

Abstract
Attempts to make a diamino disulfonic acid derivative of an aza-BODIPY showed it was difficult to add BF2 to a disulfonated azadipyrromethene, and sulfonation of an aza-BODIPY resulted in loss of the BF2 fragment. We conclude the electron-deficient character of aza-BODIPY dyes destabilizes them relative to BODIPY dyes. Consequently, sulfonation of the aza-BODIPY core is not a viable strategy to increase water solubility. This assertion was indirectly supported via stability studies of a BODIPY and an aza-BODIPY in aqueous media. To afford the desired compound type, an aza-BODIPY with two amino and two sulfonic acid groups was prepared via modification of the aryl substituents with cysteic acid.
Relative to most BODIPY dyes, fluors in the aza-BODIPY series1–3 tend to have longer wavelength absorption and fluorescence emission maxima (Fig. 1).4–6 This characteristic is advantageous for many potential applications of these materials as probes in biological systems, but it comes at a cost. Red-shifted absorption and emission in aza-BODIPY dyes seems to depend on the presence of aryl substituents in the 1,3,5,7-positions making the heterocycles highly hydrophobic and inclined to aggregation in aqueous media.
Fig. 1.

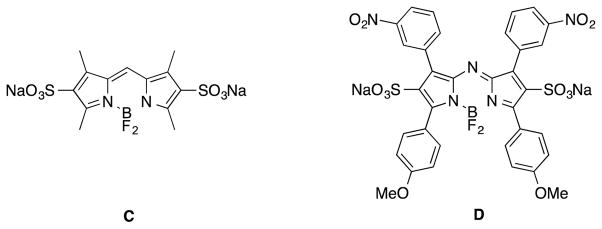
Structures of BODIPYs and aza-BODIPYs.
The literature describes some efforts to increase the hydrophilicity of aza-BODIPY dyes based on functionalization of the aryl substituents.7 Modified aza-BODIPYs resulting from those efforts feature ammonium salts,8 oligoethylene glycol fragments,9 sulfonic and carboxylic acids,8 and carbohydrate derivatives.10 We required a hydrophilic aza-BODIPYs with amino functionality on the aryl group that could be coupled with activated carboxylic acids to produce derivatized optical imaging agents. Negatively charged dyes were also attractive to us because they are intrinsically repelled by negative polar head-groups on cell surfaces.
Sometime ago we refined11 a procedure12, 13 for adding sulfonic acid groups into the BODIPY 2- and 6-positions, that gave the water-soluble BODIPY dye C. Consequently, it seemed logical to apply the same approach to obtain the aza-BODIPY disulfonic acid D. This Communication describes why that approach does not work, and an alternative that enables sulfonic acid and amino functionalities to be introduced into the aza-BODIPY system, via functionalization of the aryl groups.
Treatment of the dinitro-aza-dipyrromethene (aza-DIPY; see SI) 1 under the sulfonation conditions for BODIPY dyes,11 gave predominantly the mono- or di-sulfonated DIPY systems 2a and 2b, according to the equivalents of chlorosulfonic acid used (Scheme 1). Sulfonate 2a was isolated by chromatography on silica gel, whereas 2b was obtained more straightforwardly by precipitation from dichloromethane. Unfortunately, treatment of the DIPY systems 2 under a variety of conditions (Table S1) gave no trace of the corresponding aza-BODIPY dyes as monitored via fluorescence spectroscopy in water (with and without Triton X-100 to increase water solubility) or via 11B NMR of the crude material.
Scheme 1.
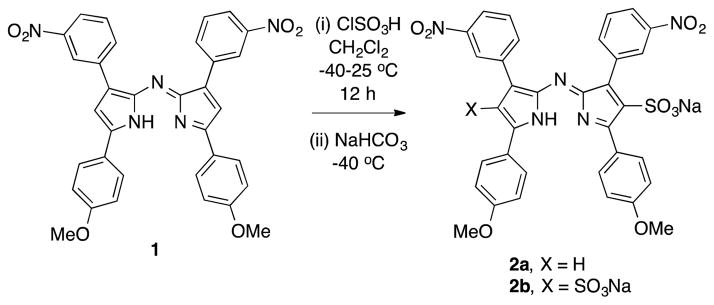
Synthesis of disulfonated aza-DIPY.
Scheme 2 describes an alternative approach to compound D, that was also unsuccessful. Treatment of the aza-BODIPY 3 with excess chlorosulfonic acid led to the aza-DIPY 2b corresponding to loss of the BF2 fragment. 11B NMR, fluorescence and absorption spectroscopy (and ESI-MS), of the crude product from this reaction showed no evidence that an aza-BODIPY was present, indicating loss of the BF2 fragment was not caused by the work-up procedure (Fig. S1). Several variations of the conditions for sulfonation of 3 also gave the same results, ie formation of the aza-DIPY 2 and not the anticipated aza-BODIPY D.
Scheme 2.
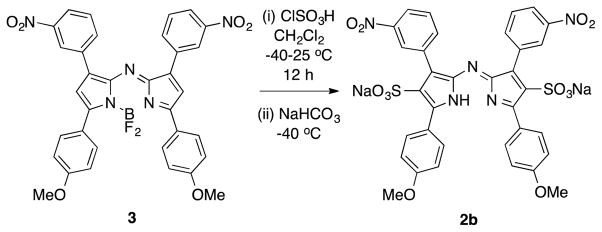
Attempt to sulfonate aza-BODIPY.
Based on the observations above, we decided to re-focus on obtaining the cysteic acid derivative 4 via the procedure outlined in Scheme 3. Thus hydrogenation of 1 then reaction with BF3 gave the diamine 5 which was conveniently coupled with BOC-protected cysteic acid to give the disulfonic acid 6 (isolated by MPLC on a reverse phase column) then the diamino-disulfonic acid 4 after a deprotection step (in which no further purification procedure was necessary).
Scheme 3.

