

Abstract
Soft fluorescent nanomaterials have attracted recent attention as imaging agents for biological applications, because they provide the advantages of good biocompatibility, high brightness, and easy biofunctionalization. Here, we provide a survey of recent developments in fluorescent soft nano-sized biological imaging agents. Various soft fluorescent nanoparticles (NPs) (including dye-doped polymer NPs, semiconducting polymer NPs, small-molecule organic NPs, nanogels, micelles, vesicles, and biomaterial-based NPs) are summarized from the perspectives of preparation method, structure, optical property, and surface functionalization. Based on both optical and functional properties of the nano-sized imaging agents, their applications are then reviewed in terms of in vitro imaging, in vivo imaging, and cellular-process imaging, by means of specific or nonspecific targeting.
1. Introduction
Optical imaging plays a major role in basic life science research and clinical applications because it is noninvasive, rapid, highly sensitive and inexpensive. In order to create the most accurate representation of biological objects and/or processes, many cutting-edge imaging technologies have emerged with the aim to increase the resolution of optical imaging. For example, stimulated emission depletion microscopy (STED), stochastical optical reconstruction microscopy (STORM), photoactivated localization microscopy (PALM) and total internal reflection fluorescence microscopy (TIRF) have been developed to image biological molecules with greater sensitivity or beyond the diffraction limit of light. 1-4 These high-resolution or super-resolution imaging technologies have been applied to visualize cellular structures in an in vitro setting with fantastic results, but the temporal resolution still needs to be further improved so as to image dynamic events that occur in a live cell. For these new imaging modalities, fluorescent probes with high brightness and photostability are in great need because the spatial resolution is often proportional to the number of photons collected from the probes.
Meanwhile, substantial effort has also been devoted to designing various imaging agents, among which fluorescent nanomaterials have been gaining special attention. When compared to the common organic dyes and fluorescent proteins, nano-sized fluorescent imaging agents are brighter and more photostable. Every nanoparticle (NP) of an imaging agent can carry a large amount of fluorescent species that is insulated by the particle matrix from complex biological environments. Moreover, the large surface-to-volume-ratio of NPs provides a convenient platform for surface modification and bioconjugation. Surface modification and bioconjugation of NPs are fundamentally necessary for biological and biomedical applications in order to label or enter into live cells for in vitro or in vivo imaging. 5
Nano-sized imaging agents can be broadly classified into two categories based on their chemical nature and likely behavior in biological environments 6: (i) hard nanomaterials (e.g. quantum dots, 7-9 dye-doped silica NPs, 10, 11 lanthanide-doped NPs, 12 carbon NPs13, 14 and metal nanoclusters 15, 16) and (ii) soft nanomaterials (e.g. polymeric NPs, nanogels and micelles). The former nano-sized imaging agents are prepared from inorganic elements, usually under harsh conditions, through multiple processes. Although they have been widely used for in vitro fluorescent imaging after surface modification, the inorganic nature and potential bio-incompatibility hampers their practical applications for in vivo imaging. 17-19 To date, only one kind of hard nano-sized imaging agent—nonporous silica NPs—has been approved by the U.S. Food and Drug Administration (FDA) for phase I clinical trial. 20 In contrast, soft optical nano-sized imaging agents are fabricated from less toxic organic molecules under mild conditions. Furthermore, many FDA-approved biocompatible molecules are available for the development of nano-sized imaging agents, such as the fluorescent dye indocyanine green (ICG) 21 and the biodegradable polymers poly(methyl methacrylate) (PMMA), poly(ethylene glycol) (PEG), poly(lactic acid) (PLA), and poly(lactic-co-glycolic acid) (PLGA) derivatives. 22
There have been a number of reviews about soft fluorescent NPs that summarize their recent developments and applications, such as in biological imaging. Those reviews have been focused on metal-complex-dyed polymer NPs, 23 metal-free polymeric NPs, 24 conjugated polymer NPs 25, 26 and small-molecule organic NPs. 27, 28 In this review, a broader range of soft fluorescent NPs are summarized, encompassing dye-doped polymer NPs, semiconducting polymer NPs, small-molecule organic NPs, hydrogels, micelles, and vesicles. The design rules and synthesis methods are discussed from the perspectives of particle structure, optical property, and surface functionalization. Their applications are reviewed in terms of in vitro imaging, in vivo imaging, and cellular-process imaging. We hope readers will gain knowledge about the construction of soft fluorescent nano-sized imaging agents, and selection of the appropriate fluorescent organic NPs with the correct nanostructure, fluorophores, and targeting groups for their biological molecule of interest. We want to point out that the scope of this review is limited to fluorescent organic NPs in the context of biological imaging; fluorescent nanocomposites (e.g. composed of an inorganic core and an organic shell) and fluorescence-based multi-functional organic NPs are excluded. We also confine our discussions to fluorescent organic NPs that have regular shape, controllable particle size, and known surface chemistry; thus, we omit discussions on fluorescent nano-aggregates that lack defined morphological information or surface chemistry. The time span of this review is approximately limited to reports from the last 5 years.
