

Abstract
Rheumatoid arthritis (RA) is associated with the presence of autoreactive CD4 T cells that produce pro-inflammatory cytokines. The role of genetic factors in the predilection to develop RA is strongly supported by the increased presence of certain HLA class II molecules in patients. The HLA class II genes are highly polymorphic and are critical for generating an immune response to clear infections. Production of Th1 and Th17 response by the CD4 T cells helps to clear infections. HLA-DQ8 is a promiscuous binder and presents many peptides generating immune response and producing a Th17 response. DRB1*0401 is associated with the production of both IL-17 and IFN-γ. Thus both DR4 and DQ8 can clear infections by producing TH1/Th17 cytokines, but their presence increases the risk of developing RA. Using transgenic mice expressing human HLA genes, we have shown that HLA polymorphism determines the cytokine profile. DRB1*04 molecules modulate the DQ8-restricted response and determine the outcome of arthritis in mice carrying DR4/DQ8 haplotype. Thus, interaction between DQ and DR molecules determines the cytokine milieu and propensity of the HLA haplotype to predispose to autoimmunity.
Rheumatoid arthritis (RA) is an autoimmune disease caused by the inflammatory changes in the immune system. While the causative antigen of RA is unknown, an infectious etiology has been suggested based on an improvement in patients treated with antibiotics [1]. Many infectious agents like EBV and parvovirus, among others, have been implicated in the pathogenesis of RA although the mechanism by which pathogens cause pathology is unknown. One proposed mechanism by which infectious agents and other environmental factors are involved in causing autoreactivity is called “molecular mimicry”. During infection the body generates a response to clear infection but a cross reactive response to epitopes of the infectious agents that are similar to self-protein can cause autoreactive T cells to expand. Even after clearance of infection, this autoreactive response may continue, due in part, to the availability of the self-protein. Modification of proteins occurs in normal healthy state to generate immune response. However during the process of post translational modifications, cryptic epitopes sharing sequences with viral or bacterial proteins may become available. There is some evidence that suggests that certain modified peptides bind the HLA-DR molecules better than naïve peptides [2].
The HLA molecules are encoded on chromosome 6 and are crucial in clearing infections by generating an immune response to pathogens. The class I and class II genes are the two major classes of the HLA loci that are involved in fighting infections. HLA genes encoded in the class I- B loci and class II –DRB1 loci are the most polymorphic. The polymorphism of HLA genes is attributed to the selective pressures of pathogens. Thus the HLA alleles that generate a response against most pathogens by activating CD4+ T cells and producing cytokines, resulting in clearance of infections, have been preserved. This is supported by a recent meta-analysis showing association of certain HLA alleles with an effective clearance of infections [3].
According to the paradigm, presentation of a peptide via class I molecules activate CD8 T cells while class II molecules activate CD4 T cells. Activated T cells produce cytokines to clear infections. While both class I and class II alleles generate responses to infectious agents, only class II molecules have been associated with a predisposition to autoimmunity. Several hypotheses have been put forth to explain the HLA association with autoreactivity, however, the mechanism by which class II molecules predispose to autoimmunity still remains an enigma. Positive and negative T cell selection in the thymus by the HLA molecules provides one mechanism. The other is the HLA-mediated antigen presentation to CD4 T cells and subsequent cytokine production. However, the immune response generated via class II molecules may also lead to bystander damage which, in certain conditions, causes pathology.
Cytokines and class II genes in infections
The major function of class II molecules is to clear infections through the adaptive immune response. Presentation of the MHC-peptide complex by antigen presenting cells to CD4 T cells leads to production of cytokines, Th1, Th2, Th9 and Th17. For clearing intracellular bacterial and viral infections, IFN-γ production by Th1 cells leads to a cellular response, differentiation of CD4 T cells into Th1 cells and the activation of macrophages which produce Th1 cytokines. IFN-γ also increases expression of MHC class II molecules, thereby increasing MHC-restricted antigen presentation and adaptive immune response. Production of IFN-γ suppresses the Th2 and Th17 response, skewing the response to be Th1.
Clearance of extracellular bacterial and fungal infections requires Th17 cells. Among Th17 cytokines, IL-17 is one of the most studied. IL-17 is a pleiotropic cytokine that acts on many immune and epithelial cells [4]. Binding of IL-17 to its receptor activates production of other pro-inflammatory cytokines, IL-1β, IL-6, and TNF-α as well as various chemokines resulting in the recruitment of neutrophils, macrophages and lymphocytes. IL-6 in conjunction with TGFβ amplifies differentiation of Th17 cells in a positive feedback loop. IL-17 also promotes a humoral response that is essential for clearing extracellular pathogens. Recently, an important role of Th17 cells has been shown in mucosal associated infections in lungs and guts[5]. However, in some individuals, an amplified immune response to clear infectious agents leads to tissue damage ensuing inflammatory autoimmune diseases. Inflammatory diseases including RA have increased numbers of IL-17 producing cells [6, 7].
