms of VEGF-A (VEGF164, VEGF120, VEGF188) takes place when pericytes transform form their normal to myofibroblast type phenotype [40]. In the kidney, pericytes to myofibroblast transition is characterized by predominance of VEGF120 and VEGF188 that brings about the microvascular rarefaction. Schrimpf et al. have identified two genes, tissue inhibitor of metalloproteinase 3 (TIMP3) and a disintegrin and metalloproteinase with thrombospondin motifs-1 (ADAMTS1), involved in pericyte detachment and regulation [36]. TIMP3, known to promote vascular stability, is lost in activated pericyte no longer attached to the endothelium loses that is known to promote. TIMP3, an endogenous inhibitor of ADAMTS-1, hastens the loss of microvasculature by breaking down the capillary basement membrane and other components of extracellular matrix. Additionally, TIMP3 also inhibits the signaling of EC receptor VEGFR2 thus preventing pericyte differentiation to myofibroblasts. Kida and colleagues reported another mediator, ephrin B4 receptor (EphB4) and its membrane bound ligand ephrinB2, became activated in the injured kidney [42]. Lack of ephrinB2 resulted in impaired angiogenesis, fibrosis and microvasculature rarefaction. Interestingly, both primary renal pericytes and primary renal microvascular ECs showed reduced migration and proliferation in response to VGEF-A in ephrinB2 knockouts compared to wildtype. Thus, bidirectional signaling between pericytes and the endothelium is indispensable in maintaining normal functioning microvasculature [43].
Endothelium in sepsis
The renal vascular endothelium is known to be a primary target in several disease processes including glomerular nephritis, vasculitis, lupus nephritis, preeclampsia, hemolytic uremic syndrome, ischemic acute renal failure, renal transplantation rejection, and chronic progressive kidney disease. During sepsis the immune system is first activated then hyper-activated leading to an uncontrolled over-whelming immune reaction, which may harm rather than protect the organism. In other words, in sepsis we may have “too much of a good thing” [44]. Apart from immune cells, ECs are also part of the first line of defense and are “activated” in various patho-physiological conditions. ECs are both “victim and perpetrator” in the sepsis. First, they are hyper-activated in the presence of pro-inflammatory cytokines and then later ECs undergo structural and functional changes that further sustain the ongoing septic process.
An increase in the renal vascular resistance (RVR) is another key hemodynamic factor associated with sepsis-induced AKI as increased in RVR have been shown to alter global and regional blood flow [45]. Increase RVR can be induced by vasoconstriction, leaky endothelium, tissue edema, leukocyte adhesion to ECs and microthrombosis [46]. The kidney is a unique organ where two capillary beds, glomerular and peritubular, are joined by an arteriole. Thus the kidney can be viewed as many different vascular compartments connected in series with separate circuits for each compartment (Figure 3). Injury events, at any given place in this series can act as a bottleneck and contribute significantly to the RVR. This also suggests that the renal microcirculation and macro-circulation are inherently connected and dependent on each other. Increases in either cortical or medullary vascular resistance may play role in observed the microvascular alterations. Doppler waveform has been used during the initial evaluation of renal dysfunction [47]. The Doppler resistive index (RI) described by Pourcelot is widely used as indicative of vascular resistance.
Figure 3.
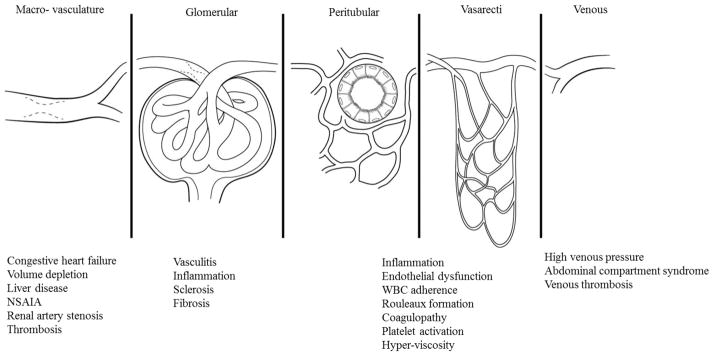
Kidney depicted as multiple vascular compartments connected in series with parallel components in each compartment. The total vascular resistance is equal to the sum of vascular resistances within the different compartments. Also shown, the associated disease conditions/syndromes within each compartment.