Synthesis of aza-BODIPY containing cysteic acid.
Disulfonic acid 4 appears to give a homogeneous bright green solution in water or 1 mM PBS buffer. The UV absorbance of the dye is broad and there is no significant fluorescence (Fig. 2a). However, the UV absorbance of this dye sharpens considerably, and it becomes significantly more fluorescent, when 0.1 % Cremophor (CrEL) is added (Φ 0.34 ± 0.010, Fig 2b); CrEL is a non-ionic surfactant that is commonly used as an excipient in pharmaceutical formulations. Near-IR concentration dependence of 4 in PBS buffer (without additives) was used to probe for further evidence of aggregation, but no significant dependence on the fluor concentration was observed (Fig. S3).
Fig. 2.

a UV and b fluorescence (excitation λ 650 nm) spectra of compound 4 in pH 7.4 PBS + 0.1 % CrEL. c Solubility profile of 3, 4 and 5 in PBS at pH 7.4 (analyses performed in triplicate for each compound).
A UV-based method from the literature14 was used to compare the aqueous thermodynamic equilibrium solubilities of dyes 3 – 5. Thus 0.1 M stock solutions of the test compounds in DMSO were diluted with PBS to 0 – 100 μM in a 96-well plate, so that the highest concentration contained only 1 % DMSO. The plate was shaken horizontally for 6 h at 25 °C and kept overnight for equilibration. Thereafter, the plate was centrifuged at 1000 rpm for 20 min, the supernatant was pipetted into a 96-well UV-transparent plate and analyzed using a microplate reader, λ = 680 nm, vs a blank (Fig 2c).
Data from the experiments described above show the featured sulfonic acid 4 is significantly more hydrophilic than the dinitro- and diamino-compound 3 and 5 (Fig. 2c). In fact, the disulfonic acid 4 is soluble in PBS pH 7.4 up to 200 μM and even more soluble (more than 1 mM) in carbonate buffer pH 9 (Fig. S4a). At pH 4, the solubility of 4 decreases (<20 μM) whereas 5 is more soluble (<50 μM, Fig. S4b).
Compound 5 is only very weakly fluorescent, and no staining was observed via confocal microscopy when this solution was incubated with 4T1 murine breast cancer cells. After acylation to obtain disulfonic acid BODIPY 4, bright fluorescent was observed in 0.1 % CrEL/PBS, and the fluor was clearly internalized into lysosomes of the same cells (Fig. 3).
Fig. 3.

a Compound 4 is readily internalized by murine 4T1 breast cancer cells. A lysotracker dye (b) colocalized with the intracellular fluorescence (c). d Bright field image for reference.
Failure to obtain the sulfonic acid system D led us to hypothesize the aza-BODIPY framework with 2- and 6-sulfonic acids is too electron deficient to hold the BF2 fragment. This would explain why the BODIPY disulfonic acid C is isolable whereas we were not able to obtain D. Extrapolating this logic, we also hypothesized that aza-BODIPY dyes generally would be less stable than BODIPY systems even if there were no 2-, 6-substituents. Consequently, the stabilities of representative compounds were compared (Fig. 4).
Fig. 4.