Most individuals have viral and bacterial infections at some point of their life but not all individuals who are infected develop autoreactivity. Most of the autoimmune diseases are strongly associated with certain class II alleles. Even though the HLA-DR and DQ genes are highly polymorphic, the 3 most common haplotypes present in various autoimmune diseases are DR2/DQ6, DR3/DQ2, and DR4/DQ8. These haplotypes are also present with high frequency in the healthy population. One can speculate that these class II haplotypes are promiscuous binders and are good for clearing infection. However, variability in the outcome of infectious diseases suggests a role for genetic factors. This is supported by the observations that the outcome of hepatitis C virus infection is influenced by the MHC class II genotype, both DRB1*0401 and DRB1*1501 are linked with increased clearance of hepatitis C [8]. Similarly, clearance of hepatitis B is also associated with certain HLA genotypes [3].
This led us to hypothesize that the HLA alleles that can generate a strong immune response and produce IFN-γ or IL-17 to infectious agents may have been selected and preserved over generations. However, some infectious agents may harbor epitopes that share similarities with human proteins. While these epitopes may not be available easily in normal healthy state, they may become available due to post translational modifications. Post-translational modifications like citrullination are known to open the folded structure of the protein exposing some of the cryptic epitopes. If these cryptic epitopes are available to the activated immune cells for presentation, a self-reactive response may be generated. One such example is alpha-enolase, which is a conserved molecule throughout eukaryotes and prokaryotes. Antibodies to citrullinated alpha enolase are expressed in the joints of RA patients. A recent study showed that a cross-reactive response to a bacterial enolase may prime autoimmunity in a subset of patients[9]. Citrullination is a process where the amino acid arginine in a protein is replaced with citrulline. This process has physiologic relevance as it is required for the generation of skin, hair follicles, and myelin sheaths of nerve fibers as well as for transcriptional and chromatin compaction regulation [10]. Antibodies to citrullinated proteins have been described not only in RA but in many other inflammatory diseases including multiple sclerosis and myositis [11–13]. Thus while citrullinated proteins are a part of healthy state, in individuals with certain class II alleles, a loss of tolerance to citrullinated proteins or an autoreactive response generated after clearance of an infectious agent may cause onset of disease. This could be due, in part, to a storm of cytokines produced to clear infections. This is supported by the association of HLA class II alleles like DRB1*0401 with the presence of anti-citrullinated antibodies.
Cytokine and Rheumatoid Arthritis
Cytokines have a fundamental role in causing inflammation and now it has become clear that cytokines are the prime suspects in articular destruction [14]. The role of cytokines in the pathogenesis of RA is underscored by the success of treatment with anti-TNF antibodies in RA patients. The success of TNF-inhibitor has set the stage for using inhibitors of other pro-inflammatory cytokines in RA. The rheumatoid joints have infiltrating macrophages that can secrete chemokines and cytokines of the innate and adaptive immune system. The recruitment of activated T and B cells involves secretion of chemokines by antigen presenting cells and synovial endothelium. Activated T cells produce cytokines in the joints further amplifying the inflammatory cascade. A recent study showed an increased numbers of pro-inflammatory cytokines producing T cells reactive to citrullinated Vimentin [15]. There is a preponderance of Th1 cells in rheumatoid joints with the majority of cells producing IFN-γ [16]. IFN-γ can enhance the production of other Th1 cytokines like IL-1, IL-6 and TNF-α. IL-6 is a potent pro-inflammatory Th1 cytokine that regulates hematopoiesis and is associated with differentiation of T and B cells [17]. Blocking IL-6R with an antibody has been shown to be effective in some patients [18]. Presentation of cartilage antigens in the joints can further cause perpetuation of an ongoing inflammatory cascade in the joints. RA has been suggested to be a Th17 dependent disease, IL-17 producing cells have been observed in RA synovium [6]. IL-23, a member of IL-12 family, can cause differentiation of naïve cells into IL-17 producing cell. IL-17 is a pro-inflammatory cytokine which acts on various cells and also induces osteoclast differentiation via the RANKL pathway [19]. Activation of IL-17R induces production of other inflammatory cytokines and chemokines that initiate recruitment of neutrophils, macrophages and lymphocytes. Phase I trials of anti-IL-17 antibodies have shown some efficacy in RA patients. A role of Th17 in RA is further supported by animal models of arthritis [20–22]. While Th1 and Th17 cytokines are associated with pathogenesis in RA, Th2 cytokines are associated with protection from arthritis. However, higher levels of IL-13 are present in RA patients as compared to that of healthy individuals [23]. The other cytokines produced in the joints, which include IL-15 and IL-10, may be important for preventing T and B cells from undergoing apoptosis and for T cell function [16, 24]. Chronic inflammation can lead to production of IL-10 and TGF-β that can diminish inflammatory cytokines. While both of these cytokines are immunomodulatory, TGF-β is also involved in pro-inflammatory pathways as it induces differentiation of pathogenic Th17 cells in the presence of IL-6 [25]. Thus it’s not clear if these cytokines are inhibitory in joints. These studies demonstrate an unmet need to define other biologics that may be effective for subsets of patients who do not respond to the available drugs. A recent study identified a signature cytokine profile, produced by PBMCs to various stimuli, to define myocardial dysfunction in RA patients [26]. Using DR4 tetramers, it was shown that while both RA patients and healthy individuals harbor DR4 positive T cells although T regulatory cells are diminished in patients [27]. This may explain inability of RA patients to suppress response to citrullinated antigens. Heterogeneity in RA may be associated with a heterogeneous cytokine response and that may explain why not all individuals respond to one therapy. The factors that determine the variability of cytokine response observed in patients largely remain unknown. Animal models provide some clues to the role of MHC molecules in predetermining the cytokine response.