2. Design and synthesis of soft fluorescent NPs
2.1 General rules
Particle synthesis
In forming organic NPs, the interaction forces between molecules could be either covalent or noncovalent, depending on the properties of the starting materials. If small molecules are utilized as the raw materials, they usually are required to be covalently bonded into a compact three-dimensional structure to stop the dyes from leaking out. For creating the covalent bonds, a direct polymerization method is preferable. 29 Small molecules can also form stable NPs by noncovalent forces, provided that an outer shell is added to encapsulate the small molecules. 28 For polymer macromolecules, noncovalent attractive forces sometimes are sufficient to bind them into particles. Specifically, hydrophobic polymers can form NPs by hydrophobic interactions using methods like reprecipitation; hydrophilic polyelectrolytes can form NPs via hydrogen bonding or ionic interaction by a self-assembly method. 30
Optical properties
In fluorescent organic NPs, the organic chromophores that normally are in the solid state may aggregate or interact with neighboring chromophores. As a result, their optical properties are quite different in the nanoscale from when they are in an organic solvent. The optical properties are largely dependent on the chromophores’ chemical structures as well as the nano-sized structure in which they are held. The organic chromophores are derived either from π-conjugated macromolecules or from small molecules. In conjugated semiconducting polymers, free carriers generated by photoexcitation can migrate along the polymer chain; the free carriers can either combine to form triplets to give fluorescence or are deactivated by other nonradiative processes. Hence, the optical properties of semiconducting polymer NPs are greatly influenced by the adopted chain conformations after particle formation. Semiconducting polymer NPs prepared by self-assembly usually exhibit red-shifted absorption spectra because their highly ordered conformation consists of aligned, stretched polymer chains. 31, 32 In contrast, NPs prepared by reprecipitation exhibit blue-shifted absorption spectra because they have a collapsed conformation with reduced conjugation length caused by bending, torsion, and kinking of the polymer backbone. 33 But when it comes to fluorescence, semiconducting polymer NPs made by either method display common red-shifted emission spectra because the increased inter- and intra-chain interactions favor energy transfer to low-energy-emitting sites. Thanks to their high light-harvesting capability and efficient intra-particle energy transfer, semiconducting polymer NPs commonly exhibit much brighter fluorescence than that of typical organic dyes. 34, 35
For most small-molecule dyes, too many of them in one particle can result in concentration quenching as well as a reduction in fluorescence by aggregation-caused quenching (ACQ) via π – π stacking. 36 To address the issue of fluorescence quenching, a widely adopted strategy is to reduce the doping ratio of dyes down to a level that avoids concentration quenching. The resulting fluorescent organic NPs are useful for ordinary biological imaging applications, such as cellular labeling and sensing, but the limited loading capacity and lower brightness precludes their application in ultra-sensitive imaging, such as single-particle tracking. Recently, several novel systems have been developed to produce highly bright fluorescent organic NPs. One is dye-doped semiconducting polymer NPs. The doping ratio is kept rather low, but the per-particle fluorescence brightness is greatly enhanced due to the extraordinary light-harvesting capability as well as efficient intraparticle energy transfer within semiconducting polymer NPs. 37-39 Another is nano-sized aggregates of organic fluorophores that are based on the photophysical process of aggregation-induced emission (AIE). 27, 40, 41 These organic dyes do not emit in dilute solution, but when they aggregate into particles, they give off bright fluorescence because of restricted intramolecular rotation. Lastly, organic dyes in organic solvent have been directly used as biological imaging agents after being encapsulated into a nano-sized droplet. 42, 43 The liquid state of the droplet prevents the ACQ effect of encapsulated dyes from occurring while the nano-sized core ensures a high loading capacity of dyes. As a result, highly fluorescent nano-sized droplets could be made.
