

Abstract
Connecting dynamics to structural data from diverse experimental sources, molecular dynamics simulations permit the exploration of biological phenomena in unparalleled detail. Advances in simulations are moving the atomic resolution descriptions of biological systems into the million-to-billion atom regime, in which numerous cell functions reside. In this opinion, we review the progress, driven by large-scale molecular dynamics simulations, in the study of viruses, ribosomes, bioenergetic systems, and other diverse applications. These examples highlight the utility of molecular dynamics simulations in the critical task of relating atomic detail to the function of supramolecular complexes, a task that cannot be achieved by smaller-scale simulations or existing experimental approaches alone.
1 Introduction
The essential conundrum of modern biology, namely the question of how life emerges from myriad molecules whose behavior is governed by physical law alone, is embodied within a single cell—the quantum of life. As illustrated in Figure 1, the rise of scientific supercomputing has allowed for the study of the living cell in unparalleled detail, from the scale of the atom [1, 2] to a whole organism [3, 4, 5] and at all levels in between [6]. In particular, the past three decades have witnessed the evolution of molecular dynamics (MD) simulations as a “computational microscope” [7], which has provided a unique framework for the study of the phenomena of cell biology in atomic (or near-atomic) detail.
Figure 1.

Characteristic length-scales currently associated with varying levels of description in biomolecular simulations. Ab-initio and semi-empirical quantum mechanical calculations permit the study of chemical reactions in electronic detail within single molecules and small proteins while all-atom and coarsed-grained molecular dynamics simulations allow for the study of biological phenomena from the individual protein level to large subcellular organelles, and at all levels in between.
Now, in the era of petascale computing, high-performance MD software packages such as NAMD [8], GROMACS [9], and LAMMPS [10] are being optimized for scaling to an ever-increasing number of cores on cutting-edge computing hardware [2, 11, 10], enabling the investigation of previously unfathomable biological phenomena through the use of large-scale atomistic simulations. Moreover, the development of computational tools such as molecular dynamics flexible fitting (MDFF) [12, 13] are forging an intimate connection between experiment and theory, informing the construction of atomic-level models of large-scale, supramolecular complexes through a synthesis of multi-scale experimental data from cryo-EM, NMR spectroscopy, and X-ray crystallography. Complementary to all-atom MD simulations, the development of force fields for coarse-grained MD (CGMD) simulations continues to be a popular source of techniques which favor computational efficiency over atomic and chemical accuracy, permitting simulations on even larger time and length scales [14].
This opinion will focus on the ways in which large-scale MD simulations are having a profound impact in numerous diverse scientific endeavors. From the treatment of disease and development of drugs [15, 16] to the fabrication of novel biomaterials [17] and creation of bio-based renewable energy sources [18], large-scale MD simulations are helping to achieve a fundamental understanding of living organisms. Taken together, the work reviewed here demonstrates the maturity of the MD apparatus as a tool to progress basic science and the investigation of the molecular makeup of life.
2 Large-scale MD simulations of viruses
Viruses are parasitic life-forms that replicate by hijacking resources present in the cells they infect. Because of their small size compared to cells (20 to 1500 nm scale), observation of the viral particle during different stages of the replication cycle is mostly limited to electron microscopy. Yet, virus particles are large in size for all-atom simulations (see Figure 2) and as a result most studies at the atomic level have been limited to isolated virus proteins or sub-fragments of a viral particle or capsid [19]. The satellite tobacco mosaic virus (STMV) became the first complete virus to be investigated through all-atom MD simulations [20]. Since then, MD programs have become capable of simulating systems of even larger size and complexity [1, 11][21]*1, thus allowing the study of viral particles up to two orders of magnitude larger in atom count than STMV [1].
Figure 2.

Viral particles of different sizes studied using MD simulations. The viruses were arranged in the order of increasing size with the capsid diameters given in parentheses : STMV (17 nm) [20], poliovirus (32 nm) [22], RHDV (43 nm) [15], SV40 (49 nm) [23], and HIV-1 (70–100 nm) [1]. For size comparison, HIV-1 protease, one of the most studied enzymes, is shown at the bottom right.
High resolution structures of symmetrical virus capsids like poliovirus, southern bean mosaic virus and satellite tobacco necrosis virus have been available for several years, leading to routine investigations using MD simulations [24, 22, 25, 26]. More recently, MDFF has been applied to elucidate the structures of yet larger and more complex virus capsids in their native environments [27, 15, 1, 28]. For instance, MDFF was instrumental in the structural determination of the HIV-1 core [1]** 2, a polymorphic capsid with no apparent symmetry. The 64-million-atom MD simulation of the mature HIV-1 capsid should enable the characterization of complex interactions between host cell factors and the assembled capsid lattice [29], thus providing an unexploited framework for the development of novel drugs targeting HIV-1. Similarly, MDFF’s application to the Rabbit hemorrhagic disease virus (RHDV), a 10-million-atom MD calculation, improved the overall fitting of the crystal structure to the cryo-EM density, leading to the development of a vaccine [15]* 3.
Spontaneous assembly of the immature and mature forms of the capsid from viral nucleic acids and proteins is an essential step for the replication cycle of a virus. However, the rate at which assembly occurs prevents a molecular-level description of the process by experimental means, making it an attractive task for computational modeling. The virus assembly kinetics and pathways can be computationally determined using stochastic kinetics [30], elastic networks [31], simulation-based data fitting [32], and CGMD simulations [23, 33, 30, 34]. In fact, by means of CGMD simulations, optimal configuration of the viral genome has been shown to be essential for proper assembly of the capsid [23, 33]. In the particular case of simian virus 40 (SV40), two pathways have been observed during assembly, with each of the pathways sampling different intermediate states depending on the strength of both ionic solution and protein-protein interactions [23]* 4. Coarse-grained simulations have also been performed to investigate the self-assembly process of the HIV-1 mature capsid [35, 36, 1], allowing the identification of a trimer-of-dimers structure as a fundamental step during assembly of hexagonal lattices in a crowded enviroment [35, 36]. However, due to the lack of structural information available at the time for informing the CG models, both studies neglected explicit interactions at the trimeric interface of the lattice, an interface now known to play an essential role in capsid curvature, assembly, and stability as well as infectivity in vivo [1].