MHC genes, cytokines and autoimmunity
The animal model of collagen-induced arthritis (CIA) has helped us understand the role of cytokines in arthritis. However, the MHC molecules of human and mice have differences. Rheumatoid arthritis is associated with the presence of DRB1*0401 while *0402 is not. The recent structural studies indicated that the peptide binding differences between the *0401 and *0402 molecules determines the association with RA[27]. Both subtypes of DR4 occur in linkage disequilibrium with HLA-DQ8, making it impossible to determine the role of a single gene. Mice expressing DR4:IE are susceptible to arthritis induced by citrullinated fibrinogen which is dependent on Th1 response to the antigen[28]. We generated transgenic mice which expressed RA-associated and non-associated HLA genes in the absence of endogenous class II alleles. We hypothesized that the presence of RA-susceptible HLA genes leads to a T cell repertoire that is predetermined to produce a pro-inflammatory cytokine profile. On the other hand RA- non-associated HLA genes can cause differentiation of naïve cells in to regulatory cells under polarizing conditions and also require different conditions for producing pro-inflammatory cytokines.
We tested this hypothesis by using mice expressing *0401 and *0402. Similar to humans, *0401 mice are susceptible to develop CIA while *0402 mice are resistant [29]. In addition, antigen-primed cells from *0401 mice produce higher levels of Th17 cytokines compared to *0402 mice although both strains can produce IFN-γ [30]. Observations in *0401 transgenic mice showed that naïve CD4 T cells from *0401 mice, under Th17 polarizing conditions, are more likely to differentiate into IL-17+ cells compared to *0402 mice [30, 31]. On the other hand, under Th2 polarizing conditions, *0402 leads to differentiation of naïve CD4 T cells into IL-10 producing cells. Recent studies have shown that IL-17 may be required for clearing infections, suggesting that individuals carrying *0401 may be better equipped to clear infection. However, the activated immune response in *0401 individuals may not shut off due to high expression of costimulatory molecules which are unregulated on activated cells or due to activation of other cells that produce proinflammatory cytokines. Thus even after clearance of infection, IL-17 production may continue causing direct damage to the tissue. This may also lead to production of a cascade of pro-inflammatory cytokines. We addressed the question about IL-17 production by molecules not associated with RA. Mice carrying the RA-resistant allele *0402 are not defective in producing IL-17, as they do produce IL-17 but also large quantities of IL-10 are produced thus immune response can be suppressed after initial activation. When naïve CD4 T cells from *0402 mice were cultured in polarizing conditions, the production of IL-17 was delayed as compared to *0401[30]. In addition, the conditions required for the production of IL-17 also differed between the two molecules. This data suggests that while both molecules are capable of producing IL-17 and IL-10, a difference in the kinetics of response may dictate their ability to clear infections and association with autoreactivity.