Biophysicochemical properties
As fluorescent organic NPs are designed for biological imaging, their biophysicochemical parameters, including particle size, surface property, and targeting moiety, are crucial for their journey inside the organism and imaging performance. These parameters should be carefully considered for the synthesis of nano-sized imaging agents.
For particle size, previous studies indicate that a size below 200 nm in diameter ensures efficient cellular internalization, while a size approximately smaller than 10 nm can be cleared quickly by glomerular filtration in the kidney and that larger than 100 nm is eliminated by macrophage cells of the liver and spleen. 5 Hence, from the perspective of achieving high contrast at the imaging site, the optimal size range for in vitro imaging may be <200 nm and 10-100 nm for in vivo imaging. Additionally, particle size in the range of 30-200 nm is appropriate for in vivo targeting of tumor tissues based on the enhanced permeation and retention (EPR) effect. 44 Surface chemistry also influences the efficiency of cellular uptake and blood circulation time. 45 Positively charged surfaces promote cellular internalization of NPs through adsorptive endocytosis via nonspecific electrostatic association with negative cell membrane. Positively charged surfaces also facilitate the subsequent escape of NPs from endolysosomal compartments through osmolytic effect. As hydrophobic surfaces suffer from nonspecific interaction with proteins, 46 hydrophilic surfaces can decrease the nonspecific absorption of proteins onto NPs and achieve prolonged circulation time accordingly. 47 Particularly, PEGylated NPs have been demonstrated to be stealthy because the PEG shields them from nonspecific interactions with various components of blood serum and consequently gives them greatly enhanced circulation in vivo. 48 The size of organic NPs can be controlled by manipulating the reaction parameters.
To design targeted organic NPs, two approaches can be followed. One is passive targeting, i.e. size-dependent uptake of NPs based on the EPR effect of tumor tissue. 49 The special microstructure of tumor tissue enables an enhanced extravasation of the particulate material from the surrounding vessel into the tumor, while the lack of a lymphatic system leads to a prolonged retention time of NPs. Together, these factors result in a preferential accumulation of NPs at tumor tissue However, the heterogeneity in tumor tissues undermines the specificity of EPR-based targeting, and some tumor types may not show an EPR effect. 50 The other approach is active targeting, which takes advantage of ligands, which are conjugated to the particle surface, that specifically bind to antigens or receptors expressed only on the target cells. 51 Depending on the type of targeting sites, various kinds of ligands, including antibodies, peptides, aptamers, sugars and vitamins, are available for bioconjugation.
A straightforward strategy for functionalization of NPs is to add molecules or polymers with the appropriate functional groups to the starting materials prior to the formation of NPs. More generally, surface functionalization is achieved by a two-step process. First, reactive groups (e.g. carboxylic and amino groups) are introduced onto the particle surface. Second, the reactive groups are conjugated with specific ligands or polymers via coupling reactions, such as the carbodiimide-mediated amide, the maleimide coupling reaction, or the click reaction.
