

Abstract
Melanoma is a highly aggressive disease that is difficult to treat due to rapid tumor growth, apoptotic resistance, and high metastatic potential. The MET tyrosine kinase receptor promotes many of these cellular processes, and while MET is often overexpressed in melanoma, the mechanism driving this overexpression is unknown. Since the MET gene is rarely mutated or amplified in melanoma, MET overexpression may be driven by to increased activation through promoter elements. In this report, we find that transcription factors PAX3 and ETS1 directly interact to synergistically activate MET expression. Inhibition of PAX3 and ETS1 expression in melanoma cells leads to a significant reduction of MET receptor levels. The 300 bp 5′ proximal MET promoter contains a PAX3 response element and two ETS1 consensus motifs. While ETS1 can moderately activate both of these sites without cofactors, robust MET promoter activation of the first site is PAX-dependent and requires the presence of PAX3, while the second site is PAX-independent. The induction of MET by ETS1 via this second site is enhanced by HGF-dependent ETS1 activation, thereby MET indirectly promotes its own expression. We further find that expression of a dominant negative ETS1 reduces the ability of melanoma cells to grow both in culture and in vivo. Thus, we discover a pathway where ETS1 advances melanoma through the expression of MET via PAX-dependent and independent mechanisms.
Keywords: MET receptor, ETS1, transcription, melanoma, PAX3
Introduction
ETS1 is a member of the ETS-family of transcription factors, defined by the presence of a winged helix-turn-helix DNA binding domain (1). Expression of ETS1 is absent or low in melanocytes of normal skin or within simple nevus structures (lentigo simplex); however, ETS1 is overexpressed in melanoma in situ and in invasive and metastatic primary tissues, as well as in melanoma cell lines (2, 3). While the role of ETS1 in melanoma is unclear, its major functionality likely lies in transcriptional regulation. Evidence supports that ETS1 promotes cell survival, tumor progression, and invasion. ETS1 may act as either a pro- or anti-apoptotic factor depending on the cell type. In melanoma, ETS1 plays an anti-apoptotic role, at least partially due to upregulation of MCL1 (4). In terms of tumor invasion and progression, inhibition of ETS1 leads to a decrease in expression of uPA, MMP1, MMP3, and integrin-β3 (3). In addition, ETS1 directly activates the integrin-αv promoter (5).
There are several lines of evidence supporting that ETS1 is upstream of MET, a receptor tyrosine kinase that promotes melanoma cell growth and survival (6–8). An increase in ETS1 protein levels raises MET levels, while inhibition of ETS1 decreases MET receptor expression (9–12). In addition, in esophageal cancer, levels of MET and ETS1 protein correlate significantly (13). While in silico studies predict that ETS1 is directly upstream of the MET promoter (9), this has not been proven definitively through experimentation in any cell type.
We previously identified the transcription factor PAX3 as an upstream regulator of MET in melanoma (14). During normal melanocyte development PAX3 is necessary for the regulation of genes involved in cell type specification while maintaining an undifferentiated state, proliferation, and migration (reviewed in (15)). These characteristics are mirrored in melanoma, where our group and others find PAX3 expression (16–19). Along with MET, PAX3 mediates its cellular effects in melanoma through the regulation of down-stream targets, such as BRN2 and TBX2 (20, 21). However, PAX3 is a weak transcription factor on its own, and often recruits other factors to synergistically regulate gene expression.
Here, we discover a pathway for promoting MET receptor expression by the transcription factors ETS1 and PAX3. We find that both transcription factors directly interact and synergistically drive MET expression by binding to promoter enhancer elements. The MET promoter contains two ETS1 sites, and activation through these two elements is enhanced by different mechanisms that are either PAX3- or HGF-dependent. Our data support a model for an oncogenic pathway where PAX3 and ETS1 drive MET expression, and this pathway is further driven in a feed-forward manner through the ligand for MET, HGF.
Results
PAX3, ETS1, and MET are expressed in melanoma cell lines and tumors
To determine the presence of PAX3, ETS1, and MET proteins in human melanoma cell lines, a panel of 7 independent lines was analyzed (Figure 1A). All cell lines expressed these three proteins to varying degrees. ETS1 contains a Ras-responsive site at threonine 38 (T38), and phosphorylation of this epitope strongly increases the protein’s transcriptional activity (22–25). The phosphorylation status of T38 in ETS1 was measured in the melanoma cell line panel (Figure 1B). In comparison to CIP controls or samples that were ETS1 negative, phospho-ETS1 (pETS1) levels are considered high for A375, SKMEL5, and SKMEL23 (p<0.0005), and significant for mel537 (p<0.05)(n=3). The pETS1 levels are considered undetectable for mel888 (p=0.051) and SKMEL28 (p=0.234) cells.
Figure 1.
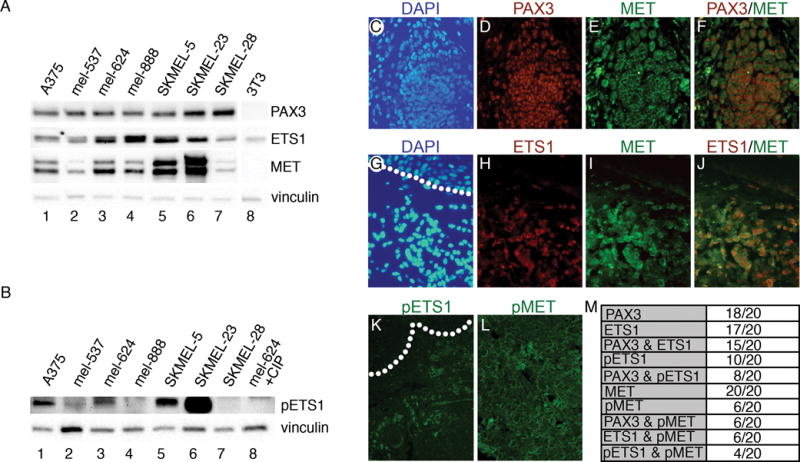
PAX3, ETS1, and MET proteins are expressed in melanoma cells and primary tumor samples. (A,B) Melanoma cell lines (lanes 1–7) express varying levels of PAX3, ETS1, MET (A), and phosphorylated ETS1 (pETS1) (B). Western blots were probed with vinculin antibody as a loading control. Cell line 3T3 (lane 8) served as negative controls for PAX3 and MET in (A), and lysate from mel-624 treated with calf-intestinal phosphatase (CIP) was a negative control for pETS1. (C–L) Immunofluorescence analysis of PAX3, ETS1, pETS1, MET, and pMET in superficial spreading melanoma primary tissue. Representative samples demonstrating PAX3 and MET (C–F) or ETS1 and MET (G–J) co-expression, with DAPI-stained nuclei (blue, C,G), PAX3 (red, D), ETS1 (red, H), MET (green, E, I), and red/green dual channel combination (F,J). Representative samples of pETS1 and pMET immunofluorescence (K,L). Dotted lines represent the dermal/epidermal junction layer. (M) Summary table of samples that expressed specific antigens.
Expression of PAX3, ETS1, MET, pETS1 and phosphorylated-MET (active form of MET receptor, pMET) was measured in twenty superficial spreading melanoma primary tissue samples. Representative results for PAX3 and MET, or ETS1 and MET immunofluorescence and co-expression are shown in Figure 1C–J,M. PAX3 and ETS1 expression is predominantly nuclear, while MET is principally located in the membrane with focal regions of pan-cellular expression. The majority of samples express PAX3 and ETS1. No melanoma samples in the panel were both PAX3 and ETS1 negative. All twenty samples expressed MET receptor. Half of the samples expressed pETS1, while 6/20 samples presented with pMET (Figure 1K–M).