In order to become infectious, entire viral particles undergo a global structural rearrangement known as maturation. Such a maturation process is thought to follow two different pathways: disassembly-reassembly or displacive transition [37, 38]. Mechanistic insights into the continuous displacive transition pathway were offered by investigating the swelling process of the cowpea chlorotic mottle virus capsid [39, 40] in which the process was identified to be symmetry-breaking. Normal mode analysis of entire viral capsids has been used to probe mechanical properties of the particles [41, 42, 31, 43], and should be a valuable tool in studying large conformational transitions associated with viral processes (e.g. maturation) [19, 31]. In a remarkable study, the pH-induced maturation of bacteriophage HK97 was investigated by Brooks and coworkers using constant pH MD simulations [44, 45]** 5, revealing a novel intermediate state stabilized by key residues that undergo significant pKa shifts during maturation. Although limited presently to a smaller subsytem of bacteriophage HK97, constant pH simulations could be applied in the future to investigate the maturation process of whole capsids. To our knowledge, however, no MD simulation has described yet the disassembly-reassembly process of a virus.
In summary, large scale MD simulations have proven to be essential to the structural determination of viral particles, in particular by the unique potential to combine information from multiple experimental sources and by the ability to probe viral protein dynamics in native environments. However, processes during virus replication that are inherently slow and involve large systems, such as virus assembly and budding [46], membrane fusion, virus maturation, uncoating of viral capsids, and nuclear entry, represent the current threshold for MD simulations of viruses. In particular, studies on host-pathogen interactions involving large numbers of proteins [47], are still scarce, yet essential to the investigation of virus replication cycles, as well as for the development of drugs and the engineering of viruses for gene therapy.
3 Large-scale MD simulation of ribosomes
As another important pharmaceutical target, the ribosome is the most ubiquitous and complex molecular machine in living cells, responsible for decoding the genetic code into functional proteins. MD simulation has been successfully applied to ribosomal translation [48]. Here, we review the most recent advances in large simulations involving complete ribosome structures.
Simulations of the ribosome have advanced greatly in recent years. A key subject of such studies has been the process of translocation, which moves mRNA ahead by three bases and shifts tRNAs from A to P to E sites. Translocation is still a challenge for MD simulations because the process involves a collective motion of both ribosome subunits as well as of tRNA substrates [52, 53] (see Figure 3A), the description of which requires simulation of a complete solvated ribosome that amounts to ~2.5 million atoms. By combining cryo-EM data and crystallographic structures, Grubmüller and coworkers [49]**6 performed MD simulations of complete ribosome structures at 13 intermediate states of the translation process. The simulations revealed an atomic-level description of time-resolved translocation, transition rates linking translocation sub-states and the underlying molecular forces, all properties essentially inaccessible to experiment. Another MD simulation of a complete ribosome performed by Sanbonmatsu and coworkers [54] captured the small subunit rotation, which is essential for moving the tRNA forward [55]. The simulation allowed the authors to identify reaction coordinates that capture the rotation and to obtain estimates of the free energy barriers associated with translocation.
Figure 3.

A. Translocating ribosome at the pre-translocation state with an A-site tRNA (red) and a P-site tRNA (green) [49]. A red arrow shows the direction of tRNA’s traversal motion. B. Insertion of a nascent protein by the ribosome into a nanodisc [50] membrane working with the SecYE translocon [51]. The nascent protein and P-site tRNA are shown in green. A red arrow shows the direction of the nascent protein’s insertion motion. C. Bacterial ribosome with the antibiotic drug erythromycin (in red circle) shown at its binding site inside the ribosome [16].
The ribosome is a target of many antibiotic drugs [56]. Simulations have played a key role in elucidating antibiotic action in the ribosome [57, 58, 59, 60, 61]. For example, MD simulations have shown that drug-regulated base-flipping of the ribosome underlies the action of small antibiotic compounds [57, 58]. A recent MD simulation involving a complete bacterial ribosome and the clinically-important macrolide drug erythromycin (ERY) [62, 63], shown in Figure 3C, has revealed a new perspective of how the family of macrolide antibiotic drugs act on bacterial ribosomes [16]. It was previously believed that the presence of the nascent protein is necessary for the action of macrolide drugs [64, 65, 66]. However, the MD simulations have shown that ERY acts on ribosomes without a nascent protein present, by inducing base flipping of critical nucleotides at the ribosomal catalytic center [16]* 7. The change of ribosomal RNA bases predicted by the MD simulations was later supported by a cryo-EM reconstruction of a ribosome-ERY complex [67].
Computational studies of the ribosome have begun to simulate the complete ribosome over increasing time-scales [49, 54, 16, 68] and to further incorporate experimental data [48]. In addition, ribosome simulations have begun to capture the heterogeneous nature of translation. For example, modeling and simulations were key in determining the translocon-bound ribosome structure with a nanodisc [50] membrane (shown in Figure 3B) and allowed the characterization, in atomic detail, of how nascent proteins are inserted into a membrane [51]. The availability of the high-resolution structure for the human ribosome [69] together with simulations will have a significant impact on antibiotic drug development, as drugs that kill bacteria, but leave human ribosomes unharmed, can be designed through computational guidance.
4 Large-scale MD simulations of bioenergy systems
Development of new pharmaceuticals is not the only target of large simulations, indeed, biotechnological applications related to bioenergy are also been studied by multi-million atom MD simulations. Applications range from studies of energy conversion by complex interlocking mechanisms of several proteins in photosynthetic systems, to production of biofuels out of agricultural waste.