2.2 Polymerization method
Polymerization is a popular strategy to synthesize soft organic NPs using two different routes. 25, 30, 52 One route is direct polymerization including dispersion polymerization and emulsion polymerization, which in situ yields polymeric NPs by coupling low-molecular-weight monomers into macromolecular polymers that don't dissolve in the starting reaction medium. The polymerization process to make nano-sized droplets occurs in water with surfactants present as stabilizers to give rise to soft fluorescent NPs. The other route is post-polymerization or reprecipitation (or nanoprecipitation). It takes advantage of commercially available (or pre-synthesized) polymers/dyes dissolved in an organic solvent as the starting solution; the starting solution is then dispersed into a solvent in which the polymers/dyes have poor solubility, such as water, to generate NPs. Dispersion of the polymer solution is usually enhanced by ultrasound waves or other catalysts. If the starting organic solvent is water-immiscible, the polymer solution needs to be emulsified into sufficiently small droplets first followed by evaporation of the organic solvent. If a water-miscible organic solvent is used, the polymer solution (in a dilute concentration) can be injected and mixed in water directly; the organic NPs precipitate because of a sudden decrease in solubility and hydrophobic interaction.
2.2.1 Direct polymerization
The merits of direct polymerization are (i) the covalent crosslinking of the incorporated fluorophores with the particle matrix can effectively avoid their leakage from NPs, especially in complex biological environments and (ii) fine-tuning of both the fluorescent property and surface chemistry of NPs by adding functional monomers to the reaction, which facilitates the synthesis of the appropriate biological imaging agents. Based on this chemical approach, a broad range of fluorescent biocompatible NPs has been prepared as nano-sized imaging agents (Table 1). These organic NPs are prepared from fluorescent conjugated polymer monomers, 53-55as well as from nonfluorescent materials such as PMMA, 56, 57 polystyrene (PS), 56-59 hydrogels 60-66 and biodegradable materials. 67-69 For the latter cases, fluorescent moieties were additionally introduced to label the polymeric NPs, except for alginate 70 and polydopamine NPs 69 which formed label-free NPs that self-fluoresce.
Table 1.
Fluorescent soft nano-sized imaging agents prepared by direct polymerization method*
| Particle type | Structure properties |
Optical properties | Applications | ||
|---|---|---|---|---|---|
| size (nm) | composition | surface + ligands | |||
| Conjugated polymer NPs | 60-120 | diethynyl pyrrolo-pyrrole (or diethynyl fluorenon) in poly(arylene ethynylene) | Non specific | tunable flu. λex = 398 nm; λem, max= 470-585 nm | nonspecific in vitro imaging (HeLa cells)53 |
| 60 ±14 | cyanovinylene backboned polymer | Tween 80 (surfactant) | multicolor flu. (blue to NIR) NIR-NPs (λem = 693 nm, QY= 21%) | in vivo mapping of sentinel lymph node (in mice)54 | |
| ca.40 | polyacrylonitrile | NH2 + anti-ErbB2 (antibody) | λex = 268 nm; λem =350-500 nm (broad band) | specific in vitro imaging (SK-BR-3 cells)55 | |
| Dye-doped polymer NPs | 93 | Rhodamine B in PMMA | Tween 80 | λex = 572 nm; λem = 628 nm | in vivo tracking of stem cells (hAFCs) in mouse brain56 |
| 20-300 | Rhodamine B in PMMA | Tween 80 | λex = 405 nm; λem = 546 nm | in vitro evaluation of cellular uptake of NPs (4T1 cells)57 | |
| 200-500 | fluorescein in PS | aminomethyl + H2N-PEG3-OH | λex = 488 nm; λem = 515-545 nm | nonspecific in vitro imaging (HEK293T and HeLa cells)58 | |
| 100-150 | n-(2,6-diisopropylphenyl)-perylene-3,4-dicarbonacidimide (PMI) in PS | NH2; COOH | λem = 530 nm | in vitro labeling of immature dendritic cells (iDCs cells)59 | |
| nanogels | 462 ±17 | fluorescein isothiocyanate in poly(amidoamine)-alginate | Non specific (negatively charged) | λex = 495 nm; λem = 519nm | in vitro imaging of drug delivery (CAL-72 cells)62 |
| 90-135 | abietane | PEG; NH4 + folic acid (electrostatic interaction) | self flu. λex = 360nm; λem = 450 nm | specific in vitro imaging (MCF-7 cells) and drug delivery70 | |