The MET proximal promoter contains potential PAX3 and ETS1 binding sites
The transcriptional start site of the MET gene has been determined and potential ETS family binding sites have been predicted through in silico analysis (9, 26). While ETS family members all bind to a core sequence of GGA(A/T) (27), each factor has longer high affinity DNA binding sites. High affinity recognition motifs for ETS1 have been experimentally determined to be C(C/A)GGA(A/T)G(T/C) (28), AC(C/A)GGA(A/T)(G/A) (29), and (G/C)CGGAAGT (30). Combining these studies produces a high affinity consensus site of C(A/C)GGA(A/T)(G/A), or CMGGAWR. Within a short segment of the MET promoter (297 bp 5′ proximal to the transcriptional start site, and 25 bp of the 5′ UTR), there is one CMGGAWR ETS1 consensus site located −85 to −79 from the transcriptional start (Figure 2A). While ETS1 binds to CCGGAA with 100 fold higher affinity than to CCGGAG, ETS1 recruitment to CCGGAG is enhanced 1,000 fold when ETS1 is in complex with PAX5 (31, 32). In contrast, converting this CCGGAG ETS/PAX site to an optimum site for independent ETS1 binding (CMGGAWR) reduces the ability of PAX5 to interact with DNA (33, 34). A CCGGAG ETS/PAX site is located in the proximal MET promoter at −241 to −236 (reverse strand, Figure 2A). This ETS/PAX site is adjacent to a previously characterized PAX3 site (14, 35).
Figure 2.

The MET promoter 5′ proximal to the transcriptional start contains putative PAX and ETS sites. (A) Sequence from the MET locus, including 297 bp 5′ upstream to the transcriptional start site (arrow), and 25 bp of 5′ UTR that includes a SMAD SBE site (48), MITF site (39, 40), and HES/NOTCH site (49). A PAX site is indicated −752 to −746 by a black box. Putative ETS sites are shown −241 to −236 on the reverse strand of the sequence (ETS/P (E1), box) and −85 to −79 (ETS (E2), underline). (B) Nomenclature and schematics of MET reporter constructs containing the promoter sequence shown in (A) driving the reporter gene lucifierase. Constructs contain wild-type PAX (P) and ETS (E1 and E2) sites as shown in A or the sites that are mutated shown schematically with an X.
PAX3 and ETS1 activate the proximal MET promoter
PAX3 drives expression of MET in melanoma and in muscle cells (14, 35). To test the ability of PAX3 and ETS1 to activate the MET promoter alone or together, a DNA fragment containing 297 base pairs of the MET 5′ proximal promoter and 28 base pairs of 5′ UTR was sub-cloned into the reporter vector pGL2-Luciferase (Figure 2A, B). Separately, PAX3 or ETS1 drive reporter expression 2.9±0.1 fold and 4.0±0.4 fold over light levels of reporter alone (Figure 3A, set 1). Together, PAX3 and ETS1 activated the MET promoter synergistically 17.0±2.8 fold (p<0005).
Figure 3.
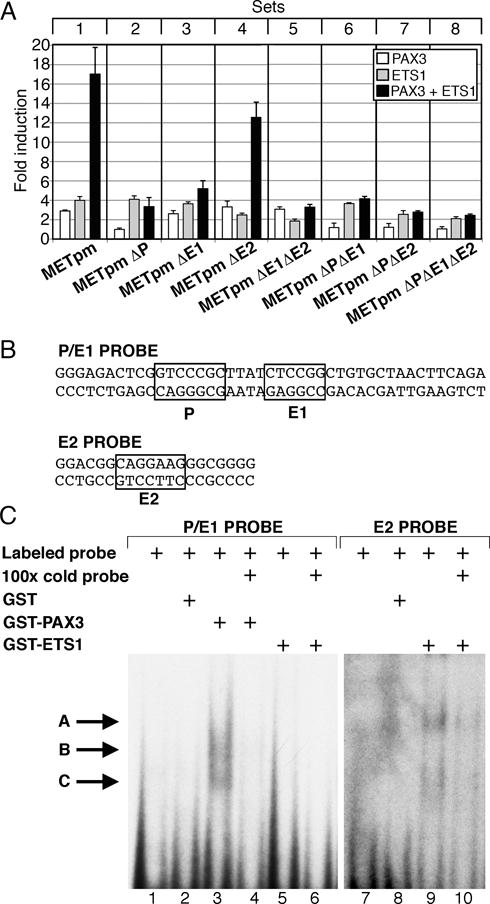
PAX3 and ETS1 activate the proximal MET promoter. (A) MET promoter reporter constructs, shown schematically in Figure 2, were transfected into 293T cells with PAX3 (white bars), ETS1 (grey bars), or both (black bars). Each bar represents n=9, with standard error of the mean as shown. Differences between synergistic activation of set 1 and the other sets are significant (p<0.05 set 4, p<0.005 other sets). (B) P/E1 and E2 probe sequences utilized in EMSA analysis. (C) PAX3 and ETS1 bind to elements within the MET promoter. Two EMSA probes were utilized, containing either the P and E1 sites (lanes 1–6) or the E2 site (lanes 7–10). Lanes contain either probe alone (lanes 1,7), with the addition of GST (lanes 2,8), PAX3 (lanes 3,4), or ETS1 (lanes 5,6,9,10) proteins. Cold probe was added in 100 times excess as a specific competitor (lanes 4, 6,10). Slower migrating bands are seen with the addition of PAX3 (lane 3, arrows B, C) or ETS1 (lane 9, arrows A, C).
To test the importance of the putative PAX and ETS sites, METpm was utilized as a template to create reporter constructs containing single, double, or triple mutations in these sites (Figure 2B). The constructs were transfected into HEK-293T cells in the presence or absence of PAX3 and/or ETS1 expression constructs. Any mutation of the PAX site alone (Figure 3A, set 2) or in concert with ETS mutations (sets 6, 7, and 8) completely abrogated the ability of PAX3 to drive reporter expression (p<0.005). Mutation of the PAX site did not disrupt the ability of ETS1 to drive reporter expression, but the synergistic activation was abolished. Mutation of either ETS site did not block the ability of ETS1 to activate the MET promoter (grey bars, set 3, 3.6±0.8 fold, set 4, 2.5±0.2 fold). Mutation of both sites reduced the ETS1 activation significantly (set 5 grey bar, 1.8±0.2 fold, p<0.005). Synergistic activation of the MET promoter by PAX3 and ETS1 was lost when the E1 site was mutated (Figure 3A, set 3,5 black bar, p<0.005), or loss of the functional PAX site (sets 2,6,7,8 black bars, p<0.0005). However, if both the PAX and E1 sites were intact (Figure 3A, sets 1,4, black bars) PAX3 and ETS1 activated the MET promoter synergistically (p<0.0005). EMSA assays were performed in order to determine if these factors bind directly to the MET promoter. Utilizing radiolabeled oligonucleotide probes containing the P and E1 sites (Figure 3B), a slower migrating band was seen in EMSA assays with the addition of PAX3 protein (Figure 3C, arrows B,C, lane 3) but not with ETS1 alone (lane 5). A slower migrating band was also seen when ETS1 was added to a probe containing the E2 site (Figure 3B,C arrows A,C, lane 9). All slower migrating bands were significantly reduced with the addition of 100 fold excess cold probe (lanes 6,10). Our findings support that PAX3 and ETS1 bind to and activate the MET promoter through specific enhancer sites.
PAX3 and ETS1 directly interact and promote MET expression in melanoma cells
PAX3 and ETS1 are co-expressed in melanoma and activate the MET promoter more robustly together than either factor alone (Figures 1,3A). Immunoprecipitation assays were performed to determine if these proteins interact within melanoma cells. Immunoprecipitation using an anti-ETS1 antibody yielded a band when probed for PAX3 by western analysis in A375 and mel-624 melanoma cells (Figure 4A, lanes 3,6). No PAX3 was detected with beads alone or with a non-specific IgG antibody. To determine if ETS1 and PAX3 directly interact, in vitro immunoprecipitation experiments were preformed utilizing recombinant ETS1 and PAX3 proteins. PAX3 and ETS1 directly interacted (Figure 4B) only when both proteins were present (Figure 4B, lane 6). We find that PAX3 and ETS1 directly interact in A375 and mel-624 melanoma cells.
Figure 4.