HLA haplotype determines cytokine response in arthritis
HLA molecules are critical for the positive and negative selection of T cells in the thymus. Since self peptides are presented for this selection, we used DQ8 transgenic mice to determine if DQ8 can present DR-derived peptides. Our data suggested that DQ8 mice generate a robust response to DR-restricted peptides derived from the resistant allele but the RA-susceptible *0401-derived peptide is presented weakly [21]. The expression of *0402 deletes more cells in the thymus of transgenic mice than *0401suggesting that *0402 may be able to negatively select autoreactive cells [32]. In transgenic mice carrying both DR*0401/DQ8 and *0402/DQ8, only the former generates a response to self-peptide, suggesting selection of a T cell profile with autoreactivity. This data suggests that a haplotype determines the T cell repertoire which may change the cytokine profile. To prove the role of a haplotype, we tested the antigen-specific cytokines produced in CIA. *0401 mice produce high levels of IL-12 (p70) required for production of IFN-γ in response to type II collagen (CII) while DQ8 mice produce high levels of TNFα and IL-12 (p40), a chain involved in IL-12 and IL-23 cytokines (Fig 1). However, introduction of *0401 in DQ8 led to a change in the cytokine profile, *0401/DQ8 mice produced high levels of IFN-γ, IL-17, IL-12 (p40) and IL-23 as compared to DQ8 mice suggesting *0401 can modulate the immune response and change the profile. A comparison of the *0401/DQ8 and *0402/DQ8 mice showed low levels of IL-17 and IL-23 but higher TGFβ production in the later in response to CII [21]. Both strains produced similar levels of IL-12 (p40). These observations support the hypothesis that DR polymorphism modulates the response of a haplotype and determine the final outcome of an immune response. Among all the class II loci, DRB1 has evolved and encodes for the most polymorphic class II genes, followed by DQB1. The selective pressure of the pathogens has generated more than 400 alleles for the DRB1 gene while the DQB1 gene has less than 100 alleles. This clearly suggests an important role of DR molecules in cytokine production and clearance of infectious agents. Our data in transgenic mice supports the contention that DQ8 is a promiscuous molecule as it can present many epitopes of CII [33], and may also be able to present many epitopes derived from a pathogen thus generating a robust response. The DRB1 polymorphism may generate and modulate immune response to clear infections, but a bystander effect of that response is autoreactivity. While this may not cause development of disease, a follow up activation of those T cells may finally break the tolerance causing onset of pathology. On the other hand, *0402 associated with resistance to autoimmunity modulates immune response to dampen Th17 response while still being able to produce Th1 cytokines, thus controlling the Th17 dependent autoimmunity.
Fig 1.
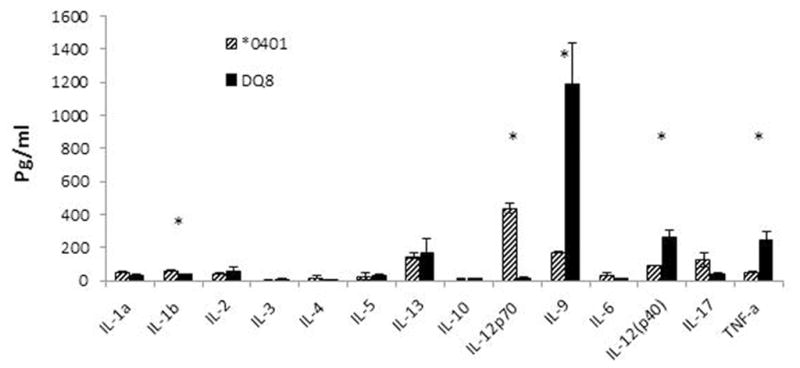
Cytokine profile is dependent on the HLA alleles. Transgenic mice were immunized with type II collagen and cytokines were tested in sera 3 weeks post-immunization. * P<0.05 *0401 compared to DQ8.
One reason for association of *0401 molecule with autoimmunity could be the presence of autoreactive cells. To determine if mice have autoreactive cells that can differentiate into Th17 cells when activated, we cultured memory CD4 T cells from naïve mice with dendritic cells (DCs) loaded with self-derived peptide, encoded by 3rd hypervariable region encompassing aa 65-79 of the DRB1 allele, in Th17 polarizing conditions. While memory T cells from *0401 mice differentiated into IL-17 producing cells when self-peptide was presented by DCs in vitro, *0402 mice did not differentiate into IL-17 producing cells. A role of IL-17 has been implicated in transgenic mice as well as other experimental model of arthritis as shown by the resistance of IL-17 deficient mice to develop arthritis [21, 22, 34]. On the other hand, IFN-γ has been suggested to be protective as arthritis is exacerbated in mice deficient in IFN-γ [35]. Our studies with a HLA haplotype expressing mice suggest that both IL-17 and IFN-γ have a role in pathogenesis though the time at which they are pathogenic may be different. As shown by *0401 and *0402 mice, kinetics of cytokines produced to an antigen may determine the protective or pathogenic nature of the immune response. Interestingly, observations in *0401/DQ8 and *0402/DQ8 mice showed that they can produce similar levels of chemokines suggesting that HLA class II alleles have a major impact on the cytokine profile [36]. Our recent observations showing a much higher TLR4-mediated response by *0401 compared to *0402 mice supports our hypothesis that *0401 has been selected for its ability to mount response to infections [30]. These observations validate the notion that T cell selection by the HLA molecules and the cytokines produced by them have a significant role in autoimmunity and provide an explanation for strong association of HLA class II with autoimmunity.
Sex-bias in cytokine production is HLA dependent
Most of the autoimmune diseases occur predominantly in women. RA occurs two to three times more often in women than in men with about 70% of patients being women. A role of hormones is underscored by the sex-bias in arthritis and its remission during pregnancy, although data in humans and animal models of RA has been controversial [37]. A gender difference in cytokine production in vitro has been suggested to lead to differential immune responses in men and women [38, 39]. Although the mechanism of sex-bias is not understood completely, penetrance of the HLA shared-epitope genotype and onset of RA is influenced by sex. Women carrying *0401 genotype show a higher penetrance with an earlier onset of arthritis compared with men [40, 41].