| 51±5 | Pd-tetra- (4-carboxyphenyl) tetrabenzoporphyrin dendrimer (G2) in polyacrylamide (PAA) | membrane penetrating peptides (TAT-Cys) or tumor specific peptides (F3-Cys) | λex = 630 nm; λem = 804 nm | (non) specific in vitro oxygen imaging (C6 glioma, MDA-MB-435 and MCF-7 cells)71 | |
| 400 | 3-hydroxyflavones in poly N-isopropylacrylamide (PNIPAM) | Non specific | ratiometric flu. λex = 355 nm, λem = 423-436 nm (↓) = 508-538 nm (↑) | temperature imaging (33- 41 °C)64 | |
| ca. 9 | DBD-AA (water-sensitive dye) in poly-N-n-propylacrylamide (NNPAM) | Non specific | λex = 456 nm, λem = 565 nm (↑); lifetime resolution 0.18 – 0.58 °C (~5-7.5 ns) | intracellular temperature mapping (COS7 cell, 29-39 °C)65 | |
| 165 - 241 | 5-aminofluorescein; dextran-poly(acrylic acid) | COOH + poly-l-lysine | λex= 492 nm; λem = 516 nm | nonspecific in vitro imaging of stem cells (ADSCs)66 | |
| Biodegradable NPs | 44 | cyanine dye (1.47%) and HPPH (0.69%) in polyacrylamide | NH2 + PEG; NH2 + F3-Cys (tumor targeting peptide) | λem = 843 nm | specific in vitro imaging and drug delivery (MDA-MB-435 cell)67 |
| 160 ± 13 | cyanie 7 in bisphosphonate (BP) | PEG; BP groups (chelated to bone mineral) | λex = 750 nm, λem = 773 nm | specific in vivo imaging of bone (chicken embryo model)68 | |
| plate-like NPs (in tens nm) | polydopamine | Non specific | excitation-dependent Flu. λex = 360-500 nm λem, max = 494-550 nm | nonspecific in vitro imaging (NIH-3T3 cells)69 | |
Notes: Size is in hydrodynamic diameter; composition mainly shows the fluorescent imaging agents and particle matrix; surface + ligand describe surface chemistry and (+) conjugated ligands; optical properties give both the excitation (λex) and emission wavelength (λem), wherein fluorescence is abbreviated as flu., and quantum yield is QY. Those are applicable to the following tables.
Organic NPs prepared by direct polymerization usually have a narrow size distribution. The particle size can be adjusted by controlling various parameters, such as reaction time, ratio of solvent/non-solvent and type of surfactants. But very small particle sizes, e.g. below 20 nm, are difficult to obtain by this method. In addition, this method can produce NPs with surface functional groups by directly adding amino-, carboxy-functionalized monomers during the process of polymerization, which are ready for further conjugation with other biomolecules. Surfactants such as PEG and Tween 80 also are used to stabilize the polymerized organic NPs which give good biocompatibility for nonspecific imaging. 54, 56, 57
Like many chemistry-based methods, the requisite expertise in organic and polymer synthesis may be an obstacle for scientists who are not chemists. Moreover, many of the polymerization processes take place in mixed organic solvents catalyzed with metal salts. Rigorous washing is therefore necessary to collect the organic NPs. But even then, there is still a high probability for organic solvents, which are toxic to cells, to be left inside NPs. However, for some polymers that are difficult to dissolve in many solvents, direct polymerization may be the only choice. 54
2.2.2 Post-polymerization
Miniemulsion method
The miniemulsion method was initially used by the Landfester group to prepare fluorescent semiconducting polymer NPs for photovoltaic and light-emitting devices. 72, 73 In order to adapt the semiconducting polymer NPs to be biocompatible and functionalized for targeting specific molecules for biological imaging, the miniemulsion technique was further developed. In an earlier work, Howes and coworkers used PEG as the surfactant to prepare PEG-capped semiconducting polymer NPs (ca. 13 nm). 74 Although the fluorescent NPs were biocompatible, the fluorophore loading was very low (0.57 wt.%). Subsequently, Howes and coworkers replaced the surfactant PEG with 1,2-diacyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and described four semiconducting polymers conjugated to phospholipids: PPE, BEHP-PPV, PF and MEH-PPV. 75 The PEG-phospholipid encapsulation allowed for stable conjugated polymer NPs as well as enhanced loading efficiency of up to 8.5 wt.%. Moreover, the phospholipid-encapsulated NPs could be readily functionalized by substituting a proportion of DSPE-PEG with carboxylic acid functionalized ones, which gave them the ability to be conjugated with biological molecules such as bovine serum albumin.