4.1 Photosynthetic apparatus
Photosynthesis is key for life on Earth, converting light energy into various forms of chemical energy, for example into the synthesis of ATP from ADP. The conversion takes place in an apparatus containing extensive membrane-protein systems. Even in case of the simplest photosynthetic systems, that of so-called purple bacteria, hundreds of proteins work in concert to turn sunlight into ATP [70]. Input from several experimental techniques has been combined to construct models of entire photosynthetic membranes, which are presently being explored through quantum mechanical calculations and MD simulations. The largest photosynthetic membrane simulation published thus far is that of a 23-million-atom membrane patch from the bacterium Rsp. photometricum [71]*8 as depicted in Figure 4; a much larger, 100-million-atom MD simulation of a chromatophore from Rb. sphaeroides [72] is presently underway. Topographical data from atomic force microscopy provided the initial positions of the light harvesting complexes, which were then placed in a lipid bilayer along with quinone molecules, crucial for completing the needed series of energy conversion steps. The 150 ns MD simulation on the membrane patch gave insight into the packing of the proteins and the mobility of the quinone molecules, which must diffuse through the narrow lipid phase separating closely packed protein complexes.
Figure 4.
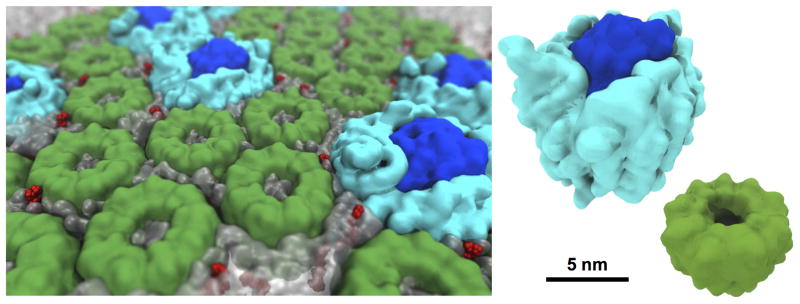
Photosynthetic membrane studied by molecular dynamics simulations. MD simulations provide insight into local and global mechanisms of light harvesting and energy conversion. The photosynthetic membrane pictured consists of three types of protein complexes, light harvesting complex 1 (light blue) and 2 (green) and photosynthetic reaction center (dark blue) as well as quinone molecules (red), which are surrounded by a phospholipid membrane (translucent gray). The full system simulated contains ~23 million million atoms [71]*.
MD simulations do not only support understanding of in vivo behavior, but are also capable of explaining in vitro processes and support the bioengineering of energy conversion devices. A remarkable example is the 200 ns MD simulation of the 3-million-atom system comprising the trimeric photosynthetic pigment-protein complex, photosystem I (PSI), embedded in a detergent belt of n-dodecyl-β-D-maltoside (DDM). The study performed by Frymier and coworkers [73]*9 gave atomic resolution insight into the impact of the detergent on conformational dynamics of the system and its possible implications on PSI functionality.
4.2 Cellulose and second-generation biofuels
Producing energy is not only essential for biological cells, but also for human society. Computer simulations are assisting in the development of cost-effective biofuels from agricultural waste, by studying enzymes and enzymatic complexes employed [18, 74, 75] in cellulose degradation as well as biomass recalcitrance [76, 77]. To avoid the recalcitrance problem, biomass pretreatment methods employ various chemical and thermal conditions to modify the molecular interactions and association of the different biomass components in favor of cellulose accessibility and digestibility [78]. Studying two of the main components of the biomass, namely lignin and cellulose in aqueous solution, Smith and coworkers carried out a 3.8-million-atom, 750 ns MD simulation. They suggested that the recalcitrance of crystalline cellulose to hydrolysis arises not only from the inaccessibility of inner fibers, but also from lignin adhesion [79]**10. Consequently, a pretreatment method which maximizes hydrophilicity of cellulose should benefit biofuel synthesis through an increase in digestibility and accessibility. Lignin, the component of biomass that is most difficult to degrade, was found to strongly associate with itself and also with cellulose. However, noncrystalline regions of cellulose were observed to have a lower tendency to associate with lignin than crystalline regions, due to stronger hydration of the noncrystalline chains.
MD simulations of large biological complexes are essential in bioenergy studies. Already reaching the 100-million-atom milestone, simulations of an entire chromatophore organelle are underway to investigate the overall mechanism of a large energy-converting system. Also taking advantage of large biomolecular complexes, biofuel production would certainly be bolstered by MD simulations of realistic plant cell wall models and efficient large multi-enzyme cellulosome complexes [75], making production more cost-effective.
5 Diverse applicability of large-scale simulations
Besides the aforementioned prominent applications to pharmaceutically relevant systems and energy conversion, MD simulations have been successfully employed to study a wide variety of large and complex systems. The scope ranges from fundamental biological processes like cell motility or essential membrane processes to biotechnological applications like biomaterial development.
5.1 Actin filament branch junction
Cell motility is organized mainly by coordinated regulation of microtubule and actin filament dynamics [80]. Actin networks are organized by actin-related protein 2/3 (Arp2/3) complexes that form geometrically regular junctions between two actin filaments [81]. Branched junction structures were proposed based on a 26 Å resolution electron microscopy (EM) tomogram [82], addressing questions about the interaction between the Arp2/3 domain and the mother actin filament. Gathering 31 protein subunits and including ~3 million atoms for almost 200 ns, MD simulations performed by Voth and coworkers suggest that the branched structure is remarkably stable [83]**11. Furthermore, the MD results were able to show that Arp2/3 complexes and the mother actin filament contain a large number of dynamic salt bridges and hydrophobic contacts that can form or break on the nanosecond timescale. MD simulations improved the fits of individual complex subunits to the EM density maps by a vast test with thousands of conformations docked by rigid body fitting into the EM map. It was possible to suggest targets for future experimental studies by examining the identified contacts between Arp2/3 complexes and actin filaments.