Collagen-induced arthritis has been used as a model for more than 3 decades. Even though we have learnt a lot from the CIA model in mice, most of the CIA models only reproduce a small segment of phenotype seen in human rheumatoid arthritis. A major difference between human RA and CIA in mice has been the lack of sex-bias and production of autoantibodies like rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPAs) in the latter. We used DQ8, DRB1*0401 and DRB1*0401/DQ8 mice to understand the sex bias of RA [29, 42–44]. The transgenic mice develop arthritis that mimics human disease, as the arthritis in mice is associated with the production of RF and ACPAs. In humans, a subset of CD4 T cells also express class II molecules and it is anticipated that these CD4 T cells may be able to present antigen and generate a proinflammatory response. In the HLA transgenic mice, a subset of CD4 T cells (5–10%) expresses HLA class II molecule and activated T cells can present antigen in these mice, confirming a role of CD4 cells in exacerbating the inflammatory response [22]. Further, our data suggested that arthritis in these mice is T and B cell dependent while CD8 T cells may be regulatory [45, 46]. Since the experimental arthritis in transgenic mice mimics human disease so closely, we determined if there is a sex-bias in predisposition. Our observations showed that DQ8 transgenic mice do not develop gender biased CIA. However, *0401 and *0401/DQ8 mice develop CIA predominantly in female mice with an incidence similar to that observed in humans suggesting DR4 may be associated with the sex-bias of disease.
This led us to address the question of how DR4 skews predisposition towards sex-bias. Since HLA molecules are required for antigen presentation, we compared antigen presentation by DR4 and DQ8 in transgenic mice. DR4-restricted CII-peptide was robustly presented in female *0401 and *0401/DQ8 mice compared to males [21, 22]. The immune response was dependent on the APC and the MHC molecule, DR or DQ, in both sexes and resulted in a different cytokine profile. Presentation of peptides via DCs led to the production of proinflammatory cytokines IL-6, IL-17, TNF and IFN-γ and anti-inflammatory IL-10 in males while females produced IL-2, IL-17 and IL-12 (p40). As CD20 depletion treatment is efficacious in RA patients, we also tested if antigen-presentation by B cells was different between sexes [21]. Our data showed that B cells are hyperactive in DR4 and DR4/DQ8 females and present the immunodominant DR4-restricted CII peptide 254-273 with higher efficiency than males. Antigen presentation via B cells resulted in activation of different pathways or subsets of T cells in both sexes as reflected in the cytokine profiles produced by *0401 mice. While females produced B cell modulating cytokines like IL-4 and IL-13 in significantly higher amounts in response to CII-peptide 254-273, males produced a mixed cytokine profile, IL-1b, TNF-α and IL-10. Production of higher levels of IL-10 in males was associated with increased numbers of B regulatory cells which might also explain why females generate a higher response to the CII-peptide. Increased levels of the Th2 cytokines, IL-4 and IL-13, in females is particularly interesting as according to the general paradigm Th2 cytokines are protective for autoimmune diseases. In RA patients high levels of IL-13 have been associated with ACPA positivity [23]. Our study confirmed this association as female transgenic mice produced higher levels of antibodies and IL-13. Similarly, an experimental model of multiple sclerosis has also shown an association of IL-13 production with gender variation [47].
Androgens are known as negative regulators of immune response and their deprivation can cause an increase in antigen-specific proliferation of T cells [48]. In support of this study, we observed an increase in antigen-specific proliferation with a decrease in regulatory cells in *0401 castrated mice [21]. Exogenous supply of androgen has been shown to reverse the autoimmunity in NZB/NZW mice [49]. On the other hand, estrogen is known to modulate B cell function by increasing their survival as well as by modulating pro-inflammatory cytokine production [50, 51]. In fact, *0401 male mice implanted with estradiol generate an enhanced immune response and autoreactivity leading to an increase in the incidence of arthritis [21]. Estrogen also modulates the expression of enzymes required for citrullination of antigens. This can lead to an increased presentation of the modified peptides by B cells further increasing the pro-inflammatory cascade. Thus estrogen can impact disease development and severity via B cell survival and production of autoantibodies, and differential antigen presentation by the DR molecule resulting in a cytokine milieu that decides the final outcome. Male patients with RA have higher levels of estrogen than healthy controls [52] which may contribute to an over active immune response.