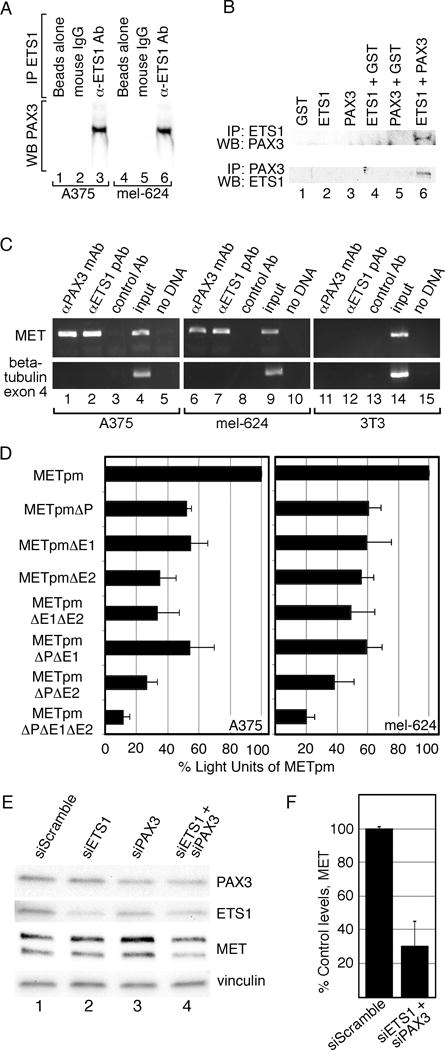
PAX3 and ETS1 activate MET in melanoma cells. (A) PAX3 and ETS1 interact in melanoma cells. A375 and mel-624 cell lysates were immunoprecipitated without (lanes 1,2,4,5) or with ETS1 antibodies (lanes 3,6). Immunoprecipitants were probed by western analysis for the presence of PAX3. (B) PAX3 and ETS1 directly interact. Recombinant proteins were immunoprecipitated ETS1 antibody then probed for PAX3 expression (top row) or immunoprecipitated with PAX3 antibody then probed for ETS1 expression (bottom row). (C) PAX3 is located on the endogenous MET promoter in A375 and mel-624 cells. Chromatin immunoprecipitation (ChIP) analysis was performed with primers specific for the MET promoter (top gels) or exon 4 of the beta tubulin gene (bottom gels, negative control) in A375 (lanes 1–5), mel-624 (lanes 6–10), and 3T3 (11–15, negative control) cell lysates. Antibodies utilized for immunoprecipitations were against PAX3 (lane 1,6,11), ETS1 (lane 2,7,12), or normal mouse IgG (negative control, lane 3,8,13). Input DNA (positive control, lane 4,9,14) and water-blank without template DNA (negative control, lane 5,10,15) acted as PCR controls. Input DNA was collected for each sample after cell sonication but before immunoprecipitation. (D) PAX and ETS sites are active in A375 and mel-624 melanoma cells. MET promoter reporter constructs, shown schematically in Figure 2, were transfected into cells with the wild-type MET promoter sequence, or with the P, E1 and/or E2 sites mutated. Percent light units was calculated by dividing the light units generated from each set by the light units of the wild-type MET promoter, then multiplying by 100. Each bar represents n=9, with standard error of the mean as shown. Differences between wild-type METpm and all of the tested mutant constructs were significant for both cell lines, p<0.005. (E, F) Inhibition of PAX3 and ETS1 expression in mel-624 cells leads to a reduction of MET levels. Cells were transfected with scrambled control siRNA (lane 1) PAX3 or ETS1 gene specific siRNA alone (lanes 2,3), or together (lane 4). Graphs shown in F are quantified densitometry readings of MET bands from western analysis from E, (n=3). All densitometry readings were normalized against vinculin loading controls. MET expression was reduced significantly (p<0.02) to 30%±14.7% in PAX3 and ETS1 siRNA transfected cells compared to siScramble.
To determine if endogenous PAX3 and ETS1 bind to the MET promoter, Chromatin Immunoprecipition (ChIP) assays were performed on melanoma cell lysates as well as cells that do not express MET (3T3 cells). The MET promoter sequence was amplified from DNA precipitated with proteins bound to antibodies specific for PAX3 or ETS1 (Figure 4C, lanes 1,2,6,7) but not with control IgG antibody (lane 3,8) in both A375 and mel-624 melanoma cells. These bands were absent when 3T3 cell lysates were utilized. These experiments support that PAX3 and ETS1 are located on the MET promoter in melanoma cells.
To determine if the PAX3 and ETS sites discovered in the MET promoter are active in melanoma cells, METpm vector or constructs with engineered mutations in the PAX, E1 and E2 sites (Figure 2B) were transfected into A375 and mel-624 melanoma cells (Figure 4D). Mutation of any of the sites resulted in a significant decrease (p<0.005) in reporter activity in both cell lines. Mutation of all three sites resulted in the most dramatic decrease in luciferase activity, 11.78%±3.98% (A375, p<0.0005), or 19.75%±5.74% (mel-624, p<0.0005). These findings demonstrate that these binding sites within the MET promoter are functional in melanoma cells.
Blocking PAX3 and ETS1 expression in melanoma cells lead to a reduction of MET levels. Inhibiting PAX3 alone reduced MET expression in some but not all melanoma cell lines (14). Reduction of PAX3 or ETS1 levels by less than half of control cells did not significantly decrease MET levels in mel-624 cells (Figure 4E). Reduction of both PAX3 and ETS1 leads to a significant loss (p<0.02) of MET expression to 30.0%±14.7% of control levels (Figure 4F). These data demonstrate that expression of MET is at least partially dependent on PAX3 and ETS1 in mel-624 cells.
ETS1 synergistically activates the MET promoter with either PAX3 or Hepatocyte Growth Factor (HGF)
ETS1 may be activated by HGF via the RAS signaling pathway (22). This supports a positive feed-forward loop model wherein MET is activated by HGF, which indirectly activates ETS1 and consequently its own promoter. The ability of HGF to synergistically activate the MET promoter was tested in cells expressing the MET receptor (HEK-293T) and cells lacking this protein (3T3)(Figure 5A). In HEK-293T cells, HGF synergistically activated the MET reporter construct significantly (16.9±3.7 fold, p<0.005) at levels parallel to ETS1/PAX3 activation (Figure 5B). In the MET-deficient 3T3 cells, the ability of ETS1 to activate the MET promoter was similar without (2.2±0.3 fold) or with (2.2±0.7 fold) the addition of HGF (Figure 5C). The addition of HGF did not significantly increase luciferase levels (p=0.48). ETS1 and PAX3 still yielded a significant synergistic activation of the MET promoter in the 3T3 cells (p<0.005).
Figure 5.