Fluorescnce studies to monitor stabilities of the fluors. The t1/2 of aza-BODIPY 4 is 50 min at 90 °C and 2 min at 100 °C, whereas BODIPY A gave less than 10 % decomposition under the same conditions.
Heating the tetramethyl BODIPY A and our hydrophilic aza-BODIPY 4 in non-deoxygenated 1:1 dioxane:water (Fig. 4) proved BODIPY A persisted at 90 and 100 °C, but the aza-BODIPY system showed significant decomposition after 30 min at 90 °C, and was mostly decomposed under the same conditions but at 100 °C.
Conclusions
Our failure to obtain the disulfonic acid D illustrates a difference between the chemistry of BODIPY and aza-BODIPY dyes; BODIPY systems are more electron-rich and can support strong electron withdrawing groups attached directly to the core, whereas similar electron withdrawing groups may destabilize aza-BODIPY systems with respect to bonding to the BF2 fragment. Consistent with this, the diformyl BODIPY E and the formyl aza-BODIPY F are known, but same authors have reported being unable to form the diformyl aza-BODIPY G.15 Our interpretation of these observation is that the diformyl aza-BODIPY G may be unstable, even though the corresponding BODIPY is known. The fact that the monoformylated aza-BODIPY F was isolated was possible even though monosulfonation was not, because formyl groups are less electron withdrawing.16
Parenthetically, we note that, consistent with the observations above, attempts to derivatives of 3 with para- rather than meta-nitro groups failed because the BF2 fragment could not be inserted.
The negatively charged cysteic acid derivative 4 is significantly more hydrophilic than its parent diamine 5, and it has amino groups for functionalization with carboxylic acid derivatives. Use of neutral carboxylic acid derivatives for such functionalizations will give products with two negative charges overall. This is desirable because such negatively charged fluorescent derivatives tend to have an intrinsic repulsion for negative head groups on the cell membrane. Intrinsic repulsion reduces non-specific binding of conjugates and facilitates studies of aza-BODIPYs designed to selectively bind certain cell surface receptors.
Supplementary Material
Fig. 5.
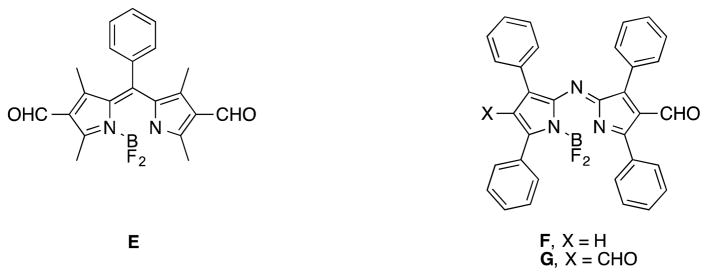
The diformyl BODIPY E and the mono-formyl aza-BODIPY F are known, but the diformyl aza-BODIPY has been proven difficult to prepare.
Acknowledgments
We thank The National Institutes of Health (GM087981), The Robert A. Welch Foundation (A-1121), and High Impact Research (HIR (UM.C/625/1/HIR/MOHE/MED/17 & UM.C/625/1/HIR/MOHE/MED/33) from the Ministry of Higher Education, Malaysia, for financial support. The NMR instrumentation at Texas A&M University was supported by a grant from the National Science Foundation (DBI-9970232) and the Texas A&M University System. The Olympus FV1000 confocal microscope acquisition was supported by the Office of the Vice President for Research at Texas A&M University.
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic procedures and characterization of compounds, spectroscopic data, cell culture and imaging methods. See DOI: 10.1039/c000000x/
Notes and references
- 1.Rogers MAT. J Chem Soc. 1943:590–596. [Google Scholar]
- 2.Gorman A, Killoran J, O’Shea C, Kenna T, Gallagher WM, O’Shea DF. J Am Chem Soc. 2004;126:10619–10631. doi: 10.1021/ja047649e. [DOI] [PubMed] [Google Scholar]
- 3.Hall MJ, McDonnell SO, Killoran J, O’Shea DF. J Org Chem. 2005;70:5571–5578. doi: 10.1021/jo050696k. [DOI] [PubMed] [Google Scholar]
- 4.Ziessel R, Goze C, Ulrich G. Synthesis. 2007;6:936–949. [Google Scholar]
- 5.Loudet A, Burgess K. In: Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine. Kadish K, Smith K, Guilard R, editors. World Scientific; 2010. p. 203. [Google Scholar]
- 6.Lu H, Mack J, Yang Y, Shen Z. Chem Soc Rev. 2014;43:4778–4823. doi: 10.1039/c4cs00030g. [DOI] [PubMed] [Google Scholar]
- 7.Fan G, Yang L, Chen Z. Front Chem Sci Eng. 2014;8:405–417. [Google Scholar]
- 8.Tasior M, Murtagh J, Frimannsson DO, McDonnell SO, O’Shea DF. Org Biomol Chem. 2010;8:522–525. doi: 10.1039/b919546g. [DOI] [PubMed] [Google Scholar]
- 9.Collado D, Vida Y, Najera F, Perez-Inestrosa E. RSC Adv. 2014;4:2306–2309. [Google Scholar]
- 10.Murtagh J, Frimannsson DO, O’Shea DF. Org Lett. 2009 doi: 10.1021/ol902140v. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Han J, Nguyen B, Burgess K. J Org Chem. 2008;73:1963–1970. doi: 10.1021/jo702463f. [DOI] [PubMed] [Google Scholar]
- 12.Shah M, Thangaraj K, Soong ML, Wolford L, Boyer JH, Politzer IR, Pavlopoulos TG. Heteroat Chem. 1990;1:389–399. [Google Scholar]
- 13.Wories HJ, Koek JH, Lodder G, Lugtenburg J, Fokkens R, Driessen O, Mohn GR. Recl Trav Chim Pays-Bas. 1985;104:288–291. [Google Scholar]
- 14.Bharate SS, Vishwakarma RA. Bioorg Med Chem Lett. 2015;25:1561–1567. doi: 10.1016/j.bmcl.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Jiao L, Yu C, Li J, Wang Z, Wu M, Hao E. J Org Chem. 2009;74:7525–7528. doi: 10.1021/jo901407h. [DOI] [PubMed] [Google Scholar]
- 16.McMurry J. Organic Chemistry. 7. Brooks/Cole; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.