Sepsis involves activation of both innate immune cells such as natural killer cells, natural killer T cells, neutrophils, macrophages, and dendritic cells as well as adaptive immune CD4+ T cells. Most of the immune cells are detrimental but recent reports have suggested protective role of CD4+FoxP3+ regulatory cells, also called as Tregs [48–50]. During inflammation, pro-inflammatory mediators secreted by immune cells interact with EC and change their physiologic functions. During infectious conditions, activated leukocytes must transmigrate to infectious sites to engulf, phagocytose, and destroy invading microbes. This is an active process not only for the migrating leukocytes but also for the ECs. During this process numerous pro and anti-inflammatory mediators lead to activation of the endothelium and result in the expression of various adhesion molecules, which in turn facilitate or inhibit the transmigration of activated leukocytes into tissues. In addition, ECs can generate their own inflammatory mediators and express adhesion molecules. The hallmark of these disease processes is glomerular and renal microvascular EC injury with associated apoptosis. Microvascular endothelial damage is the salient feature associated with acute vascular rejection and, in chronic allograft nephropathy, the single most common cause of long term graft loss [8]. Lipopolysaccharide (LPS) and Toll-Like-Receptor (TLR) interaction are important mediators in sepsis. LPS and its cognate LPS-binding protein (LBP), exert their biological activity via CD14. LPS/LBP/CD14 complexes are known to activate ECs [51]. Regulated by a variety of pro- and anti-inflammatory mediators, EC express TLR4, which, when bound to LPS, leads to activation of ECs during the course of sepsis. Further studies have revealed binding of β2 integrins (CD11/CD18) to LPS [52]. This complex, consisting of CD11/CD18-LPS, can further activate ECs.
Under physiologic conditions EC perform several functions to maintain homeostasis. ECs normally inhibit blood coagulation and minimize the adherence of circulating cells. Further, by producing prostacyclin, nitric oxide (NO) and other vasoactive substances, EC inhibit platelet aggregation and regulate the tone of arterioles and venules, there by regulating the microcirculation. Tight regulation of the immune and inflammatory system is crucial for maintaining the balance between protective and tissue-damaging responses. Sepsis is characterized by a loss of this balance leading to hyperactive and often followed by hypoactive immune and inflammatory responses. During inflammatory states ECs come in contact with a variety of proinflammatory mediators that change their physiologic functions profoundly. This is collectively considered as “EC activation ”. As a result an interaction with and adhesion of leukocyte with ECs is facilitated, ECs switch from an anticoagulant state to procoagulant state, and an altered barrier function of ECs leads to increased permeability and impaired vasomotor tone. These changes are discussed in detail in following sections.
Activation of Inflammation
One of the main underlying mechanisms leading to a state of immune system hyperactivation involves the production and release of pro- and anti-inflammatory mediators (chemokines and cytokines). Chemokines are involved in attracting neutrophils to the site of inflammation, activating leukocytes after the initial rolling, and facilitating their transendothelial migration. Endothelial and epithelial cells, as well as neutrophils, macrophages, and lymphocytes, produce large amounts of proinflammatory cytokines (e.g., TNF-α, IL-6, IL-1β, and IL-8).
TNF-α, which appears to be mainly synthesized in activated macrophages/monocytes, is a very early proinflammatory cytokine produced during sepsis and other renal disease state. Binding to its receptor leads to cell activation via nuclear factor (NF)-κB transcription factor. TNF-α stimulates neutrophils and ECs to release a variety of proinflammatory mediators [53]. The IL-1 family (IL-1α, IL-1β) represents another important series of cytokines produced during sepsis. IL-1 and TNF-α lead synergistically to further activation of the inflammatory cascade and upregulation of vascular adhesion molecule. The chemokines IL-8 and monocyte chemoattractant protein (MCP)-1 are additional proinflammatory components during sepsis [54]. Released by ECs, IL-8 plays a key role in neutrophil chemotaxis and degranulation, whereas EC-derived MCP-1 is involved in attracting monocytes. Taken together, production of strong proinflammatory mediators, and increased expression of vascular adhesion molecules, lead to a state of endothelial hyperactivation. Intracellular pathways become activated, more proinflammatory cytokines and chemokines are released, and neutrophils are attracted to the site of inflammation. Neutrophils release granular enzymes, ROS (such as H2O2), and NO, that are capable of causing increased vascular permeability, tissue damage and, ultimately, organ injury.
Structural changes
During inflammation specific adhesion molecules are expressed on WBC, ECs and platelets. Three families of cell adhesion molecules (CAM), the selectins (E, P, L), the integrins ( 1, 2), and the immunoglobulin superfamily intercellular adhesion molecule (ICAM-1) and vascular adhesion molecule (VCAM-1) are expressed on leukocytes, ECs, and platelets play important roles in leukocyte-endothelial interactions. Sepsis-induced AKI is associated with reduced blood flow enhancing leukocyte to margination and rolling along the surface of blood vessels. Rolling and subsequent loose adhesion are facilitated by two interactions. First, L-selectin present on leukocytes and its respective ligands on endothelial cells including CD34 and glycosylation dependent cell adhesion molecule (GlyCAM-1). Second, E-selectin present on endothelial cells and its corresponding ligand, sialyl Lewis X compounds (sLeX) (NeuAc 2–3Gal 1–4[Fuc 1–3]GlcNAc 1 on leukocytes. L-selectins are constitutively expressed on leukocytes and shed upon EC activation by membrane bound serine proteases. On the contrary, E-selection are expressed only upon activation of EC and facilitates the adhesion of leukocytes to ECs. Synthesis of E-selectins are mediated by pro-inflammatory mediators like TNF- and IL-1 or lipopolysaccharides (LPS). Under physiological conditions, P-selectins are stored intracellularly in blood platelets and within Weibel-Palade bodies in endothelial cells. Upon EC activation, Weibel-Palade bodies fuses with cell membrane and P-selectins move to the cell surface membrane rapidly. P-selectins of EC bind to their corresponding ligand, P-selectin glycoprotein ligand (PSGL-1) facilitating adhesion of leukocytes to ECs.