5.2 Realistic cell membrane
The cell membrane, which is the environment for manifold cellular processes, consists of many different lipid species. The minor lipid species that are present in very low concentration, yet essential in regulating membrane protein functions, can now be studied in large system simulations. Important examples of minor lipid species are polyunsaturated phospholipids that can enhance membrane fission induced by dynamin and endophilin [84]* 12. In a remarkable study, Marrink and coworkers performed CGMD simulations of a large plasma membrane that mimics the lipid composition of a cellular membrane [85]* 13. The plasma membrane model consisted of ~20,000 lipids with 63 lipid species. Due to the size of the membrane patch, built employing the coarse-grained MARTINI lipid model [86], it was possible to include minor lipid species in a large number. After 40 μs of simulation, the plasma membrane established an asymmetrical distribution of cholesterol and formed transient domains with liquid-order character, a finding which agrees with behaviors in a plasma membrane observed experimentally [86].
5.3 Membrane sculpting
Large-scale simulations, complementing recent advances in structural biology techniques, provide unique opportunities to study protein-induced membrane sculpting [88]. Complementing atomistic simulations, CG methods offer a feasible approach to studying relevant large protein-membrane assembly structures due to the long timescale associated with membrane morphology transitions and the large size of the protein-membrane complex [89]. Recently, large MD simulations have been carried out to study membrane tubulation induced by lattices of membrane sculpting proteins, namely the F-BAR domains [87]* 14. The dynamic process of F-BAR-driven tube formation from an initially flat membrane, as shown in Figure 5, was illustrated by employing ~350 μs CG simulations based on a shape-based coarse-grained model previously developed [90, 91]. The N-BAR domains are another example of membrane sculpting protein complexes. Voth and coworkers described the self-assembly mechanism of tubular networks by N-BAR domains starting from lipid vesicles [92]. Combining previous experiments [93] and CG simulations of a 26-site N-BAR domain model, the authors concluded that such tubular networks were formed by interactions between the membrane and amphipathic helices. Simulations of large protein-membrane assemblies at the sub-cellular scale serve as a stepping stone towards future whole-cell modeling at the molecular level.
Figure 5.
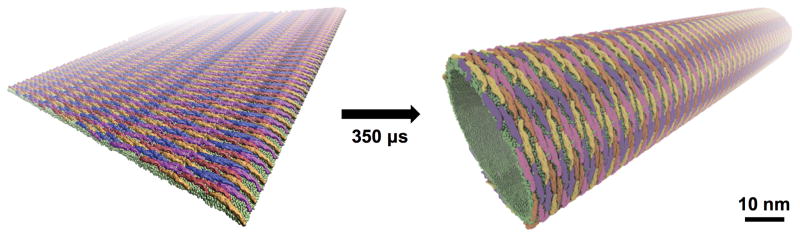
Membrane sculpting by F-BAR domains. Shape-based coarse-grained model simulations identified optimal lattice structures that maximize membrane curvature [87]*. Membrane lipids are shown in green and individual F-BAR domain within the lattice by the remaining colors seen.
5.4 Nuclear pore complex
Another large protein-membrane assembly at the sub-cellular scale is the nuclear pore complex (NPC) [94, 95], located in the double bilayers of the nuclear envelope mediating particle exchange between nucleus and cytoplasm. Nsp1, a nuclear pore protein, is the major component of NPC. MD simulations, performed at all-atom and coarse-grained levels, showed that an Nsp1 array forms dynamic, brush-like structures of multiprotein bundles [96]. More recently, microsecond-long CGMD simulations were performed on the entire transport channel of the NPC, the description of which required a 16-million-particle CGMD calculation. Although NPC has a physical opening of 60 nm in diameter, the cross-linking between the Nsp1 creates a sieve-like structure that limits the size of freely diffusing molecules passing through the NPC to < 9 nm [94]* 15. Larger molecules require the assistance of transport factors to be transported through the NPC.
5.5 DNA-functionalized nanoparticles
Even though widely applied to pure biological systems MD simulations are not restricted to them, and MD use in the development of biomaterials [97] and bio-functionalized materials with enzymes [98] has been widely effective. An important application concerns new systems for drug delivery. Gold nanoparticles (AuNPs) functionalized with DNA have been intensively exploited as a drug carrier even though little was known about molecular details of the complex. Performing MD simulations of systems of up to 5 million atoms, Vashishta and coworkers were able to construct two significant supercrystal conformations of AuNPs showing that DNA becomes capable in the presence of the supercrystals of smooth changes that do not arise for DNA in solution [17]*16. The differences resulted in a contraction of DNA by changing its base-base stacking. The simulations permitted an estimate of system properties, such as elasticity.
6 Conclusion
Through the combination of state-of-the-art computing hardware and advanced software development[11, 99] a true computational microscope has emerged [7]. Molecular dynamics simulation has established itself as a reliable tool to view the structure and dynamics of large protein complexes within realistic cellular environments. While all-atom MD remains the gold standard for MD simulations, CGMD approaches present a complementary technique to probe extended time and length scales [14], applicable in case one can be certain that the simplified force field captures the physical and chemical mechanism underlying the process investigated. Future physics-based multi-scale models, incorporating biochemical information and systems biology will enable the study of cellular-scale processes involving thousands of proteins, as exemplified by the pioneering work of Feig et. al, related to the atomistic description of the bacterial cytoplasm [5].
Advances in computational microscopy are moving the atomic resolution descriptions of biological systems into the 1 million (10 nm) to 1 billion (100 nm) atom regime, where Ångstrom-scale interactions bring about 100-nm scale concerted dynamic processes, crucial for numerous cell functions. This regime is rich in exciting biological questions and characteristic of large biomolecular systems [100, 101, 102, 103] beyond those described here.
In this opinion, we reviewed the progress driven by large-scale molecular dynamics simulations in applications ranging from cell biology to health sciences to biofuel production. Considering the success of these applications, we forsee large-scale simulations playing an ever-increaseing role. Combined, simulation and experiment are helping to bridge the gap between atomic level properties and whole cell behavior, an endeavor which cannot be accomplished by either approach alone.
Highlights.
Advances in MD and imaging enables structural determination of large complexes.
Large-scale MD is essential to the atomic level description of cell-scale processes.
MD enables the study of local and global dynamics of multi-million atom complexes.