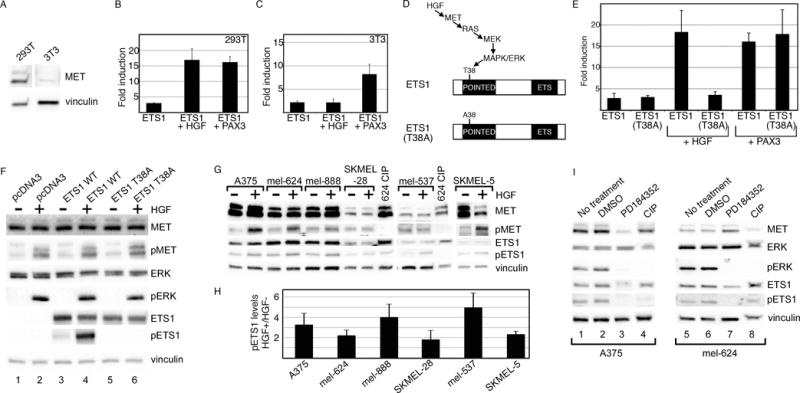
ETS1 synergistically activates MET with PAX3 or HGF. (A) MET is expressed in 293T cells but not in 3T3 cells. Western analysis for MET and vinculin (loading control) in 293T and 3T3 cells. (B, C) ETS1 and PAX3 activate MET synergistically in 293T and 3T3 cells, while a synergistic effect of ETS1 and HGF is only seen in 293T cells. Luciferase assays of 293T (B) and 3T3 (C) cells transfected with the wild-type METpm (shown schematically in Figure 2B) and ETS1, with the addition of PAX3 or exogenous HGF. Increased levels of luciferase were significant for all samples in comparison to vector alone (293T: p<0.005, 3T3: p<0.05), and between ETS1 + HGF in 293T cells and ETS1 + PAX3 in both cell lines in comparison to ETS1 alone (p<0.05). The levels of luciferase did not increase significantly in 3T3 cells with the addition of HGF and ETS1 in comparison to ETS1 alone (p=0.475). (D) Schematic of the HGF/MET/MAPK phosphorylation site on epitope T38 of ETS1. In the ETS1(T38A) mutant protein, a mutation of T38 to an alanine abrogates the ability of MAPK to phosphorylate ETS1 (22–25). (E) The ETS1(T38A) mutant synergistically activates MET expression with PAX3 but not with HGF. Luciferase assays of 293T cells transfected with either ETS1 or ETS1(T38A), with or without the addition of HGF or PAX3. Each bar represents n=9, with standard error of the mean as shown. The addition of HGF or PAX3 to ETS1, or HGF to ETS1(T38A) transfected cells led to a significant fold increase in luciferase versus ETS1 proteins alone (p<0.005). Conversely, HGF was unable to increase overall luciferase levels in comparison to ETS1(T38A) alone (p=0.172). (F) Mutation of ETS1(T38A) abrogates ETS1 phosphorylation after HGF treatment. 293T cells transfected with empty vector (pcDNA3), ETS1 wild-type, and ETS1(T38A) expression constructs are grown in the absence (−) or presence (+) of exogenous HGF. Western blots were probed with antibodies against MET, phosphorylated MET (pMET), ERK, pERK, ETS1, and pETS1. Vinculin antibody was included as a loading control. (G, H) HGF increases the levels of pETS1 in 5/6 melanoma cell lines. Protein lysates from cells without (−) or with (+) the addition of HGF treatment were tested for the expression of MET, pMET, ETS1, pETS1 and vinculin. Lysate from mel-624 treated with calf-intestinal phosphatase (CIP) was a negative control for phosphorylated proteins. Band intensity for pETS1 was measured by densitometry for three independent experiments (shown graphically in H). Levels for pETS1 post HGF treatment, in comparison to untreated cells, increased significantly (p<0.05) in all cell lines except for SKMEL-28 (p=0.218). (I) Treatment with a MEK-specific inhibitor, PD184352, decreases pETS1 levels. Protein lysates from A375 and mel-624 cells untreated, mock treated (DMSO), or treated with PD1184352 were tested for the expression of MET, ERK, pERK, ETS1, pETS1, and vinculin. Lysates treated with CIP served as a negative control for phosphorylated proteins. All Western analyses shown are representatives of three independent experiments.
HGF activates ETS1 by a RAS signaling pathway, leading to phosphorylation of threonine 38 (T38) within the ETS1 Pointed domain (Figure 5D)(22–25). Mutation of T38 blocks the ability of the RAS pathway to phosphorylate ETS1. HEK-293T cells were transfected with ETS1 or a mutant form of ETS1, ETS1(T38A) (Figure 5D) and were expressed with or without HGF or PAX3 (Figure 5E). Both versions of ETS1 activated the MET promoter to similar levels (ETS1, 2.8±1.1 fold, ETS1(T38A), 3.1±0.4 fold). Both proteins synergistically activated the MET promoter with PAX3, while only the wild-type protein was able to have the same effect with HGF (all groups p<0.005 except for ETS1(T38A) + HGF, p=0.172). To verify that the ETS1(T38A) protein is not phosphorylated by HGF, protein levels of MET, ERK, and ETS were measured in transfected HEK-293T cells. While all cell sets responded to HGF through the phosphorylation and activation of MET and ERK, the wild-type ETS1 protein but not the mutant was phosphorylated (Figure 5F). Here, we find that exogenous wild-type ETS1 protein is phosphorylated concurrently to ERK activation due to HGF stimulation, and is able to promote MET expression when the MET receptor is present.
To determine if HGF activates endogenous ETS1 protein within melanoma cells, HGF was added to a panel of six melanoma cell lines and MET and ETS1 protein levels were measured. Levels of phosphorylated ETS1 (pETS1) were significantly higher in 5/6 cell lines after HGF treatment in comparison to untreated cells (Figure 5G, H, p<0.05). SKMEL-28 cells also had an increase in pETS1 levels post HGF treatment, but not to a significant level (p<0.218). The increase in pETS1 levels corresponded to a parallel rise in pMET levels. To determine if ETS1 phosphorylation levels are dependent on MEK and ERK activity, HGF induced melanoma cells were treated with a specific inhibitor of MEK, PD184352. Levels of pETS1 decreased in A375 and mel-624 cells in the presence of PD184352 but not in untreated or mock treated cells (Figure 5I). These data support a model where MET receptor activation via HGF activates ETS1 and consequently its own promoter.
The proximal MET promoter contains two ETS binding sites that function synergistically with PAX3 or through HGF-activation
To determine if the E1 and E2 enhancers where necessary for ETS1-dependent synergistic activation of METpm with PAX3 or HGF, MET promoter reporter and ETS1 expression constructs were transfected into HEK-293T cells in the presence or absence of PAX3 or HGF. Reporter constructs contained intact or mutated E1 and E2 sites (Figure 2,6A). ETS1 was able to synergistically activate METpm vector with the addition of either PAX3 or HGF (Figure 5B,6A, set 1). A mutation in the E1 site resulted in a loss of synergistic activation with PAX3, but not with HGF treatment (Figure 6A, set 2). Conversely, mutation of the E2 site did not significantly effect ETS1-PAX3 activation of the MET reporter vector, but the robust activation of the reporter after HGF treatment was lost (Figure 6A, set 3). Loss of both E1 and E2 sites abrogated both the ETS1-PAX3 and ETS1-HGF activation (Figure 6A, set 4). These data support a model of two distinct ETS binding sites in the MET promoter (Figure 6B). The E1 site, along with an adjacent PAX site, comprises an “ETS-PAX enhancer.” Optimum activation of this site requires the presence of both PAX3 and ETS1. The other site, E2, contains the ideal binding sequence for independent ETS1 binding (CCGGAWR). The ability of ETS1 to drive expression from this “ETS-PETS1 enhancer” is heightened by ETS1 phosphorylation as a result of HGF signaling.
Figure 6.
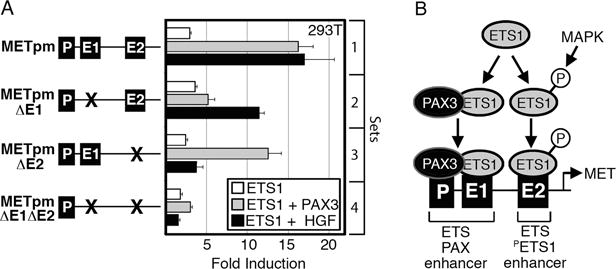
The MET promoter contains two ETS-responsive enhancers that work synergistically with either PAX3 or HGF stimulation. (A) ETS1 synergistically activates the MET promoter with PAX3 through enhancer E1, and HGF via enhancer E2. Luciferase assays of 293T cells transfected with MET promoter constructs with wild-type E1 and E2 enhancer sequence (set 1), E1 mutated (set 2), E2 mutated (set 3), or with both E1 and E2 mutated (set 4). The reporter constructs were transfected with ETS1 (white bars), ETS1 and PAX3 (grey bars), or ETS1 with exogenous HGF (black bars). Fold induction was calculated by measuring luciferase activity in arbitrary light units, normalized against beta-galactosidase activity, then divided by the measurements obtained for reporter vector alone (n=9). (B) Schematic summarizing ETS1 activation of the MET promoter through the E1 and E2 elements. The E1 site has a PAX-dependent ETS1 binding sequence with a PAX site directly proximal; ETS1 synergistically activates the MET promoter with PAX3 through E1. The E2 site contains an ideal ETS1 binding site, and ETS1 synergistically activates the MET promoter through this site with exogenous HGF.