Leukocytes after firmly adhering to the endothelium, transmigrate through ECs aided by platelet-endothelial cells adhesion molecule (PEACM)-1. Platelets and ECs expressed PECAM-1 is mostly localized at the junction of ECs. Antibodies against PECAM-1 has been shown to be effective in the migration of leukocyte in vitro [55] and in vivo [56]. Thereafter, leukocytes finally migrate into the surrounding interstitial tissue space.
Coagulation
Under physiological conditions ECs inhibit blood coagulation by activation of protein C, expression of thrombomodulin [57], specific proteoglycans [58] and release of tissue plasminogen activator [59]. Activation of coagulation is central to microcirculatory dysfunction. Activated EC under septic conditions play key role in the coagulation. Endothelium cell protein C receptor (EPCR) binds to protein C (PC) on the EC and presents it to the thrombin-thrombomodulin complex. PC is cleaved and activated by thrombin aided by thrombomodulin [60]. Activated protein C (aPC) possesses profibrinolytic, anti-inflammatory, and antiapoptotic properties and it counter-acts the effect of thrombin and pro-inflammatory cytokines helping to restore homeostasis [61, 62]. During inflammation expression of naturally occurring anticoagulant and mediators of the PC pathway decrease in turn resulting in decreased anti-inflammatory effects. Exposure to either bacterial components (e.g., lipopolysaccharide) and/or pro-inflammatory cytokines (e.g., TNF-, IL-1) damages ECs, which later undergo apoptosis, thus further amplifying the coagulation cascade. In concert with decreased activation and expression of protein C, the extrinsic pathway of coagulation also becomes activated. This results in increased microvascular coagulation and a further dysfunctional endothelium. Activated protein C has been shown to mitigate LPS-induced AKI effects and down regulate renal inducible nitrogen synthase (INOS) and angiotensin 2 [62]. Dysfunctional microvascular perfusion, increased vascular permeability and coagulation have been shown to be ameliorated by administration of soluble thrombomodulin [63]. During sepsis-induced AKI and ischemia reperfusion injury the complement pathway undergoes activation and the plasma level of complement inflammatory mediators, C3a and C5a, is increased. Both C3a and C5a bind to their respective C3a and C5a receptors, belonging to the rhodopsin family of G-protein coupled receptors, on renal tubular epithelial cells and granulocytes [64]. This interaction leads to the upregulation of proinflammatory cytokines promoting ischemic injury and further bringing more leukocytes in to the interstitium. In addition, C3a and c5a causes renal EC activation especially in the S3 segments of the cortical medullary region [65]. C5a has been shown to induce the expression of P-selectin on the surface of human umbilical vein ECs (HUVECs) and binds to ECs as well [66, 67]. Laudes et al. have shown the ability of C5a to bind to ECs [66]. Furthermore, they showed that exposure of mouse dermal microvascular ECs (MDMECs) to LPS, IL-6 and interferon (IFN)-resulted in increase C5aR, and incubation with C5a and IL-6 led to increased levels of proinflammatory mediators. These data indicate tht C5aR can be upregulated on ECs and that C5a in the copresence of additional agonists may mediate proinflammatory effects of the endothelium.
Permeability
The endothelium is natural barrier between intravascular and extravascular spaces. ECs are not a passive control system but they are actively engaged in blood-tissue exchange of plasma fluid, proteins and cells. They are also involved in maintaining vasomotor tone and vascular permeability.
Dysfunctional microvascular permeability results from injury to endothelial cells, actin cytoskeleton alteration, endothelial cell-cell junctions dissociation, and up-regulation of leukocyte-endothelial interactions.