Acknowledgments
We thank Dr. Yanxin Liu for providing the figure of the RHDV virus. The authors gratefully acknowledge funding from the National Institutes of Health (NIH, 9P41GM104601, P01-GM067887, U54 GM087519), the National Science Foundation (NSF, MCB-1157615, PHY1430124), and Energy Biosciences Institute (EBI, 231 UCB BP 2014OO4J01). TR is supported by the Alexander von Humboldt Foundation.
Footnotes
[21] * The authors present a computational model of an Influenza A virion. The model enables the study of biophysical properties of the viral particle that may be related to the fitness of the virus.
[1]** The authors solve two different structures for the HIV-1 mature capsid. The atomic capsid model derived from cryo-electron microscopy, computational modeling, coarse-grained Monte Carlo and MD simulation provides unprecedented atomic detail of the trimeric interface, critical for infectivity.
[15]* The authors obtain an improved virus capsid model using multimillion-atom MDFF calculations that combine the structural information from cryo-EM and X-ray crystallography.
[23]* This paper describes various assembly pathways of a non-enveloped DNA virus under different ionic concentrations by using coarse-grained Brownian dynamics simulation.
[45]** The work is the first constant pH MD simulation on the maturation process of a virus. The methods and approach presented in this paper could be used to investigate many large scale pH-dependent processes.
[49]** Combination of cryo-EM data and MD simulations reveal a full picture of ribosomal translocation at atomic-detail.
[16]* MD simulations reproducibly see the antibiotic drug erythromycin inducing conformational changes at the catalytic center of the bacterial ribosome.
[71]* This study consolidates several experimental results in a MD simulation of a large multi-protein-membrane system, which served as input to study the quantum process of photosynthetic light harvesting.
[73]* MD simulations provide insight into the in vitro behavior of photosystem I (PSI) in detergent.
[79]** The landmark paper describes the structure of the cellulose-lignin complex, showing that recalcitrance of biomass results from lignin adhesion to cellulose.
[83]** MD simulations identified the dynamics of the contact area between actin filament and Arp2/3.
[84]* The authors showed that polyunsaturated phospholipids, even though they arise only as a small precentage of membrane lipids, enhance membrane fission induced by dynamin and endophilin.
[85]* The authors performed the first MD simulation on a membrane bilayer with realistic lipid composition.
[87]* The authors demonstrated that the F-BAR domains generate membrane sculpting via the scaffolding mechanism; the protein lattice structure optimal for producing membrane curvature was determined.
[94]* The authors studied nuclear pore complex structure and function by means of residue-based CGMD simulations, one of the largest simulations of the kind ever carried out.
[17]* This paper reveals the applicability of MD simulations to study the interaction between inorganic materials and biological molecules.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronen-born AM, Schulten K, Aiken C, Zhang P. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mei C, Sun Y, Zheng G, Bohm EJ, Kalé LV, Phillips JC, Harrison C. Enabling and scaling biomolecular simulations of 100 million atoms on petascale machines with a multicore-optimized message-driven runtime. Proceedings of the 2011 ACM/IEEE conference on Supercomputing; Seattle, WA. 2011. [Google Scholar]
- 3.Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Jr, Assad-Garcia N, Glass JI, Covert MW. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts E. Cellular and molecular structure as a unifying framework for whole-cell modeling. Current opinion in structural biology. 2014;25:86–91. doi: 10.1016/j.sbi.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Feig M, Harada R, Mori T, Yu I, Takahashi K, Sugita Y. Complete atomistic model of a bacterial cytoplasm for integrating physics, biochemistry, and systems biology. Journal of Molecular Graphics and Modelling. 2015;58:1–9. doi: 10.1016/j.jmgm.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou HX. Theoretical frameworks for multiscale modeling and simulation. Current opinion in structural biology. 2014;25:67–76. doi: 10.1016/j.sbi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee EH, Hsin J, Sotomayor M, Comellas G, Schulten K. Discovery through the computational microscope. Structure. 2009;17:1295–1306. doi: 10.1016/j.str.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. Gromacs 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:1–10. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plimpton SJ. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics. 1995;117:1–19. [Google Scholar]
- 11.Phillips JC, Sun Y, Jain N, Bohm EJ, Kalé LV. Mapping to irregular torus topologies and other techniques for petascale biomolecular simulation. Proceedings of the International Conference on High Performance Computing, Networking, Storage and Analysis SC ’14; IEEE Press; 2014. pp. 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular Dynamics Flexible Fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingólfsson HI, Lopez CA, Uusitalo JJ, de Jong DH, Gopal SM, Periole X, Marrink SJ. The power of coarse graining in biomolecular simulations. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2014;4:225–248. doi: 10.1002/wcms.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Xu F, Liu J, Gao B, Liu Y, Zhai Y, Ma J, Zhang K, Baker TS, Schulten K, Zheng D, Pang H, Sun F. Atomic model of rabbit hemorrhagic disease virus by cryo-electron microscopy and crystallography. PLoS Pathogens. 2013;9:e1003132. 14. doi: 10.1371/journal.ppat.1003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sothiselvam S, Liu B, Han W, Klepacki D, Atkinson GC, Brauer A, Remm M, Tenson T, Schulten K, Vázquez-Laslop N, Mankin AS. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proceedings of the National Academy of Sciences, USA. 2014;111:9804–9809. doi: 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngo VA, Kalia RK, Nakano A, Vashishta P. Supercrystals of dna-functionalized gold nanoparticles: a million-atom molecular dynamics simulation study. Journal of Physical Chemistry C. 2012;116:19579–19585. [Google Scholar]