Expression of a dominant-negative ETS1 protein inhibits MET induction and melanoma cell growth
To determine if disruption of normal ETS1 function would affect MET expression and melanoma cell growth, cells were transfected with a dominant-negative ETS1 expression construct (DN-ETS1). A truncated ETS1 protein containing only the C-terminal DNA-binding ETS domain acts as a dominant-negative against wild-type activity by competitive DNA binding (Figure 7A)(36–38). The presence of DN-ETS1 significantly (p<0.005) inhibits activation of MET by PAX3 and ETS1 (Figure 7B). In addition, melanoma cells demonstrated significant inhibition of growth both in cellulo and in vivo (Figure 7C,D,E).
Figure 7.
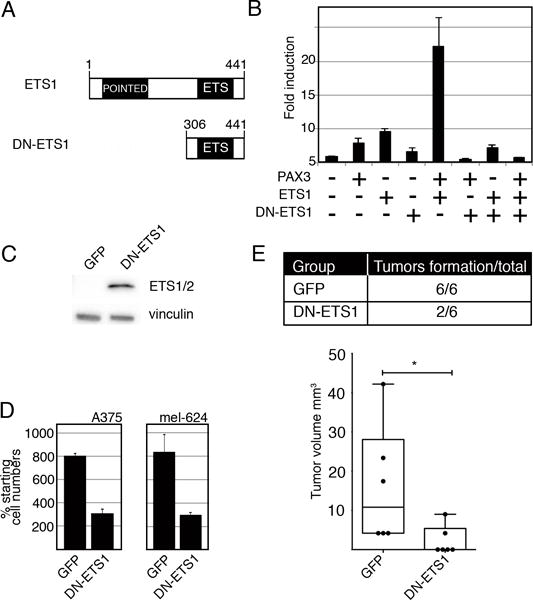
Expression of a dominant-negative ETS1 (DN-ETS1) protein inhibits MET induction and melanoma cell growth. (A) A schematic of wild-type ETS1 and DN-ETS1 proteins. (B) DN-ETS1 inhibits the activation of the MET promoter by both PAX3 and ETS1. Luciferase assays of 293T cells transfected with METpm, in the presence (+) or absence (−) of PAX3, ETS1, and/or DN-ETS1 expression vectors. Fold induction was calculated by measuring luciferase activity in arbitrary light units, normalized against beta-galactosidase activity, and divided by the measurements obtained for reporter vector alone (n=9, p<0.005). (C) A western analysis detects DN-ETS1 expression in A375 melanoma cells utilizing an antibody that recognizes the C-terminal of ETS1 (ETS1/2 antibody). (D) DN-ETS1 attenuates melanoma cell growth. A375 and mel-624 cells were transfected with either a GFP or a dual DN-ETS1/GFP expressing construct, and green cells were counted after cells were transfected and at 48 hours post transfection. The “% starting cell numbers” were calculated as GFP-expressing cell numbers at 48 hours post-transfection divided by starting cell numbers, then multiplied by 100 (n=3, p<0.005). (E) DN-ETS1 attenuates tumor formation in vivo. A375 cells were transfected with either a GFP or a dual DN-ETS1/GFP expression construct and transplanted into the flanks of nu/nu mice. While all A375 cells transfected with GFP formed tumors ten days after transplantation, only 2/6 of DN-ETS1 formed any palpable tumors with significant inhibition of tumor formation (p<0.05). For each group, n=6.
A375 and mel-624 melanoma cells, transfected with either a control GFP-expressing vector or a dual DN-ETS1/GFP expressing construct, demonstrated a significantly (p< 0.005) attenuated growth at 48 hours post-transfection when DN-ETS1 was present (Figure 7D). The transfected A375 cells, when transplanted into athymic Nu/Nu mice, grew tumors for all GFP transfected cells ten days post transplantation, but only 2/6 times when the DN-ETS1 was present (Figure 7E).
In summary, ETS1 drives expression of MET in melanoma cells, either through a PAX-dependent enhancer, or a feed-forward PAX-independent mechanism via ETS1 phosphorylation and activation downstream of HGF.
Discussion
While it is evident that MET overexpression drives tumor proliferation, survival, and progression in melanoma, the mechanism for this overexpression is unknown. Our group and others have previously found PAX3, MITF, and SOX10 upstream of MET (14, 39, 40). While PAX3 and MITF are able to promote MET expression independently, SOX10 alone was unable to drive expression from the proximal promoter element utilized in this study. However, SOX10 was able to function as a cofactor with either PAX3 or MITF. While PAX3 is able to drive expression and bind to the MET promoter in a number of different melanoma cell lines, inhibition of PAX3 lead to a significant reduction of MET in SK-MEL23 and SK-MEL28 but not in A375 cells ((14) and Figure 4E). These earlier studies suggest that PAX3 is a regulator of MET in melanoma cells, but that the mechanism for activation was not the same for all melanoma cells.
Here, we identify a pathway wherein transcription factors ETS1 and PAX3 promote excess MET protein expression. We find that PAX3 and ETS1 are commonly expressed in melanoma (Figure 1), efficient at driving MET promoter expression (Figure 3A), and bind directly the MET promoter (Figure 3C,4B). The PAX and ETS enhancer sites are important for promoter expression in melanoma, and inhibition of these factors lead to a reduction of MET levels (Figure 4). In this report, we discover that ETS1 drives MET expression in melanoma, and ETS1 activity is increased multifold with the addition of HGF or PAX3.
Our group and others find that PAX3 is expressed in melanoma, where it actively promotes migration and invasiveness (20). These qualities are the most deadly in melanoma, and they are reacquired during resistance to the small molecule BRAF inhibitor, vemurafenib. Since PAX3 promotes migration and metastasis, it may be an active player in vemurafenib resistance. Indeed, recent reports find that PAX3 is overexpressed in vemurafenib resistant cell lines, and PAX3 inhibition restores drug sensitivity in these cells (21). One major mechanism for melanoma progression and drug resistance may be through PAX3-dependent MET expression.
An added mechanism of PAX3-related resistance may be through its interaction with ETS1, a protein activated through pathways downstream of BRAF. Our findings suggest that ETS1 activates MET both in a PAX3-dependent and independent manner (Figure 5,6). This is the first report to find that ETS1 directly interacts with the transcription factor PAX3, and that these factors synergistically activate the expression of MET (Figures 3,4,6). ETS1 also binds to the PAX3-related protein PAX5 and the PAX5 epitopes that directly bind to ETS1 are also found in PAX3 (31, 34, 41). It is likely that ETS1 drives the expression of many genes together with PAX factors. In this report, we find the first example of a PAX3-ETS1 regulated gene in melanoma, the tyrosine kinase receptor MET.
We find that ETS1 promotes melanoma progression, and the implementation of a dominant-negative ETS1 (DN-ETS1) protein inhibits MET promoter expression and melanoma cell growth (Figure 7). This DN-ETS1 protein also inhibits tumor growth, invasion and migration in other tumor models (37, 42). DN-ETS1 has also been previously expressed in melanoma cells to study in vivo invasiveness (43). This group found that DN-ETS1 enhances invasion and metastasis in one overexpressing clone (TM4) but not in another (TM5). Our findings, coupled to these prior discoveries, support that ETS1 may have differential roles in melanoma progression in terms of growth and metastasis. This falls in line with recent findings for MITF and BRN2, where both factors promote melanoma progression, but higher levels of each protein drives either cellular proliferation or migration. (44, 45). The precise role of ETS1 in melanoma initiation, establishment, and metastasis still needs to be elucidated.
In our report, we discover a pathway for MET to promote its own expression by HGF-dependent induction of ETS1 (Figure 5, 6). HGF signals through the MET receptor, which in turn activates ETS1 downstream of the MAPK pathway. This has potential long-term clinical implications as well, since HGF has been found to be a major driver of resistance against mutant BRAF inhibitors in melanoma therapy (46). This resistance may be supported, at least in part, by ETS1 activity. It is not known if the consequential phosphorylation and activation of ETS1 contributes to the progression of this tumor, or how dependent the cells are on ETS1 activity. Our report further supports that ETS1, along with PAX3, drives the expression of oncogenic genes that promote tumor progression.