Using similar phospholipid-encapsulation strategy, other semiconducting polymers76, 77 and dye-conjugated biocompatible polymers78, 79 have been developed as fluorescent nano-sized imaging agents. So far, the biocompatible polymers are mainly derived from PLGA or DSPE-PEG, with functionalized terminals such as amino and carboxyl groups 76, 78 and folate. 77, 79 DSPE-PEG usually yields NPs with smaller diameters (<100 nm) than NPs made from PLGA because DSPE-PEG has a higher amphiphilicity and a lower molecular weight. For more detailed information about polymer-encapsulated fluorescent organic NPs, please refer to a recent review by Liu and co-workers. 28
The NPs described so far have solid cores. In contrast, a type of nanoemulsion with a liquid core has been developed by nanoemulsification, a modified miniemulsion method. 42, 43, 80 In this method, a water-immiscible solvent is preserved as the oily core of the nano-sized droplets rather than be evaporated. This oily core functions as a reservoir for encapsulation of lipophilic dyes. The nanoemulsions are of particular interest for biological imaging because (i) they are composed of nontoxic components which are biodegradable and/or readily eliminated from the body and (ii) they are highly fluorescent because of the high loading capacity of dyes (dissolved in oily core) without formation of nonfluorescent aggregates. For example, a cationic cyanine dye (DiI-TPB) with high solubility in oil (Labrafac) was synthesized and subsequently encapsulated into nano-sized droplets, with the assistance of a surfactant (Cremophor ELP®) and the sudden addition of water. The DiI-TPB dye, due to its exceptional oil solubility, could be loaded at 8 wt% concentration into nano-sized droplets. The extremely bright droplets (90 nm) containing ~12,000 cyanine molecules, were 100-fold brighter than quantum dots. 43
Table 2 shows the representative nano-sized imaging agents prepared by the miniemulsion method. In this method, the amphiphilic polymers function as the surfactant to form stable droplets but they also participate in the formation of particles. Therefore, the concentration of the loaded fluorophores is relatively limited. Furthermore, the percentage of hydrophobic segments in amphiphilic polymers greatly influences the compactness of the particle core through the hydrophobic interaction between encapsulation matrix and incorporated fluorophores. Because the density of particle core affects both the efficiency of intra-particle energy transfer and the insulation from outside quenching species, the quantum yield of the NPs is highly dependent on the nature of the amphiphilic polymers.
Table 2.
Fluorescent soft nano-sized imaging agents prepared by miniemulsion method
| Particle type | Structure properties |
Optical properties | Applications | ||
|---|---|---|---|---|---|
| size(nm) | composition | surface + ligands | |||
| Conjugated polymer NPs | ca.90 | MEH-PPV / PF / BEHP-PPV / PPE in DSPE-PEG and DPPC | PEG; COOH +BSA | λem = 593 nm (QY:1.3%), 542 nm (26.9%), 484 nm (1.26%) and 418 nm (18.98%) | nonspecific in vitro imaging (SH-SY5Y neuroblastoma and HeLa cells)75 |
| ca. 250 | POSS-PFV in PLGA | PEG; NH2 + trastuzumab (antibody) | λex = 405 nm λem = 479 (0.19%) |
specific in vitro detection of HER2-positive cancer cells (SKBR-3 against MCF-7, NIH-3T3)76 | |
| PFBTDBT10 in DSPE-PEG-(folate) | PEG; folate | λex = 467 nm λem = 700 nm (27%) |
specific in vivo imaging of hepatoma H22 tumor (mouse)77 | ||
| Dye-doped polymer NPs | 200 | NIR dye (DY-700) in PLGA | PVA | λex = 700 nm λem = 725 nm |
nonspecific in vitro (HUVEC, NIH/3T3 and J774.A1 cells), in vivo imaging (mice)78 |
| 110 | FP547 / FP682 in PLA-b-PEG | PEG; biotin/ anisamide/ folic acid/ (FA) | λem = 547 nm and 682 nm | specific in vitro imaging (PC-3, KB-3-1 and KB-3-1* cells)79 | |
| Nanoemulsion | 30-90 | cationic cyanine dye (DiI-TPB) in oil (Labrafac) | Cremophor ELP® (surfactant) | λex = 553 nm λem = 590 nm (3.5%) |
in vivo imaging of blood flow in zebrafish embryo43 |
Reprecipitation method
The representative nano-sized imaging agents prepared by the reprecipitation method are listed in Table 3. This method was first developed by the Masuhara group to prepare fluorescent conjugated polymer NPs, 81 and then modified by McNeill and coworkers. 33, 82 Because of its merits of easy fabrication, low cost and high-throughput, the reprecipitation method has been used to make many fluorescent semiconducting NPs 26 and dye-doped polymer NPs 83 from hydrophobic polymers. They are highly fluorescent and small-sized (ranging from about 5 to 30 nm), which are desired for biological imaging. But the lack of surface functionalization restricts them from being used in more complicated biological applications. To address this issue, several strategies have been adopted to complement the reprecipitation method.