Normal ECs form a continuous semipermeable barrier whereas activated ECs may loose their barrier function resulting in increased permeability. Much of our current knowledge regarding the endothelial cell-cell junctions comes from in-vitro studies. Two photon in-vivo studies have shown the role of MMP-2 and MMP-9 in maintaining the endothelial barrier. Activation of these two metalloproteinase is correlated with an increase in microvascular permeability [68]. Two pathways, transcellular and paracellular, defines the endothelial permeability. Transcellular permeability, mediated via a specialized vesicles containing caveolin-1 called caveoli, contributes to the basal permeability of endothelium. The paracellular pathway, allowing flow of plasma, proteins and cells through endothelial cell-cell junctions, is a major contributor under pathophysiological conditions. The integrity of endothelial barrier is primarily defined by tight and adherens junctions. Briefly, tight junctions are made up of three major transmembrane proteins, claudins, occludins and zonular occludins (ZO-1 and ZO-2) which form a zipper like structure at the junctions. Adherens are made up of a transmembrane protein, vascular endothelial cadherin (VE-cadherin), consisting of an extracellular domain that binds homophilically with another VE-cadherin and intracellular domain associated with actin binding proteins called catenins (α, β, γ and p120 catenins). Catenins also serve as mediators in the intracellular signaling pathway which modulate the cytoskeletal structure. Numerous intracellular signaling mediators including cytosolic calcium, protein kinase C, tyrosine kinases, Rho GTPases, cAMP and phosphoinositides play key roles in regulation of the actin cytoskeleton [69–74]. Depletion of adenosine triphosphate (ATP) and exposure of oxidants like H2O2 to ECs have been demonstrated to alter the actin cytoskeleton. [75–77]. Depletion of ATP results in disruption of basal and normal level of F-actin in ECs [78, 79]. Similar observations have been made in renal tubular epithelial cell where depletion of ATP results in breakdown of larger cortical F-actin to smaller non-functional aggregates [79]. Interestingly, in activated EC, paracellular permeability has been shown to increase through contraction of actin stress fiber [80, 81]. Dysregulation of focal adhesion transmembrane proteins, focal adhesion kinase (FAK), talin and paxillin, which tethers the ECs to the extracellular matrix also contributes to endothelial permeability [82]. Dysfunctional endothelium in pathophysiological conditions results in increased permeability allowing hemoconcentration due to the outflow of plasma water that leads to stasis and compromised perfusion especially in corticomedullary junction [83]. Hemoconcentration, in turn, increases the endothelial-leukocyte interactions which initiate inflammatory cascade leading to further dysfunction of the endothelial permeability barrier[84]. Although an interaction between EC and leukocyte has been shown to play a key role in endothelium dysfunction, surprisingly, increased endothelial permeability during ischemia-perfusion has been demonstrated to occur in the absence of leukocytes [85].
Therapeutics
Sepsis accounts for more death in United States than myocardial infarction [86]. Sepsis combined with acute kidney injury is associated with a 70% mortality rate [87]. Though substantial progress has been made in the understanding of the sepsis-related acute kidney injury, mortality remains unacceptably high. Recent strategies include the neutralization of bacterial products, targeting undesirable proinflammatory responses, addition of immuno-stimulatory agents to restore the immune system, reducing endothelial cells and leukocyte interactions, and correction of abnormal coagulation system. Here we focus on therapeutic strategies directed towards microvascular endothelial cell activation and dysfunction. Restoring a functional endothelial barrier is of utmost importance in sepsis related renal injury.
VEGF upon binding with it cognate receptor VEGFR2 is well known to increase vascular permeability resulting in a leaky endothelium. An inhibitory VEGFR2 antibody has been shown to decrease hantavirus-directed EC permeability in vitro [88]. Two FDA approved VEGF2 inhibitors, pazopanib and dasatinib, decrease vascular permeability via internalization of the surface adhesion molecule, VE-cadherin [89]. Anti-VEGF antibodies to neutralize VEGF are being evaluated in an ongoing clinical trial [90]. Sphingosine-1-phosphate (S1P) binds to the G-protein coupled receptor, activates integrins, increases cortical actin formation thus contributing significantly towards the stability of the microvasculature [91–95]. The S1P receptor agonist, FTY720 has been shown to induce β-catenin and localize VE-cadherin and improve EC permeability [96–99]. Jones et al. have recently shown that binding of an endothelial receptor, Robo4, to its cognate ligand Slit stabilizes the vascular network by inhibiting endothelial permeability and angiogenesis [100]. Further, it was demonstrated that blockade of Robo4 dependent Slit signaling inhibits inflammation induced endothelial permeability [101]. Slit-Robo4 modulates endothelial permeability by blocking Arf6 activity [102] and regulating the actin cytoskeleton [103]. Very recently, Zhu et al. elucidated a novel cytokine mediated pathway involved in EC barrier stability. They showed that interleukin receptor-1β activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability [104]. SecinH3 which targets the MYD88-ARNO-ARF6 pathway, has been shown to ameliorate leaky endothelium [100]. Angiopoietin-1 has been identified as an anti-permeability and anti-inflammatory agent. Ang-1 strengthens the endothelial cell junctions and downregulates the expression of cell surface adhesion molecule like VCAM-1 and E-selectin [105–107]. Ang-1 and its endothelial specific receptor tyrosine kinase, Tie2, axis have been actively investigated as potential drug targets to decrease the complications associated with inflammation. Kim et.al have demonstrated that COMP-Ang-1 decreases LPS induced AKI [108]. A synthetic Tie2 agonist, vasculotide, protects against vascular leakage and brings down the mortality rate in murine abdominal sepsis [109]. Excess of Angiopoietin-2 (Ang-2), a functional antagonist of Ang-1, has been shown to contribute significantly to pulmonary vascular leakage during sepsis [110], AKI and multiple organ dysfunction syndrome [111]. Ang-2 has been postulated to “sensitize” ECs to TNFα thus aiding in induction of inflammation [112]. In CLP and LPS induced Ang-2+/− heterozygous septic mice were relatively protected against AKI, leaky endothelium and acute lung injury compared to wild type littermates [113]. Use of statins and HMG-CoA reductase inhibitors, has been explored to downregulate P-selectin [114] and ICAM-1 expression by endothelial cell [115] and NFκB dependent gene expression to reduce inflammation [116]. Our group, using hypoperfusion model of ischemic kidney injury and two-photon microscopy, have shown that administration of a variant soluble thrombomodulin (sTM F376L) significantly improved microvascular erythrocytes flow rates, reduced microvascular endothelial leukocyte rolling and attachment, and minimized endothelial permeability[63].