Materials and Methods
Cell culture
Human melanoma lines (A375, mel-537, mel-624, mel-888, SKMEL-5, SKMEL-23, and SKMEL-28), HEK293T, and 3T3 cells (ATCC, Manassas, VA and University of Chicago Comprehensive Cancer Center Core Facilities) were cultured in DMEM/10% FBS (Sigma-Alrich, St. Louis, MO, USA). Morphology, growth curve analysis, and melanoma-marker testing verified melanoma cell identity. All cells were negative for the presence of mycoplasma. For Hepatocyte Growth Factor (HGF) treatment, cells were grown in serum-free media, supplemented with 0.5% bovine serum albumin (BSA) and 30 ng/ml HGF (EMD-Millipore, Temecula, CA) for 7.5–10 minutes prior to cell collection and analysis. For PD184352 (Sigma-Alrich) treatment, 1 μM was added for the length of the experiment.
Antibodies
Primary antibodies utilized for western analysis, immunohistochemistry, ChIP and immunoprecipitation were against PAX3 (University of Iowa Hybridoma Bank, Iowa City, IA, USA), ETS1 (Santa-Cruz Biotechnology, Santa-Cruz, CA), 1:200 pETS1 (Sigma-Alrich), ETS1/2 (Santa-Cruz Biotechnology), MET (Cell-Signaling Technology, Danvers, MA), pMET (1:500, Invitrogen Co., Carlsbad, CA), or vinculin (Sigma-Alrich) antibody.
Western blots
Cells were lysed in RIPA buffer and 50μg total protein was separated on 4–12% Bis-Tris gels, transferred to nitrocellulose membranes then probed with 1:200 Pax3, 1:500 ETS1, 1:200 pETS1, 1:200 ETS1/2, 1:6,000 MET, or 1:1,000,000 vinculin antibody. Control lysates to serve as negative controls for phosphorylated protein were treated with calf intestinal phosphatase (CIP, New England BioLabs, Ipswich, MA) at 0.54 units/μg of total protein at 37°C for 1 hour.
Immunofluorescence analysis of PAX3, ETS1, and MET expression
Archival paraffin-embedded primary melanoma specimens were obtained through the University of Chicago Hospital Dermatology tissue bank following approved IRB and Clinical Trials Committee protocols. For antigen retrieval, 5μm tissue sections were boiled in EDTA buffer (pH 8.5). Samples were blocked in 1% normal goat serum then incubated with primary antibodies PAX3 (1:250) and ETS1 (1:500), pETS1 (1:500), MET (1:1000), or pMET (1:500). Samples were washed in PBS buffer, and incubated with goat anti-rabbit fluorescein or goat anti-mouse dyl647 secondary antibody (1:1000, Pierce Biotechnology/Thermo Scientific, Rockford, IL). Staining was scored as “positive” or “negative” with a positive score ≥25% of the tumor tissue expressing the antigen.
Plasmid construction
The MET reporter construct (METpm) contains the genomic DNA fragment shown in Figure 2A and cloned as described (14). Mutations in putative PAX and ETS sites were created by site directed mutagenesis. The sites were mutated by changing the PAX site from GTCCCGC to ACTAGTC, the E1 site from CTCCGG to CTCGAG, and the E2 site from GCAGGAAG to GCTAGCAG. The PAX3 expression construct was created as described (47). The pcDNA3-ETS1 expression construct was provided by Eric Svensson (University of Chicago, Chicago, IL). ETS1(T38A) was created by site directed mutagenesis, changing codon 38 from ACT (Threonine) to GCT (Alanine). The pGex2T-PAX3 vector was a kind gift from Jonathan Epstein (University of Pennsylvania, Philadelphia, PA). The coding sequence of ETS1 was cloned into pGex2T (GE Healthcare, Uppsala, Sweden) for overexpression in bacteria and purification using primers AACCCGGGTATGAAGGCGGCCGTCGATCTCAA and TTCCCGGGTCACTCGTCGGCATCTGGCTT. The pcDNA3-DN-ETS1-IRES-GFP construct contains amino acids 306–441 of ETS1 which encodes the DNA binding domain only and functions as a dominant negative protein against wild-type ETS1 activity (36–38). This region was amplified using primers ATAAGCTTATGGACTATGTGCGGGACCGTGCT and ATGGATCCTCACTCGTCGGCATCTGGCT then cloned into pcDNA3 (Invitrogen). The internal ribosomal entry site (IRES) was amplified from the pIRES-hrGFP-1a vector (Agilent Technologies, Santa Clara, CA) with primers TAGATATCCTTGGGTTACCCCCCTCTCCCT and TACTCGAGGCGGCCGCCATTATCATCGTGTTTTTCA then cloned into the previous construct. To add the green fluorescent protein (GFP) gene to this construct, Primers AAGCGGCCGCAATGGTGAGCAAGGGCGAGGAG and AATCTAGATTACTTGTACAGCTCGTCCAT were used to amplify the GFP gene of the pEGFP-N1 vector (Clontech Laboratories, Inc., Palo Alto, CA). The pcDNA3-GFP vector was constructed by amplifying the EGFP gene and cloning it into pcDNA3.
Luciferase assays
Cells were transfected with MET promoter luciferase reporter constructs, an internal control beta-galactosidase expressing construct pCMV (Clontech), PAX3 and ETS1 expression constructs. Cells were transfected with Lipofectamine 2000 (Invitrogen), incubated for 48 hours, then luciferase and beta-galactosidase levels were measured (Promega Biosciences, Inc., Madison, WI). For the calculation of fold induction, luciferase activity was measured in arbitrary light units, normalized against beta-galactosidase activity, and divided by the measurements obtained for reporter vector alone (or with HGF, if the experimental group also had HGF treatment.) All experiments were performed in at least triplicate.
siRNA Inhibition
Melanoma cell line mel-624 was transfected with siRNA against PAX3 (GAAACACCGUGCCGUCAGU), ETS1 (GAAAUGAUGUCUCAAGCAU), both, or siScramble (100pmol, Dharmacon, Lafayette, CO) with Lipofectamine-2000 (Invitrogen) according to manufacturer’s recommendations. Cells were transfected for two consecutive days, then the media was changed to media containing 30ng/ml HGF (EMD Millipore) 48 hours after the initial transfection.
Chromatin Immunoprecipitation (ChIP) assays
Cells were fixed in 1% formaldehyde, quenched in 0.125M glycine, then lysed and sonicated in SDS lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-HCl pH 8.1). Protein-DNA complexes were incubated with antibodies against PAX3 or ETS1. Normal IgG (Sigma-Alrich) was used as a negative control against non-specific DNA precipitation by an antibody. PCR was performed with primers to the MET enhancer, with TCCGCCTCTAACAATGAACTCC (F,human) or TTGTCTGTGACAATGAGCGCC (F,mouse) and AAGGTGAAACTTTCTAGGTGG (R). All ChIP samples were tested for false positive PCR amplification using beta-tubulin gene sequence primers. The nested primer set for this control was AAAGGCCACTACACAGAGGG (F) and TACCAACTGATGGACGGAGAGG (R,human) or AACCAACTGATGGACAGACAGG (R,mouse).
Co-immunoprecipitation of PAX3 and ETS1 proteins
Proteins collected from melanoma cells were obtained via sonication in 2X GS buffer (40mM HEPES, 100mM KCl, 40% glycerol, 2mM 2-mercaptoethanol) with protease inhibitor cocktail (Sigma-Aldrich) and 1mM PMSF. GST, GST-ETS1, and GST-PAX3 were expressed in BL21 bacteria and purified with Glutathione-Sepharose 4B beads according to the manufacturer’s protocol (GE Healthcare). Protein was incubated with either PAX3 or ETS1 antibodies for 2 hours at 4°C, then with Protein A/G agarose beads (EMD Millipore) for an additional 2 hours. The resulting precipitates were washed 3 times in lysis buffer, resolved on 10% SDS-PAGE gels, and evaluated by standard Western blot analysis. Normal mouse IgG antibody was used as controls.