Table 3.
Fluorescent soft nano-sized imaging agents prepared by reprecipitation method
| Particle type | Structure Properties |
Optical properties | Applications | ||
|---|---|---|---|---|---|
| size (nm) | composition | surface + ligands | |||
| Conjugated polymer NPs | 15 | PFBT; PS-PEG-COOH | PEG; COOH+streptavidin / IgGs | λex = 460 nm; λem = 540 nm (30 %) | specific in vitro imaging (MCF-7 cells), single-particle imaging84 |
| 15 | PFBT; PSMA | COOH + NH2-PEG; COOH + NH2-N3; COOH+ NH2-C≡CH | λex = 460 nm; λem = 540 nm | specific bioorthogonal in vitro imaging (MCF-7 cells)88 | |
| 15 | PFBT; PFDBT5; PSMA | COOH + NH2-PEG; COOH + CTX (peptide ligand) | λex = 488 nm; λem = 650 nm (56%) | specific in vitro (MCF-7 cells) and in vivo imaging of brain tumor (mice)35 | |
| 16-18 | BODIPY-PFO; PS-PEG-COOH | PEG; COOH+ streptavidin | multicolor and narrow emission (λem = 520 nm, 600 nm and 690 nm), FWHM 40-55 nm | specific in vitro imaging (MCF-7 cells)86 | |
| rod or ellipsoidal, 16 | fluorinated PFDPBT; PS-PEG-COOH | PEG; COOH+ streptavidin | λex = 410 nm; λem = 510 nm (49%) | specific in vitro imaging (MCF-7 cells)87 | |
| 21 | COOH (side-chain)-PFBT | COOH+ streptavidin | λex = 460 nm; λem = 540 nm | specific in vitro imaging (Her2-positive SK-BR-3 and MCF-7 cells).101 | |
| Dye-doped conjugated polymer NPs | ca. 33 | NIR dyes; PFBTDBT | COOH-terminated polydiacetylene shell + streptavidin | λex = 450 nm; λem = 653 nm (22.5%), 695 nm (22.2%) and 775 nm (20.1%) | specific in vitro imaging (MCF-7 cells)102 |
| 160 (22) | RhB-PS-NH2; PFBT (or PFPV) | NH2 | ratiometric flu. λem = 540 nm (ref.) = 573 nm (decreased by ~1%/°C) | in vitro temperature imaging (HeLa cells)38 | |
| ca.26 | PPE | SH + FITC | ratiometric flu. λem = 440 nm (ref.) = 513 nm (increased by 0.37/pH) | in vitro pH sensing (HeLa cells)39 | |
| 16 | spiropyran (photo chromic)-PFBT; PS-COOH-PEG | PEG; COOH+ streptavidin | Photoswitchable flu. λex = 460 nm; λem = 540 nm (decreased by 86% under UV, recovered under visible light) | specific in vitro imaging (MCF-7 cells)88 | |
| Dye-doped polymer NPs | 167-204 | Poly[(2-(4-vinylphenyl)ethene-1,1,2-triyl)tribenzene] | Non specific (negatively charged) | AIE flu. λex = 350 nm; λem = 470 nm | nonspecific in vitro imaging (HeLa cells)83 |
| Small-molecule organic NPs | 33 | AIE fluorogen (BTPEBT); | PEG | AIE flu. λex = 318 nm, 425 nm λem = 547 nm (62 ± 1%) |
nonspecific in vivo imaging of blood vasculature (mice)41 |
| 31-60 | DSPE-PEG 200 hydrophobic dye (Spiro-BTA) | multidentate ligands (C18PMH-PEG) + folic acid | No ACQ λex = 471 nm; λem = 591 nm (42%) | specific in vitro imaging (KB cells) | |
| Dye-doped biodegradable NPs | 5.8±1.1 | dye (IR-1061); PAA; DSPE-mPEG | PEG | λex = 808 nm; λem = 920 nm = 1064 nm (1.8%) | nonspecific in vivo imaging of inner organs, blood vessels (mice)93 |
| 200 | NIR dye (DY-675) – PLGA; simil-opioid glycopeptide (g7)-PLGA | g7 | λex = 674 nm; λem = 699 nm (11%) | specific in vivo imaging of brain (mice)94 | |