Conclusion
The kidney microvasculature is a complex series of linked domains with extreme environmental variations best demonstrated by changes in oxygenation and osmolality from the cortex to the outer medullary region. It serves to deliver oxygen and substrates and covey reabsorbed compounds and hormonal responses from the kidney. It is positioned anatomically and functionally as a crossroad for communication between the tubular epithelium and immune systems. The overall complexity has limited our understanding to date, but it is well recognized to play major primary or secondary roles in the events leading to initiation, maintenance and recovery of acute kidney injury from multiple conditions. Multiple molecular pathways have been identified as potential therapeutic targets in animal models, yet no translation of this has occurred to man. Furthermore, loss of the microvasculature network has been linked to progressive loss of kidney function. Therefore, a further understanding of the complexity, responses to stimuli and potential therapeutic agents of the kidney microvasculature is necessary if progress in AKI and CKD is to be made.
Figure 2.

Schematic representation of the different forms of ECs found within the peritubular microvasculature of the kidney in the different zones of the kidney. (Reprinted with permission from Pallone TL, Turner MR, Edwards A, Jamison RL. Countercurrent exchange in the renal medulla. Am J Physiol Regul Inter Comp Physiol. 2003;284:R1153-R1175)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Casellas D, Mimran A. Shunting in renal microvasculature of the rat: a scanning electron microscopic study of corrosion casts. Anat Rec. 1981;201(2):237–48. doi: 10.1002/ar.1092010204. [DOI] [PubMed] [Google Scholar]
- 2.Pallone TL, et al. Countercurrent exchange in the renal medulla. Am J Physiol Regul Integr Comp Physiol. 2003;284(5):R1153–75. doi: 10.1152/ajpregu.00657.2002. [DOI] [PubMed] [Google Scholar]
- 3.Rops AL, et al. Isolation and characterization of conditionally immortalized mouse glomerular endothelial cell lines. Kidney Int. 2004;66(6):2193–201. doi: 10.1111/j.1523-1755.2004.66009.x. [DOI] [PubMed] [Google Scholar]
- 4.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108( Pt 6):2369–79. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 5.Kitamoto Y, Tokunaga H, Tomita K. Vascular endothelial growth factor is an essential molecule for mouse kidney development: glomerulogenesis and nephrogenesis. J Clin Invest. 1997;99(10):2351–7. doi: 10.1172/JCI119416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YG, et al. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000;58(6):2390–9. doi: 10.1046/j.1523-1755.2000.00422.x. [DOI] [PubMed] [Google Scholar]
- 7.Kang DH, et al. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol. 2001;12(7):1448–57. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 8.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 9.Ballermann BJ. Glomerular endothelial cell differentiation. Kidney Int. 2005;67(5):1668–71. doi: 10.1111/j.1523-1755.2005.00260.x. [DOI] [PubMed] [Google Scholar]
- 10.Eremina V, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–16. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen S, et al. Aquaporin-1 water channels in short and long loop descending thin limbs and in descending vasa recta in rat kidney. Am J Physiol. 1995;268(6 Pt 2):F1023–37. doi: 10.1152/ajprenal.1995.268.6.F1023. [DOI] [PubMed] [Google Scholar]
- 12.Sims DE. Diversity within pericytes. Clin Exp Pharmacol Physiol. 2000;27(10):842–6. doi: 10.1046/j.1440-1681.2000.03343.x. [DOI] [PubMed] [Google Scholar]
- 13.Park F, et al. Evidence for the presence of smooth muscle alpha-actin within pericytes of the renal medulla. Am J Physiol. 1997;273(5 Pt 2):R1742–8. doi: 10.1152/ajpregu.1997.273.5.R1742. [DOI] [PubMed] [Google Scholar]
- 14.Crawford C, et al. Extracellular nucleotides affect pericyte-mediated regulation of rat in situ vasa recta diameter. Acta Physiol (Oxf) 2011;202(3):241–51. doi: 10.1111/j.1748-1716.2011.02310.x. [DOI] [PubMed] [Google Scholar]
- 15.von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Exp Cell Res. 2006;312(5):623–9. doi: 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314(1):15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy-Lydon TM, et al. Renal pericytes: regulators of medullary blood flow. Acta Physiol (Oxf) 2013;207(2):212–25. doi: 10.1111/apha.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pallone TL, Silldorff EP. Pericyte regulation of renal medullary blood flow. Exp Nephrol. 2001;9(3):165–70. doi: 10.1159/000052608. [DOI] [PubMed] [Google Scholar]
- 19.Crawford C, et al. An intact kidney slice model to investigate vasa recta properties and function in situ. Nephron Physiol. 2012;120(3):p17–31. doi: 10.1159/000339110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eglen RM, et al. Muscarinic acetylcholine receptor subtypes in smooth muscle. Trends Pharmacol Sci. 1994;15(4):114–9. doi: 10.1016/0165-6147(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 21.Edwards A, Cao C, Pallone TL. Cellular mechanisms underlying nitric oxide-induced vasodilation of descending vasa recta. Am J Physiol Renal Physiol. 2011;300(2):F441–56. doi: 10.1152/ajprenal.00499.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao C, et al. Intrinsic nitric oxide and superoxide production regulates descending vasa recta contraction. Am J Physiol Renal Physiol. 2010;299(5):F1056–64. doi: 10.1152/ajprenal.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Touyz RM. Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Rev Cardiovasc Ther. 2003;1(1):91–106. doi: 10.1586/14779072.1.1.91. [DOI] [PubMed] [Google Scholar]