Electrophoretic Mobility Shift Assay
GST, GST-ETS1, or GST-PAX3 proteins were mixed in reaction buffer (10mM Tris-HCl, pH 8.0, 150mM KCl, 0.5mM EDTA, 0.1% Triton-X 100, 12.5% glycerol, 0.2mM DTT, and 100μg/ml poly(dI-dC) for 30 minutes at 25°C followed by the addition of probe for 15 minutes at 25°C. Primers for probe sequences (Figure 3B) were annealed and labeled by end filling with DNA polymerase I Large (Klenow) fragment (New England Biolabs) then purified on Illustra ProbeQuant G-50 Micro columns (GE Healthcare). Electrophoresis was performed on 5% native gels. The gels were dried then exposed to autoradiography film.
In vivo tumor formation assays
Athymic Nu/Nu mice (6–7 month old females, Charles River Laboratories,Roanoke, IL) and treated and maintained in accordance to institutional guidelines. A375 cells were transfected with either the pcDNA3-GFP or pcDNA3-ETS1-DN-IRES-GFP vectors with TransIT-2020 Transfection Reagent (Mirus, Madison, WI) according to manufacturer’s instruction. Eighteen hours post-transfection, cells were trypsinized and resuspended at 20×106 cells/mL in DMEM media containing 20% FBS. GFP-positive cells were collected by the University of Chicago Flow Cytometry Core Facility using the FACSAria III (BD, Franklin Lakes, NJ). Cell counts and viability were confirmed using hemacytometer readings and trypan blue exclusion. Sorted cells were washed once in PBS and resuspended in a final volume of 15×106 cells/mL. Mice received subcutaneous injections of 200μL (3×106 cells) in each flank (n=6, pcDNA3-GFP; n=6, pcDNA3-ETS1-DN-IRES-GFP). Tumor volume was measured using calipers, with volume calculated as (length × width2) × (π/6).
Statistical Analysis and densitometry
For each experiment, three or more replicates were analyzed. Western band intensities were measured using ImageJ64 (http://rsbweb.nih.gov/ij/). Data presented as percentages or fold are normalized to a control group as stated. Data error bars represent standard error of the mean (SEM) for comparisons of samples within one group. One-tailed unpaired student T-test analysis determined significance for all graphical data, where all sample groups were compared to a control (Microsoft Excel). For in vivo tumor growth experiments, significance was determined by two-tailed Mann-Whitney U tests. P-values of ≤0.05 are considered significant.
Acknowledgments
We thank Drs. Ravi Salgia, Eric Svennson and Jonathan Epstein for providing material and/or intellectual support for this project. This work was supported by grants and financial support from the American Cancer Society (RSG-CSM-121505), Friends of Dermatology-University of Chicago, The Wendy Will Chase Foundation and the National Institutes of Health (R01CA130202 and R01AR062547).
Financial support: University of Chicago Cancer Center Pilot program (P30-CA014599), American Cancer Society (RSG-CSM-121505), Friends of Dermatology-University of Chicago, Wendy Will Chase Foundation, and the National Institutes of Health (R01CA130202 and R01AR062547).
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
References
- 1.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 2.Keehn CA, Smoller BR, Morgan MB. Expression of the ets-1 proto-oncogene in melanocytic lesions. Mod Pathol. 2003;16(8):772–7. doi: 10.1097/01.MP.0000082395.59356.4F. [DOI] [PubMed] [Google Scholar]
- 3.Rothhammer T, Hahne JC, Florin A, Poser I, Soncin F, Wernert N, et al. The Ets-1 transcription factor is involved in the development and invasion of malignant melanoma. Cell Mol Life Sci. 2004;61(1):118–28. doi: 10.1007/s00018-003-3337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong L, Jiang CC, Thorne RF, Croft A, Yang F, Liu H, et al. Ets-1 mediates upregulation of Mcl-1 downstream of XBP-1 in human melanoma cells upon ER stress. Oncogene. 2011;30(34):3716–26. doi: 10.1038/onc.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tajima A, Miyamoto Y, Kadowaki H, Hayashi M. Mouse integrin alphav promoter is regulated by transcriptional factors Ets and Sp1 in melanoma cells. Biochim Biophys Acta. 2000;1492(2–3):377–84. doi: 10.1016/s0167-4781(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 6.Natali PG, Nicotra MR, Di Renzo MF, Prat M, Bigotti A, Cavaliere R, et al. Expression of the c-Met/HGF receptor in human melanocytic neoplasms: demonstration of the relationship to malignant melanoma tumour progression. Br J Cancer. 1993;68(4):746–50. doi: 10.1038/bjc.1993.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, et al. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13(7):2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 8.Saitoh K, Takahashi H, Sawada N, Parsons PG. Detection of the c-met proto-oncogene product in normal skin and tumours of melanocytic origin. J Pathol. 1994;174(3):191–9. doi: 10.1002/path.1711740308. [DOI] [PubMed] [Google Scholar]
- 9.Gambarotta G, Boccaccio C, Giordano S, Ando M, Stella MC, Comoglio PM. Ets up-regulates MET transcription. Oncogene. 1996;13(9):1911–7. [PubMed] [Google Scholar]
- 10.Horikawa T, Sheen TS, Takeshita H, Sato H, Furukawa M, Yoshizaki T. Induction of c-Met proto-oncogene by Epstein-Barr virus latent membrane protein-1 and the correlation with cervical lymph node metastasis of nasopharyngeal carcinoma. Am J Pathol. 2001;159(1):27–33. doi: 10.1016/S0002-9440(10)61669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y, Xu W, Lu J, He F, Yang X. Invasiveness of hepatocellular carcinoma cell lines: contribution of hepatocyte growth factor, c-met, and transcription factor Ets-1. Biochem Biophys Res Commun. 2001;286(5):1123–30. doi: 10.1006/bbrc.2001.5521. [DOI] [PubMed] [Google Scholar]
- 12.Tomita N, Morishita R, Taniyama Y, Koike H, Aoki M, Shimizu H, et al. Angiogenic property of hepatocyte growth factor is dependent on upregulation of essential transcription factor for angiogenesis, ets-1. Circulation. 2003;107(10):1411–7. doi: 10.1161/01.cir.0000055331.41937.aa. [DOI] [PubMed] [Google Scholar]
- 13.Saeki H, Kuwano H, Kawaguchi H, Ohno S, Sugimachi K. Expression of ets-1 transcription factor is correlated with penetrating tumor progression in patients with squamous cell carcinoma of the esophagus. Cancer. 2000;89(8):1670–6. doi: 10.1002/1097-0142(20001015)89:8<1670::aid-cncr4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Mascarenhas JB, Littlejohn EL, Wolsky RJ, Young KP, Nelson M, Salgia R, et al. PAX3 and SOX10 activate MET receptor expression in melanoma. Pigment Cell Melanoma Res. 2010;23(2):225–37. doi: 10.1111/j.1755-148X.2010.00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008;21(6):627–45. doi: 10.1111/j.1755-148X.2008.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He SJ, Stevens G, Braithwaite AW, Eccles MR. Transfection of melanoma cells with antisense PAX3 oligonucleotides additively complements cisplatin-induced cytotoxicity. Mol Cancer Ther. 2005;4(6):996–1003. doi: 10.1158/1535-7163.MCT-04-0252. [DOI] [PubMed] [Google Scholar]