Conclusions
The MHC molecules clear infections by generating an immune response and the production of cytokines. The type of cytokine profile, Th1/Th2/Th17, is dependent on the HLA allele which determines the clearance of extracellular and intracellular infections. DRB1*0401 and DRB1*0402 activate Th1 and Th17 producing T cells which help in clearing intracellular and extracellular infections but predispose to autoimmunity (Fig 2). However, DRB1*0401 has autoreactive memory cells which can be activated in case of molecular mimicry causing activation of those cells and IL-17 production. On the other hand, *0402 does not produce any autoreactive response. DRB1*0401/DQ8 is associated with predisposition to RA. HLA-DQ8 is the most autoimmune prone molecule as it is associated with the most commonly observed autoimmune diseases- rheumatoid arthritis, type I diabetes, celiac disease, relapsing Polychondritis [33]. Further, sex of an individual as well as the type of APC presenting antigen(s) may influence the cytokine profile. Thus MHC has evolved to produce IL-17 and IFN-γ by different HLA molecules for clearing infections. However, certain HLA molecules can positively select autoreactive cells. If the specificity of those autoreactive cells mimics an epitope from a pathogen, it can lead to production of pro-inflammatory cytokines that can, in certain conditions, result in autoimmunity.
Fig 2.
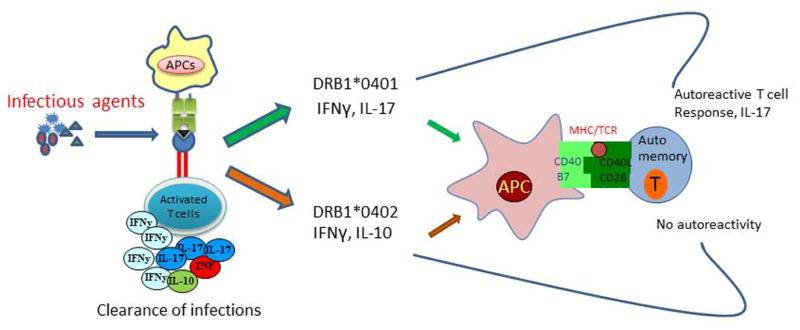
HLA molecules predetermine the cytokine profile to predispose to autoimmune diseases. DRB1*0401 and DQ8 activate CD4 T cells and produce cytokines which Th1/Th17 and immunomodulatory cytokines IL-13 and IL-10. Complementation between the HLA molecules leads to high pro-inflammatory cytokines involved in clearing infections though they predispose to autoreactivity. Studies with HLA transgenic mice suggest that when DRB1*0401gene is introduced in DQ8 mice, *0401/DQ8 mice, the cytokine profile is associated with high levels of pro-inflammatory cytokines, IFN-γ, IL-17 and IL-23, and increased incidence and severity of arthritis compared to single transgenic mice.
Highlights.
HLA alleles positively select T cells and predetermine production of TH17/TH2 under different conditions.
HLA alleles that produce TH1/TH17 cytokines and provide advantage in clearing infections have been preserved for generations.
Most studies point a significant role of molecular mimicry in autoimmunity.
Acknowledgments
The transgenic mice were generated and provided by Dr Chella David. Julie A. Hanson and her staff for animal husbandry, Michele K. Smart for the characterization of transgenic mice are greatly appreciated. This work was supported by National Institutes of Health grants AR30752 and AI 75262
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ebringer A, Rashid T, Wilson C. Rheumatoid arthritis, Proteus, anti-CCP antibodies and Karl Popper. Autoimmun Rev. 2010;9:216–223. doi: 10.1016/j.autrev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Boyle LH, Hill Gaston JS. Breaking the rules: the unconventional recognition of HLA-B27 by CD4+ T lymphocytes as an insight into the pathogenesis of the spondyloarthropathies. Rheumatology (Oxford) 2003;42:404–412. doi: 10.1093/rheumatology/keg097. [DOI] [PubMed] [Google Scholar]
- 3.Yan ZH, Fan Y, Wang XH, Mao Q, Deng GH, Wang YM. Relationship between HLA-DR gene polymorphisms and outcomes of hepatitis B viral infections: a meta-analysis. World J Gastroenterol. 2012;18:3119–3128. doi: 10.3748/wjg.v18.i24.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 5.Gill N, Finlay BB. The gut microbiota: challenging immunology. Nat Rev Immunol. 2011;11:636–637. doi: 10.1038/nri3061. [DOI] [PubMed] [Google Scholar]
- 6.Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41:84–91. doi: 10.1016/j.cyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thursz M, Yallop R, Goldin R, Trepo C, Thomas HC. Influence of MHC class II genotype on outcome of infection with hepatitis C virus. The HENCORE group. Hepatitis C European Network for Cooperative Research. Lancet. 1999;354:2119–2124. doi: 10.1016/s0140-6736(99)91443-5. [DOI] [PubMed] [Google Scholar]