- 24.Schnackenberg CG, Welch WJ, Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32(1):59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- 25.Silldorff EP, Pallone TL. Adenosine signaling in outer medullary descending vasa recta. Am J Physiol Regul Integr Comp Physiol. 2001;280(3):R854–61. doi: 10.1152/ajpregu.2001.280.3.R854. [DOI] [PubMed] [Google Scholar]
- 26.Agmon Y, Dinour D, Brezis M. Disparate effects of adenosine A1- and A2-receptor agonists on intrarenal blood flow. Am J Physiol. 1993;265(6 Pt 2):F802–6. doi: 10.1152/ajprenal.1993.265.6.F802. [DOI] [PubMed] [Google Scholar]
- 27.Zou AP, et al. Role of renal medullary adenosine in the control of blood flow and sodium excretion. Am J Physiol. 1999;276(3 Pt 2):R790–8. doi: 10.1152/ajpregu.1999.276.3.R790. [DOI] [PubMed] [Google Scholar]
- 28.de Nucci G, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc Natl Acad Sci U S A. 1988;85(24):9797–800. doi: 10.1073/pnas.85.24.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sprague RS, et al. ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol. 1996;271(6 Pt 2):H2717–22. doi: 10.1152/ajpheart.1996.271.6.H2717. [DOI] [PubMed] [Google Scholar]
- 30.Praetorius HA, Frokiaer J, Leipziger J. Transepithelial pressure pulses induce nucleotide release in polarized MDCK cells. Am J Physiol Renal Physiol. 2005;288(1):F133–41. doi: 10.1152/ajprenal.00238.2004. [DOI] [PubMed] [Google Scholar]
- 31.Lindahl P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–5. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 32.Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–61. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 33.Murakami M. Signaling required for blood vessel maintenance: molecular basis and pathological manifestations. Int J Vasc Med. 2012;2012:293641. doi: 10.1155/2012/293641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diaz-Flores L, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24(7):909–69. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- 35.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Schrimpf C, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23(5):868–83. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YT, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80(11):1170–81. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 38.Annuk M, et al. Endothelial function, CRP and oxidative stress in chronic kidney disease. J Nephrol. 2005;18(6):721–6. [PubMed] [Google Scholar]
- 39.Basile DP, et al. Low proliferative potential and impaired angiogenesis of cultured rat kidney endothelial cells. Microcirculation. 2012;19(7):598–609. doi: 10.1111/j.1549-8719.2012.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin SL, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178(2):911–23. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg JI, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456(7223):809–13. doi: 10.1038/nature07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kida Y, et al. EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol. 2013;24(4):559–72. doi: 10.1681/ASN.2012080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerety SS, et al. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4(3):403–14. doi: 10.1016/s1097-2765(00)80342-1. [DOI] [PubMed] [Google Scholar]
- 44.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redfors B, et al. Acute renal failure is NOT an “acute renal success”--a clinical study on the renal oxygen supply/demand relationship in acute kidney injury. Crit Care Med. 2010;38(8):1695–701. doi: 10.1097/CCM.0b013e3181e61911. [DOI] [PubMed] [Google Scholar]
- 46.Bougle A, Duranteau J. Pathophysiology of sepsis-induced acute kidney injury: the role of global renal blood flow and renal vascular resistance. Contrib Nephrol. 2011;174:89–97. doi: 10.1159/000329243. [DOI] [PubMed] [Google Scholar]
- 47.Tublin ME, Bude RO, Platt JF. Review. The resistive index in renal Doppler sonography: where do we stand? AJR Am J Roentgenol. 2003;180(4):885–92. doi: 10.2214/ajr.180.4.1800885. [DOI] [PubMed] [Google Scholar]
- 48.Kinsey GR, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20(8):1744–53. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gandolfo MT, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76(7):717–29. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- 50.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 51.Peters K, et al. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60(1):49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 52.El-Samalouti VT, et al. Detection of lipopolysaccharide (LPS)-binding membrane proteins by immuno-coprecipitation with LPS and anti-LPS antibodies. Eur J Biochem. 1997;250(2):418–24. doi: 10.1111/j.1432-1033.1997.0418a.x. [DOI] [PubMed] [Google Scholar]