- 17.Medic S, Ziman M. PAX3 expression in normal skin melanocytes and melanocytic lesions (naevi and melanomas) PLoS One. 2010;5(4):e9977. doi: 10.1371/journal.pone.0009977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plummer RS, Shea CR, Nelson M, Powell SK, Freeman DM, Dan CP, et al. PAX3 expression in primary melanomas and nevi. Mod Pathol. 2008;21(5):525–30. doi: 10.1038/modpathol.3801019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholl FA, Kamarashev J, Murmann OV, Geertsen R, Dummer R, Schafer BW. PAX3 is expressed in human melanomas and contributes to tumor cell survival. Cancer Res. 2001;61(3):823–6. [PubMed] [Google Scholar]
- 20.Bonvin E, Falletta P, Shaw H, Delmas V, Goding CR. A phosphatidylinositol 3-kinase-Pax3 axis regulates Brn-2 expression in melanoma. Mol Cell Biol. 2012;32(22):4674–83. doi: 10.1128/MCB.01067-12. Epub 2012/09/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Cao J, Wu J, Sullivan K, Shen J, Ryu B, et al. Stat3-targeted therapies overcome the acquired resistance to vemurafenib in melanomas. J Invest Dermatol. 2013;133(8):2041–9. doi: 10.1038/jid.2013.32. Epub 2013/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paumelle R, Tulasne D, Kherrouche Z, Plaza S, Leroy C, Reveneau S, et al. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene. 2002;21(15):2309–19. doi: 10.1038/sj.onc.1205297. [DOI] [PubMed] [Google Scholar]
- 23.Wasylyk C, Bradford AP, Gutierrez-Hartmann A, Wasylyk B. Conserved mechanisms of Ras regulation of evolutionary related transcription factors, Ets1 and Pointed P2. Oncogene. 1997;14(8):899–913. doi: 10.1038/sj.onc.1200914. [DOI] [PubMed] [Google Scholar]
- 24.Yang BS, Hauser CA, Henkel G, Colman MS, Van Beveren C, Stacey KJ, et al. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol. 1996;16(2):538–47. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seidel JJ, Graves BJ. An ERK2 docking site in the Pointed domain distinguishes a subset of ETS transcription factors. Genes Dev. 2002;16(1):127–37. doi: 10.1101/gad.950902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gambarotta G, Pistoi S, Giordano S, Comoglio PM, Santoro C. Structure and inducible regulation of the human MET promoter. J Biol Chem. 1994;269(17):12852–7. [PubMed] [Google Scholar]
- 27.Karim FD, Urness LD, Thummel CS, Klemsz MJ, McKercher SR, Celada A, et al. The ETS-domain: a new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes Dev. 1990;4(9):1451–3. doi: 10.1101/gad.4.9.1451. [DOI] [PubMed] [Google Scholar]
- 28.Nye JA, Petersen JM, Gunther CV, Jonsen MD, Graves BJ. Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 1992;6(6):975–90. doi: 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- 29.Woods DB, Ghysdael J, Owen MJ. Identification of nucleotide preferences in DNA sequences recognised specifically by c-Ets-1 protein. Nucleic Acids Res. 1992;20(4):699–704. doi: 10.1093/nar/20.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher RJ, Mavrothalassitis G, Kondoh A, Papas TS. High-affinity DNA-protein interactions of the cellular ETS1 protein: the determination of the ETS binding motif. Oncogene. 1991;6(12):2249–54. [PubMed] [Google Scholar]
- 31.Fitzsimmons D, Hodsdon W, Wheat W, Maira S, Wasylyk B, Hagman J. Pax-5 (BSAP) recruits Ets proto-oncogene family proteins to form functional ternary comlexes on a B-cell-specific promoter. Genes & Develop. 1996;10:2198–211. doi: 10.1101/gad.10.17.2198. [DOI] [PubMed] [Google Scholar]
- 32.Fitzsimmons D, Lukin K, Lutz R, Garvie CW, Wolberger C, Hagman J. Highly cooperative recruitment of Ets-1 and release of autoinhibition by Pax5. J Mol Biol. 2009;392(2):452–64. doi: 10.1016/j.jmb.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garvie CW, Hagman J, Wolberger C. Structural studies of Ets-1/Pax5 complex formation on DNA. Mol Cell. 2001;8(6):1267–76. doi: 10.1016/s1097-2765(01)00410-5. [DOI] [PubMed] [Google Scholar]
- 34.Wheat W, Fitzsimmons D, Lennox H, Krautkramer SR, Gentile LN, McIntosh LP, et al. The highly conserved beta-hairpin of the paired DNA-binding domain is required for assembly of Pax-Ets ternary complexes. Mol Cell Biol. 1999;19(3):2231–41. doi: 10.1128/mcb.19.3.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A. 1996;93(9):4213–8. doi: 10.1073/pnas.93.9.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato T, Furukawa K. Sequential action of Ets-1 and Sp1 in the activation of the human beta-1,4-galactosyltransferase V gene involved in abnormal glycosylation characteristic of cancer cells. J Biol Chem. 2007;282(38):27702–12. doi: 10.1074/jbc.M611862200. Epub 2007/07/28. [DOI] [PubMed] [Google Scholar]
- 37.Holterman CE, Franovic A, Payette J, Lee S. ETS-1 oncogenic activity mediated by transforming growth factor alpha. Cancer Res. 2010;70(2):730–40. doi: 10.1158/0008-5472.CAN-09-2090. Epub 2010/01/14. [DOI] [PubMed] [Google Scholar]
- 38.Nakano T, Abe M, Tanaka K, Shineha R, Satomi S, Sato Y. Angiogenesis inhibition by transdominant mutant Ets-1. J Cell Physiol. 2000;184(2):255–62. doi: 10.1002/1097-4652(200008)184:2<255::AID-JCP14>3.0.CO;2-J. Epub 2000/06/27. [DOI] [PubMed] [Google Scholar]
- 39.Beuret L, Flori E, Denoyelle C, Bille K, Busca R, Picardo M, et al. Up-regulation of MET expression by alpha-melanocyte-stimulating hormone and MITF allows hepatocyte growth factor to protect melanocytes and melanoma cells from apoptosis. J Biol Chem. 2007;282(19):14140–7. doi: 10.1074/jbc.M611563200. [DOI] [PubMed] [Google Scholar]
- 40.McGill GG, Haq R, Nishimura EK, Fisher DE. c-Met expression is regulated by Mitf in the melanocyte lineage. J Biol Chem. 2006;281(15):10365–73. doi: 10.1074/jbc.M513094200. [DOI] [PubMed] [Google Scholar]
- 41.Maier H, Ostraat R, Parenti S, Fitzsimmons D, Abraham LJ, Garvie CW, et al. Requirements for selective recruitment of Ets proteins and activation of mb-1/Ig-alpha gene transcription by Pax-5 (BSAP) Nucleic Acids Res. 2003;31(19):5483–9. doi: 10.1093/nar/gkg785. Epub 2003/09/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sahin A, Vercamer C, Kaminski A, Fuchs T, Florin A, Hahne JC, et al. Dominant-negative inhibition of Ets 1 suppresses tumor growth, invasion and migration in rat C6 glioma cells and reveals differentially expressed Ets 1 target genes. Int J Oncol. 2009;34(2):377–89. Epub 2009/01/17. [PubMed] [Google Scholar]
- 43.Mattia G, Errico MC, Felicetti F, Petrini M, Bottero L, Tomasello L, et al. Constitutive activation of the ETS-1-miR-222 circuitry in metastatic melanoma. Pigment Cell Melanoma Res. 2011;24(5):953–65. doi: 10.1111/j.1755-148X.2011.00881.x. Epub 2011/06/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20(24):3426–39. doi: 10.1101/gad.406406. Epub 2006/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, et al. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69(20):7969–77. doi: 10.1158/0008-5472.CAN-09-0781. Epub 2009/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum Mol Genet. 2003;12(8):937–45. doi: 10.1093/hmg/ddg107. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Yang J, Li Y, Liu Y. Both Sp1 and Smad participate in mediating TGF-beta1-induced HGF receptor expression in renal epithelial cells. Am J Physiol Renal Physiol. 2005;288(1):F16–26. doi: 10.1152/ajprenal.00318.2003. [DOI] [PubMed] [Google Scholar]
- 49.Stella MC, Trusolino L, Pennacchietti S, Comoglio PM. Negative feedback regulation of Met-dependent invasive growth by Notch. Mol Cell Biol. 2005;25(10):3982–96. doi: 10.1128/MCB.25.10.3982-3996.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]