- 9.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010;6:727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 10.Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JC, Zernicka-Goetz M, Nielsen ML, Gurdon JB, Kouzarides T. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature. 2014;507:104–108. doi: 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makrygiannakis D, af Klint E, Lundberg IE, Lofberg R, Ulfgren AK, Klareskog L, Catrina AI. Citrullination is an inflammation-dependent process. Ann Rheum Dis. 2006;65:1219–1222. doi: 10.1136/ard.2005.049403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moscarello MA, Wood DD, Ackerley C, Boulias C. Myelin in multiple sclerosis is developmentally immature. J Clin Invest. 1994;94:146–154. doi: 10.1172/JCI117300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vossenaar ER, Smeets TJ, Kraan MC, Raats JM, van Venrooij WJ, Tak PP. The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis and rheumatism. 2004;50:3485–3494. doi: 10.1002/art.20584. [DOI] [PubMed] [Google Scholar]
- 14.Astry B, Harberts E, Moudgil KD. A cytokine-centric view of the pathogenesis and treatment of autoimmune arthritis. J Interferon Cytokine Res. 2011;31:927–940. doi: 10.1089/jir.2011.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snir O, Rieck M, Gebe JA, Yue BB, Rawlings CA, Nepom G, Malmstrom V, Buckner JH. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1* 0401-positive humanized mice and rheumatoid arthritis patients. Arthritis and rheumatism. 2011;63:2873–2883. doi: 10.1002/art.30445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 17.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside.Nature clinical practice. Rheumatology. 2006;2:619–626. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 18.Mima T, Nishimoto N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. 2009;21:224–230. doi: 10.1097/BOR.0b013e3283295fec. [DOI] [PubMed] [Google Scholar]
- 19.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van de Loo FA, van den Berg WB. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- 21.Behrens M, Trejo T, Luthra H, Griffiths M, David CS, Taneja V. Mechanism by which HLA-DR4 regulates sex-bias of arthritis in humanized mice. Journal of Autoimmunity. 2010;35:1–9. doi: 10.1016/j.jaut.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taneja V, Behrens M, Mangalam A, Griffiths MM, Luthra HS, David CS. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis and rheumatism. 2007;56:69–78. doi: 10.1002/art.22213. [DOI] [PubMed] [Google Scholar]
- 23.Hitchon CA, Alex P, Erdile LB, Frank MB, Dozmorov I, Tang Y, Wong K, Centola M, El-Gabalawy HS. A distinct multicytokine profile is associated with anti-cyclical citrullinated peptide antibodies in patients with early untreated inflammatory arthritis. The Journal of rheumatology. 2004;31:2336–2346. [PubMed] [Google Scholar]
- 24.Levy Y, Brouet JC. Interleukin-10 prevents spontaneous death of germinal center B cells by induction of the bcl-2 protein. J Clin Invest. 1994;93:424–428. doi: 10.1172/JCI116977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 26.Davis JM, 3rd, Knutson KL, Strausbauch MA, Crowson CS, Therneau TM, Wettstein PJ, Roger VL, Matteson EL, Gabriel SE. A signature of aberrant immune responsiveness identifies myocardial dysfunction in rheumatoid arthritis. Arthritis and rheumatism. 2011;63:1497–1506. doi: 10.1002/art.30323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, Wijeyewickrema LC, Eckle SB, van Heemst J, Pike RN, McCluskey J, Toes RE, La Gruta NL, Purcell AW, Reid HH, Thomas R, Rossjohn J. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. The Journal of experimental medicine. 2013;210:2569–2582. doi: 10.1084/jem.20131241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill JA, Bell DA, Brintnell W, Yue D, Wehrli B, Jevnikar AM, Lee DM, Hueber W, Robinson WH, Cairns E. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. The Journal of experimental medicine. 2008;205:967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behrens M, Trejo T, Luthra H, Griffiths M, David CS, Taneja V. Mechanism by which HLA-DR4 regulates sex-bias of arthritis in humanized mice. Journal of autoimmunity. 2010;35:1–9. doi: 10.1016/j.jaut.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luckey D, Behrens M, Smart M, Luthra H, David CS, Taneja V. DRB1*0402 may influence arthritis by promoting naive CD4 T-cell differentiation in to regulatory T cells. Eur J Immunol. 2014 doi: 10.1002/eji.201344424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Almeida DE, Ling S, Pi X, Hartmann-Scruggs AM, Pumpens P, Holoshitz J. Immune dysregulation by the rheumatoid arthritis shared epitope. J Immunol. 2010;185:1927–1934. doi: 10.4049/jimmunol.0904002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taneja V, Taneja N, Behrens M, Pan S, Trejo T, Griffiths M, Luthra H, David CS. HLA-DRB1*0402 (DW10) transgene protects collagen-induced arthritis-susceptible H2Aq and DRB1*0401 (DW4) transgenic mice from arthritis. J Immunol. 2003;171:4431–4438. doi: 10.4049/jimmunol.171.8.4431. [DOI] [PubMed] [Google Scholar]