- 53.Varani J, Ward PA. Mechanisms of endothelial cell injury in acute inflammation. Shock. 1994;2(5):311–9. doi: 10.1097/00024382-199411000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118(2):503–8. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 55.Muller WA, et al. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178(2):449–60. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vaporciyan AA, et al. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262(5139):1580–2. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 57.Esmon CT. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 1995;9(10):946–55. doi: 10.1096/fasebj.9.10.7615164. [DOI] [PubMed] [Google Scholar]
- 58.Mertens G, et al. Cell surface heparan sulfate proteoglycans from human vascular endothelial cells. Core protein characterization and antithrombin III binding properties. J Biol Chem. 1992;267(28):20435–43. [PubMed] [Google Scholar]
- 59.Iversen N, et al. Binding of tissue factor pathway inhibitor to cultured endothelial cells-influence of glycosaminoglycans. Thromb Res. 1996;84(4):267–78. doi: 10.1016/s0049-3848(96)00186-7. [DOI] [PubMed] [Google Scholar]
- 60.Van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24(8):1374–83. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 61.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24(6):327–34. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 62.Gupta A, et al. Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2. Am J Physiol Renal Physiol. 2007;293(1):F245–54. doi: 10.1152/ajprenal.00477.2006. [DOI] [PubMed] [Google Scholar]
- 63.Sharfuddin AA, et al. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol. 2009;20(3):524–34. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349(6310):614–7. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- 65.Singbartl K, Ley K. Leukocyte recruitment and acute renal failure. J Mol Med (Berl) 2004;82(2):91–101. doi: 10.1007/s00109-003-0498-8. [DOI] [PubMed] [Google Scholar]
- 66.Laudes IJ, et al. Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol. 2002;169(10):5962–70. doi: 10.4049/jimmunol.169.10.5962. [DOI] [PubMed] [Google Scholar]
- 67.Foreman KE, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94(3):1147–55. doi: 10.1172/JCI117430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sutton TA, et al. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2005;288(1):F91–7. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- 69.Takenawa T, Itoh T, Fukami K. Regulation of phosphatidylinositol 4,5-bisphosphate levels and its roles in cytoskeletal re-organization and malignant transformation. Chem Phys Lipids. 1999;98(1–2):13–22. doi: 10.1016/s0009-3084(99)00014-6. [DOI] [PubMed] [Google Scholar]
- 70.Parsons JT, et al. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19(49):5606–13. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 71.Keenan C, Kelleher D. Protein kinase C and the cytoskeleton. Cell Signal. 1998;10(4):225–32. doi: 10.1016/s0898-6568(97)00121-6. [DOI] [PubMed] [Google Scholar]
- 72.Diviani D, Scott JD. AKAP signaling complexes at the cytoskeleton. J Cell Sci. 2001;114(Pt 8):1431–7. doi: 10.1242/jcs.114.8.1431. [DOI] [PubMed] [Google Scholar]
- 73.Carpenter CL. Actin cytoskeleton and cell signaling. Crit Care Med. 2000;28(4 Suppl):N94–9. doi: 10.1097/00003246-200004001-00011. [DOI] [PubMed] [Google Scholar]
- 74.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–55. [PMC free article] [PubMed] [Google Scholar]
- 75.Kevil CG, et al. H(2)O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279(1):C21–30. doi: 10.1152/ajpcell.2000.279.1.C21. [DOI] [PubMed] [Google Scholar]
- 76.Kevil CG, et al. Role of cadherin internalization in hydrogen peroxide-mediated endothelial permeability. Free Radic Biol Med. 1998;24(6):1015–22. doi: 10.1016/s0891-5849(97)00433-4. [DOI] [PubMed] [Google Scholar]
- 77.Alexander JS, et al. The role of cadherin endocytosis in endothelial barrier regulation: involvement of protein kinase C and actin-cadherin interactions. Inflammation. 1998;22(4):419–33. doi: 10.1023/a:1022325017013. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe H, et al. Macromolecule permeability of coronary and aortic endothelial monolayers under energy depletion. Am J Physiol. 1991;260(4 Pt 2):H1344–52. doi: 10.1152/ajpheart.1991.260.4.H1344. [DOI] [PubMed] [Google Scholar]
- 79.Kuhne W, et al. Disintegration of cytoskeletal structure of actin filaments in energy-depleted endothelial cells. Am J Physiol. 1993;264(5 Pt 2):H1599–608. doi: 10.1152/ajpheart.1993.264.5.H1599. [DOI] [PubMed] [Google Scholar]
- 80.Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. 1995;130(3):613–27. doi: 10.1083/jcb.130.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163(3):510–22. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 82.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 83.Vollmar B, et al. Hepatic microcirculatory perfusion failure is a determinant of liver dysfunction in warm ischemia-reperfusion. Am J Pathol. 1994;145(6):1421–31. [PMC free article] [PubMed] [Google Scholar]