- 18.Beckham GT, Bomble YJ, Bayer EA, Himmel ME, Crowley MF. Applications of computational science for understanding enzymatic deconstruction of cellulose. Current Opinion in Biotechnology. 2011;22:231–238. doi: 10.1016/j.copbio.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Roos W, Bruinsma R, Wuite G. Physical virology. Nature Physics. 2010;6:733–743. [Google Scholar]
- 20.Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure. 2006;14:437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Reddy T, Shorthouse D, Parton DL, Jefferys E, Fowler PW, Chavent M, Baaden M, Sansom MS. Nothing to sneeze at: A dynamic and integrative computational model of an influenza a virion. Structure. 2015;23:584–597. doi: 10.1016/j.str.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andoh Y, Yoshii N, Yamada A, Fujimoto K, Kojima H, Mizutani K, Nakagawa A, Nomoto A, Okazaki S. All-atom molecular dynamics calculation study of entire po-liovirus empty capsids in solution. Journal of Chemical Physics. 2014;141:165101. doi: 10.1063/1.4897557. [DOI] [PubMed] [Google Scholar]
- 23.Perlmutter JD, Perkett MR, Hagan MF. Pathways for virus assembly around nucleic acids. Journal of Molecular Biology. 2014;18:3148–3165. doi: 10.1016/j.jmb.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts JA, Kuiper MJ, Thorley BR, Smooker PM, Hung A. Investigation of a predicted n-terminal amphipathic α-helix using atomistic molecular dynamics simulation of a complete prototype poliovirus virion. Journal of Molecular Graphics and Modelling. 2012;38:165–173. doi: 10.1016/j.jmgm.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Larsson DSD, Liljas L, van der Spoel D. Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Computational Biology. 2012;8:e1002502. doi: 10.1371/journal.pcbi.1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zink M, Grubmüller H. Mechanical properties of the icosahedral shell of southern bean mosaic virus: A molecular dynamics study. Biophysical Journal. 2009;96:1350–1363. doi: 10.1016/j.bpj.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bharat TAM, Castillo Menendez LR, Hagen WJH, Lux V, Igonet S, Schorb M, Schur FKM, Kräusslich H-G, Briggs JAG. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proceedings of the National Academy of Sciences, USA. 2014;111:8233–8238. doi: 10.1073/pnas.1401455111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schur FKM, Hagen WJH, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JAG. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature. 2015;517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 29.Ambrose Z, Aiken C. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology. 2014;454:371–379. doi: 10.1016/j.virol.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rapaport DC. Molecular dynamics simulation: a tool for exploration and discovery using simple models. Journal of Physics: Condensed Matter. 2014;26:503104. doi: 10.1088/0953-8984/26/50/503104. [DOI] [PubMed] [Google Scholar]
- 31.Polles G, Indelicato G, Potestio R, Cermelli P, Twarock R, Micheletti C. Mechanical and assembly units of viral capsids identified via quasi-rigid domain decomposition. PLoS Computational Biology. 2013;9:e1003331. doi: 10.1371/journal.pcbi.1003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie L, Smith GR, Feng X, Schwartz R. Surveying capsid assembly pathways through simulation-based data fitting. Biophysical Journal. 2012;103:1545–1554. doi: 10.1016/j.bpj.2012.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perlmutter JD, Qiao C, Hagan MF. Viral genome structures are optimal for capsid assembly. eLife. 2013;2:e00632. doi: 10.7554/eLife.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapaport DC. Molecular dynamics simulation of reversibly self-assembling shells in solution using trapezoidal particles. Physical Review E. 2012;86:051917. doi: 10.1103/PhysRevE.86.051917. [DOI] [PubMed] [Google Scholar]
- 35.Chen B, Tycko R. Simulated self-assembly of the HIV-1 capsid: protein shape and native contacts are sufficient for two-dimensional lattice formation. Biophysical Journal. 2011;100:3035–3044. doi: 10.1016/j.bpj.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grime J, Voth GA. Early stages of the HIV-1 capsid protein lattice formation. Biophysical Journal. 2012;103:1774–1783. doi: 10.1016/j.bpj.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller PW, Huang RK, England MR, Waki K, Cheng N, Heymann JB, Craven RC, Freed EO, Steven AC. A two-pronged structural analysis of retroviral maturation indicates that core formation proceeds by a disassembly-reassembly pathway rather than a displacive transition. Journal of Virology. 2013;87:13655–13664. doi: 10.1128/JVI.01408-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veesler D, Johnson JE. Virus maturation. Annual Review of Biophysics. 2012;41:473–96. doi: 10.1146/annurev-biophys-042910-155407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao Y, Ortoleva PJ. Viral structural transition mechanisms revealed by multiscale molecular dynamics/order parameter extrapolation simulation. Biopolymers. 2010;93:61–73. doi: 10.1002/bip.21299. [DOI] [PubMed] [Google Scholar]
- 40.Miao Y, Johnson JE, Ortoleva PJ. All-atom multiscale simulation of CCMV capsid swelling. Journal of Physical Chemistry B. 2010;114:11181–11195. doi: 10.1021/jp102314e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dykeman EC, Sankey OF. Normal mode analysis and applications in biological physics. Journal of Physics: Condensed Matter. 2010;22:423202. doi: 10.1088/0953-8984/22/42/423202. [DOI] [PubMed] [Google Scholar]
- 42.Roos WH, Gibbons MM, Arkhipov A, Uetrecht C, Watts N, Wingfield P, Steven AC, Heck AJR, Schulten K, Klug WS, Wuite GJ. Squeezing protein shells: how continuum elastic models, molecular dynamics simulations and experiments coalesce at the nanoscale. Biophysical Journal. 2010;99:1175–1181. doi: 10.1016/j.bpj.2010.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu M, Ming D, Ma J. fSUB: Normal mode analysis with flexible substructures. The Journal of Physical Chemistry B. 2012;116:8636–8645. doi: 10.1021/jp300312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khandogin J, Brooks CL. Constant pH molecular dynamics with proton tautomerism. Biophysical Journal. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.May ER, Arora K, Brooks CL. pH-induced stability switching of the bacteriophage HK97 maturation pathway. Journal of the American Chemical Society. 2014;136:3097–3107. doi: 10.1021/ja410860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz-Herrero T, Hagan MF. Simulations show that virus assembly and budding are facilitated by membrane microdomains. Biophysical Journal. 2015;108:585–595. doi: 10.1016/j.bpj.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D’Orso I, Fernandes J, Fahey M, Mahon C, O’Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanbonmatsu KY. Computational studies of molecular machines: the ribosome. Current Opinion in Structural Biology. 2012;22:168–174. doi: 10.1016/j.sbi.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bock LV, Blau C, Schröde GF, Davydo I, Fischer N, Stark H, Rodnina MV, Vaiana AC, Grubmüller H. Energy barriers and driving forces in tRNA translocation through the ribosome. Nature Structural & Molecular Biology. 2013;20:1390–1396. doi: 10.1038/nsmb.2690. [DOI] [PubMed] [Google Scholar]