- 33.Taneja V, David CS. Role of HLA class II genes in susceptibility/resistance to inflammatory arthritis: studies with humanized mice. Immunol Rev. 2010;233:62–78. doi: 10.1111/j.0105-2896.2009.00858.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 35.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 36.Taneja V, Behrens M, Basal E, Sparks J, Griffiths MM, Luthra H, David CS. Delineating the role of the HLA-DR4 “shared epitope” in susceptibility versus resistance to develop arthritis. J Immunol. 2008;181:2869–2877. doi: 10.4049/jimmunol.181.4.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cutolo M. Sex and rheumatoid arthritis: mouse model versus human disease. Arthritis and rheumatism. 2007;56:1–3. doi: 10.1002/art.22322. [DOI] [PubMed] [Google Scholar]
- 38.Bouman A, Schipper M, Heineman MJ, Faas MM. Gender difference in the non-specific and specific immune response in humans. Am J Reprod Immunol. 2004;52:19–26. doi: 10.1111/j.1600-0897.2004.00177.x. [DOI] [PubMed] [Google Scholar]
- 39.Giron-Gonzalez JA, Moral FJ, Elvira J, Garcia-Gil D, Guerrero F, Gavilan I, Escobar L. Consistent production of a higher TH1:TH2 cytokine ratio by stimulated T cells in men compared with women. Eur J Endocrinol. 2000;143:31–36. doi: 10.1530/eje.0.1430031. [DOI] [PubMed] [Google Scholar]
- 40.Meyer JM, Han J, Singh R, Moxley G. Sex influences on the penetrance of HLA shared-epitope genotypes for rheumatoid arthritis. Am J Hum Genet. 1996;58:371–383. [PMC free article] [PubMed] [Google Scholar]
- 41.Jawaheer D, Lum RF, Gregersen PK, Criswell LA. Influence of male sex on disease phenotype in familial rheumatoid arthritis. Arthritis and rheumatism. 2006;54:3087–3094. doi: 10.1002/art.22120. [DOI] [PubMed] [Google Scholar]
- 42.Taneja V, Behrens M, Basal E, Sparks J, Griffiths MM, Luthra H, David CS. Delineating the role of the HLA-DR4 “shared epitope” in susceptibility versus resistance to develop arthritis. J Immunol. 2008;181:2869–2877. doi: 10.4049/jimmunol.181.4.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taneja V, Behrens M, Mangalam A, Griffiths MM, Luthra HS, David CS. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis and rheumatism. 2007;56:69–78. doi: 10.1002/art.22213. [DOI] [PubMed] [Google Scholar]
- 44.Taneja V, Taneja N, Paisansinsup T, Behrens M, Griffiths M, Luthra H, David CS. CD4 and CD8 T cells in susceptibility/protection to collagen-induced arthritis in HLA-DQ8-transgenic mice: implications for rheumatoid arthritis. J Immunol. 2002;168:5867–5875. doi: 10.4049/jimmunol.168.11.5867. [DOI] [PubMed] [Google Scholar]
- 45.Taneja V, Krco CJ, Behrens MD, Luthra HS, Griffiths MM, David CS. B cells are important as antigen presenting cells for induction of MHC-restricted arthritis in transgenic mice. Mol Immunol. 2007;44:2988–2996. doi: 10.1016/j.molimm.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taneja V, Taneja N, Paisansinsup T, Behrens M, Griffiths M, Luthra H, David CS. CD4 and CD8 T cells in susceptibility/protection to collagen-induced arthritis in HLA-DQ8-transgenic mice: implications for rheumatoid arthritis. J Immunol. 2002;168:5867–5875. doi: 10.4049/jimmunol.168.11.5867. [DOI] [PubMed] [Google Scholar]
- 47.Sinha S, Kaler LJ, Proctor TM, Teuscher C, Vandenbark AA, Offner H. IL-13-mediated gender difference in susceptibility to autoimmune encephalomyelitis. J Immunol. 2008;180:2679–2685. doi: 10.4049/jimmunol.180.4.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roden AC, Moser MT, Tri SD, Mercader M, Kuntz SM, Dong H, Hurwitz AA, McKean DJ, Celis E, Leibovich BC, Allison JP, Kwon ED. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol. 2004;173:6098–6108. doi: 10.4049/jimmunol.173.10.6098. [DOI] [PubMed] [Google Scholar]
- 49.Roubinian JR, Talal N, Greenspan JS, Goodman JR, Siiteri PK. Effect of castration and sex hormone treatment on survival, anti-nucleic acid antibodies, and glomerulonephritis in NZB/NZW F1 mice. The Journal of experimental medicine. 1978;147:1568–1583. doi: 10.1084/jem.147.6.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109:1625–1633. doi: 10.1172/JCI14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cutolo M, Capellino S, Sulli A, Serioli B, Secchi ME, Villaggio B, Straub RH. Estrogens and autoimmune diseases. Ann N Y Acad Sci. 2006;1089:538–547. doi: 10.1196/annals.1386.043. [DOI] [PubMed] [Google Scholar]
- 52.Tengstrand B, Carlstrom K, Fellander-Tsai L, Hafstrom I. Abnormal levels of serum dehydroepiandrosterone, estrone, and estradiol in men with rheumatoid arthritis: high correlation between serum estradiol and current degree of inflammation. The Journal of rheumatology. 2003;30:2338–2343. [PubMed] [Google Scholar]