- 84.Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Crit Care Med. 2002;30(5 Suppl):S235–40. doi: 10.1097/00003246-200205001-00011. [DOI] [PubMed] [Google Scholar]
- 85.Sakuma T, et al. Ischemia-reperfusion lung injury in rabbits: mechanisms of injury and protection. Am J Physiol. 1999;276(1 Pt 1):L137–45. doi: 10.1152/ajplung.1999.276.1.L137. [DOI] [PubMed] [Google Scholar]
- 86.Angus DC, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 87.Rangel-Frausto MS, et al. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273(2):117–23. [PubMed] [Google Scholar]
- 88.Gavrilovskaya IN, et al. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J Virol. 2008;82(12):5797–806. doi: 10.1128/JVI.02397-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gorbunova EE, et al. VEGFR2 and Src kinase inhibitors suppress Andes virus-induced endothelial cell permeability. J Virol. 2011;85(5):2296–303. doi: 10.1128/JVI.02319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pilot study of Bevacizumab (Avastin) in Patients With Septic Shock. Available at: http://clinicaltrials.gov/ct2/show/NCT01063010.
- 91.Yatomi Y, et al. Sphingosine 1-phosphate as a major bioactive lysophospholipid that is released from platelets and interacts with endothelial cells. Blood. 2000;96(10):3431–8. [PubMed] [Google Scholar]
- 92.Schaphorst KL, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L258–67. doi: 10.1152/ajplung.00311.2002. [DOI] [PubMed] [Google Scholar]
- 93.Lee MJ, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99(3):301–12. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 94.Camerer E, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119(7):1871–9. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Su G, et al. Absence of integrin alphavbeta3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med. 2012;185(1):58–66. doi: 10.1164/rccm.201108-1381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sanchez T, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278(47):47281–90. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- 97.Mandala S, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 98.Brinkmann V, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 99.Brinkmann V, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–97. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 100.Jones CA, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med. 2008;14(4):448–53. doi: 10.1038/nm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.London NR, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2(23):23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones CA, et al. Slit2-Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nat Cell Biol. 2009;11(11):1325–31. doi: 10.1038/ncb1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anand AR, et al. N-terminal Slit2 inhibits HIV-1 replication by regulating the actin cytoskeleton. Retrovirology. 2013;10:2. doi: 10.1186/1742-4690-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu W, et al. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature. 2012;492(7428):252–5. doi: 10.1038/nature11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jain RK, Munn LL. Leaky vessels? Call Ang1! Nat Med. 2000;6(2):131–2. doi: 10.1038/72212. [DOI] [PubMed] [Google Scholar]
- 106.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460–3. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 107.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–4. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 108.Kim DH, et al. COMP-angiopoietin-1 decreases lipopolysaccharide-induced acute kidney injury. Kidney Int. 2009;76(11):1180–91. doi: 10.1038/ki.2009.387. [DOI] [PubMed] [Google Scholar]
- 109.Kumpers P, et al. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care. 2011;15(5):R261. doi: 10.1186/cc10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Parikh SM, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3(3):e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumpers P, et al. Angiopoietin-2 in patients requiring renal replacement therapy in the ICU: relation to acute kidney injury, multiple organ dysfunction syndrome and outcome. Intensive Care Med. 2010;36(3):462–70. doi: 10.1007/s00134-009-1726-7. [DOI] [PubMed] [Google Scholar]
- 112.Fiedler U, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12(2):235–9. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 113.David S, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med. 2012;40(11):3034–41. doi: 10.1097/CCM.0b013e31825fdc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Omi H, et al. Statins inhibit high glucose-mediated neutrophil-endothelial cell adhesion through decreasing surface expression of endothelial adhesion molecules by stimulating production of endothelial nitric oxide. Microvasc Res. 2003;65(2):118–24. doi: 10.1016/s0026-2862(02)00033-x. [DOI] [PubMed] [Google Scholar]
- 115.Takeuchi S, et al. Cerivastatin suppresses lipopolysaccharide-induced ICAM-1 expression through inhibition of Rho GTPase in BAEC. Biochem Biophys Res Commun. 2000;269(1):97–102. doi: 10.1006/bbrc.2000.2238. [DOI] [PubMed] [Google Scholar]
- 116.Dichtl W, et al. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23(1):58–63. doi: 10.1161/01.atv.0000043456.48735.20. [DOI] [PubMed] [Google Scholar]