- 50.Shih AY, Freddolino PL, Sligar SG, Schulten K. Disassembly of nanodiscs with cholate. Nano Letters. 2007;7:1692–1696. doi: 10.1021/nl0706906. [DOI] [PubMed] [Google Scholar]
- 51.Frauenfeld J, Gumbart J, van der Sluis EO, Funes S, Gartmann M, Beatrix B, Mielke T, Berninghausen O, Becker T, Schulten K, Beckmann R. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nature Structural & Molecular Biology. 2011;18:614–621. doi: 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodnina MV, Wintermeyer W. The ribosome as a molecular machine: the mechanism of tRNA-mRNA movement in translocation. Biochemical Society Transactions. 2011;39:658–662. doi: 10.1042/BST0390658. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Cruz-Chu ER, Woodard J, Gartia MR, Schulten K, Liu L. Electrically induced conformational change of peptides on metallic nano-surfaces. ACS Nano. 2012;6:8847–8856. doi: 10.1021/nn3027408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitford PC, Blanchard SC, Cate JHD, Sanbonmatsu KY. Connecting the kinetics and energy landscape of trna translocation on the ribosome. PLoS Computational Biology. 2013;9:e1003003. doi: 10.1371/journal.pcbi.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 56.Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nature Reviews Microbiology. 2005;3:870–881. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- 57.Meroueh SO, Mobashery S. Conformational transition in the aminoacyl t-rna site of the bacterial ribosome both in the presence and absence of an aminoglycoside antibiotic. Chemical Biology and Drug Design. 2007;69:291–297. doi: 10.1111/j.1747-0285.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 58.Vaiana AC, Sanbonmatsu KY. Stochastic gating and drug-ribosome interactions. Journal of Molecular Biology. 2009;386:648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aleksandrov A, Simonson T. Binding of tetracyclines to elongation factor Tu, the Tet repressor, and the ribosome: a molecular dynamics simulation study. Biochemistry. 2008;47:13594–13603. doi: 10.1021/bi801726q. [DOI] [PubMed] [Google Scholar]
- 60.Wolf A, Baumann S, Arndt HD, Kirschner KN. Influence of thiostrepton binding on the ribosomal GTPase associated region characterized by molecular dynamics simulation. Bioorganic Medical Chemistry. 2012;20:7194–7205. doi: 10.1016/j.bmc.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 61.Small MC, Lopes P, Andrade RB, MacKerell AD., Jr Impact of ribosomal modifi-cation on the binding of the antibiotic telithromycin using a combined grand canonical monte carlo/molecular dynamics simulation approach. PLoS Computational Biology. 2013;9:e1003113. doi: 10.1371/journal.pcbi.1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laing R, Waning B, Gray A, Ford N, Hoen E. 25 years of the WHO essential medicines lists: progress and challenges. Lancet. 2003;361:1723–1729. doi: 10.1016/S0140-6736(03)13375-2. [DOI] [PubMed] [Google Scholar]
- 63.Lindenberg M, Kopp S, Dressman J. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biophar-maceutics classification system. European Journal of Pharmaceutics and Biopharmaceutics. 2004;58:265–278. doi: 10.1016/j.ejpb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 64.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 65.Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Molecular Cell. 2002;10:117–128. doi: 10.1016/s1097-2765(02)00570-1. [DOI] [PubMed] [Google Scholar]
- 66.Tenson T, Lovmar M, Ehrenberg M. The mechanism of action of macrolides, lin-cosamides and streptogramin b reveals the nascent peptide exit path in the ribosome. Journal of Molecular Biology. 2003;330:1005–1014. doi: 10.1016/s0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- 67.Arenz S, Meydan S, Starosta AL, Berninghausen O, Beckmann R, Vázquez-Laslop N, Wilson DN. Drug sensing by the ribosome induces translational arrest via active site perturbation. Molecular Cell. 2014;56:446–452. doi: 10.1016/j.molcel.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brandman R, Brandman Y, Pande VS. A-Site Residues Move Independently from P-Site Residues in all-Atom Molecular Dynamics Simulations of the 70S Bacterial Ribosome. PLoS One. 2012;7:e29377. doi: 10.1371/journal.pone.0029377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. Structures of the human and Drosophila 80S ribosomes. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- 70.Hunter CN, Daldal F, Thurnauer MC, Beatty JT, editors. The Purple Phototrophic Bacteria. Springer; 2008. Advances in Photosynthesis and Respiration. [Google Scholar]
- 71.Chandler D, Strümpfer J, Sener M, Scheuring S, Schulten K. Light harvesting by lamellar chromatophores in Rhodospirillum photometricum. Biophysical Journal. 2014;106:2503–2510. doi: 10.1016/j.bpj.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cartron ML, Olsen JD, Sener M, Jackson PJ, Brindley AA, Qian P, Dickman MJ, Leggett GJ, Schulten K, Hunter CN. Integration of energy and electron transfer processes in the photosynthetic membrane of Rhodobacter sphaeroides. Biochimica et Biophysica Acta – Bioenergetics. 2014;1837:1769–1780. doi: 10.1016/j.bbabio.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris BJ, Cheng X, Frymier P. All-atom molecular dynamics of a photosystem i/detergent complex. Journal of Physical Chemistry B. 2014;118:11633–11645. doi: 10.1021/jp507157e. [DOI] [PubMed] [Google Scholar]
- 74.Bernardi RC, Cann I, Schulten K. Molecular dynamics study of enhanced Man5B enzymatic activity. Biotechnology for Biofuels. 2014;7:1–8. doi: 10.1186/1754-6834-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schoeler C, Malinowska KH, Bernardi RC, Milles LF, Jobst MA, Durner E, Ott W, Fried DB, Bayer EA, Schulten K, Gaub HE, Nash MA. Ultrastable cellulosome-adhesion complex tightens under load. Nature Communications. 2014;5:5635. doi: 10.1038/ncomms6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hadden JA, French AD, Woods RJ. Effect of microfibril twisting on theoretical powder diffraction patterns of cellulose iβ. Cellulose. 2014;21:879–884. doi: 10.1007/s10570-013-0051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science. 2007;315:804–807. doi: 10.1126/science.1137016. [DOI] [PubMed] [Google Scholar]
- 78.Agbor VB, Cicek N, Sparling R, Berlin A, Levin DB. Biomass pretreatment: fundamentals toward application. Biotechnology Advances. 2011;29:675–685. doi: 10.1016/j.biotechadv.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 79.Lindner B, Petridis L, Schulz R, Smith JC. Solvent-driven preferential association of lignin with regions of crystalline cellulose in molecular dynamics simulation. Biomacro-molecules. 2013;14:3390–3398. doi: 10.1021/bm400442n. [DOI] [PubMed] [Google Scholar]
- 80.Bugyi B, Carlier MF. Control of actin filament treadmilling in cell motility. Annual Review of Biophysics. 2010;39:449–470. doi: 10.1146/annurev-biophys-051309-103849. [DOI] [PubMed] [Google Scholar]
- 81.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annual Review of Biophysics and Biomolecular Structure. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 82.Rouiller I, Xu XP, Amann KJ, Egile C, Nickell S, Nicastro D, Li R, Pollard TD, Volkmann N, Hanein D. The structural basis of actin filament branching by the Arp2/3 complex. Journal of Cell Biology. 2008;180:887–895. doi: 10.1083/jcb.200709092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pfaendtner J, Volkmann N, Hanein D, Dalhaimer P, Pollard TD, Voth GA. Key structural features of the actin filament Arp2/3 complex branch junction revealed by molecular simulation. Journal of Molecular Biology. 2012;416:148–161. doi: 10.1016/j.jmb.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pinot M, Vanni S, Pagnotta S, Lacas-Gervais S, Payet LA, Ferreira T, Gautier R, Goud B, Antonny B, Barelli H. Polyunsaturated phospholipids facilitate membrane deformation and fission by endocytic proteins. Science. 2014;345:693–697. doi: 10.1126/science.1255288. [DOI] [PubMed] [Google Scholar]
- 85.Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, López CA, Wassenaar TA, Periole X, De Vries AH, Tieleman DP, Marrink SJ. Lipid organization of the plasma membrane. Journal of the American Chemical Society. 2014;136:14554–14559. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- 86.Marrink SJ, Tieleman DP. Perspective on the MARTINI model. Chemical Society Reviews. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- 87.Yu H, Schulten K. Membrane sculpting by F-BAR domains studied by molecular dynamics simulations. PLoS Computational Biology. 2013;9:e1002892. doi: 10.1371/journal.pcbi.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frost A, Perera R, Roux A, Spasov K, Destaing O, Egelman EH, De Camilli P, Unger VM. Structural basis of membrane invagination by F-BAR domains. Cell. 2008;132:807–817. doi: 10.1016/j.cell.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arkhipov A, Freddolino PL, Schulten K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure. 2006;14:1767–1777. doi: 10.1016/j.str.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 90.Arkhipov A, Yin Y, Schulten K. Four-scale description of membrane sculpting by BAR domains. Biophysical Journal. 2008;95:2806–2821. doi: 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yin Y, Arkhipov A, Schulten K. Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure. 2009;17:882–892. doi: 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Simunovic M, Mim C, Marlovits T, Resch G, Unger V, Voth G. Protein-mediated transformation of lipid vesicles into tubular networks. Biophysical Journal. 2013;105:711–719. doi: 10.1016/j.bpj.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, Unger VM. Structural basis of membrane bending by the N-BAR protein endophilin. Cell. 2012;149:137–145. doi: 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gamini R, Han W, Stone JE, Schulten K. Assembly of Nsp1 nucleoporins provides insight into nuclear pore complex gating. PLoS Computational Biology. 2014;10:e1003488. doi: 10.1371/journal.pcbi.1003488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mincer JS, Simon SM. Simulations of nuclear pore transport yield mechanistic insights and quantitative predictions. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3351–3358. doi: 10.1073/pnas.1104521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miao L, Schulten K. Transport-related structures and processes of the nuclear pore complex studied through molecular dynamics. Structure. 2009;17:449–459. doi: 10.1016/j.str.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sarikaya M, Tamerler C, Jen AKY, Schulten K, Baneyx F. Molecular biomimetics: nanotechnology through biology. Nature Materials. 2003;2:577–585. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 98.Gomes DEB, Lins RD, Pascutti PG, Lei C, Soares TA. The role of nonbonded interactions in the conformational dynamics of organophosphorous hydrolase adsorbed onto functionalized mesoporous silica surfaces. Journal of Physical Chemistry B. 2010;114:531–540. doi: 10.1021/jp9083635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shaw DE, Grossman J, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; IEEE Press; 2014. pp. 41–53. [Google Scholar]
- 100.Bui KH, von Appen A, DiGuilio AL, Ori A, Sparks L, Mackmull MT, Bock T, Hagen W, Andrés-Pons A, Glavy JS, et al. Integrated structural analysis of the human nuclear pore complex scaffold. Cell. 2013;155:1233–1243. doi: 10.1016/j.cell.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 101.Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cramer P. A tale of chromatin and transcription in 100 structures. Cell. 2014;159:985–994. doi: 10.1016/j.cell.2014.10.047. [DOI] [PubMed] [Google Scholar]
- 103.Falke JJ, Piasta KN. Architecture and signal transduction mechanism of the bacterial chemosensory array: Progress, controversies, and challenges. Current Opinion in Structural Biology. 2014;29:85–94. doi: 10.1016/j.sbi.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]