| < 80 | FITC; PBLG-alendronate; PBLG-PEG | PEG; Alendronate | λex = 495 nm; λem = 519 nm | specific in vivo imaging of femur tissue (rat)95 | |
| 130 | fgBODIPY dye; PLGA; DSPE-PEG 2000 | PEG | Two factor triggered flu.: pH value and molecule binding λem = 740 nm | nonspecific in vitro (endocytosis) and in vivo imaging of tumor (mice)103 | |
One strategy is the amphiphilic polymer coprecipitation method, which utilizes amphiphilic polymers bearing functional groups to coprecipitate with the semiconducting polymers in one step to form surface-functionalized NPs. Our groups have performed a series of studies in this area. 35, 84, 85 Typically, an amphiphilic comb-like polystyrene polymer, PS-PEG-COOH, is used to functionalize highly fluorescent semiconducting NPs (Fig. 1A). 84 The NPs have an average diameter of about 15 nm and contain more than 80 percent of effective fluorophores. Moreover, this strategy produces surface-functionalized NPs with both PEG and carboxylic groups, which ensure good stability and high reactivity with biomolecules, such as antibodies or streptavidin. Similarly, we have also used poly(styrene-co-maleic anhydride) (PSMA) polymer to produce carboxy-functionalized semiconducting NPs (Fig. 1B). 85 The surface carboxylic groups can further react with small amine-containing molecules, like amino azides and amino alkynes, using click chemistry to form NPs for bioorthogonal labeling.
Figure 1.

Schematic illustration of the synthesis of surface-functionalized semiconducting polymer NPs. The fluorescent semiconducting polymer PFBT (poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1',3-thiadiazole)]) is either co-precipitated with an amphiphilic polymer, PS-PEG-COOH (A) or PSMA (B), into NPs with surface carboxyl groups. Alternatively, PFBT is first synthesized with side-chain carboxylic acid groups before precipitated into NPs (C) (Adapted with permission from ref. 84 for A (Copyright 2010, American Chemical Society), from ref. 85 for B (Copyright 2010, Wiley-VCH) and from ref. 79 for C (Copyright 2012, American Chemical Society), respectively.
The amphiphilic polymer coprecipitation method can be easily applied to any hydrophobic fluorescent semiconducting polymers. 86-88 However, a weak point of this strategy is that the functional groups are noncovalently attached to the particles through hydrophobic association. In case of complex biological environments, these functional molecules may be detached from the NPs if the polymer backbone swells or undergoes internal reorganization. 25 Our group furthered studied surface functionalization and developed a synthetic strategy to covalently link functional groups to semiconducting polymer NPs, 7989 which we named the direct functionalization method. The premise of this method is that semiconducting polymers with functional groups are synthesized prior to the formation of NPs. For example, PFBT polymers with side-chain carboxylic acid groups were synthesized and reprecipitated to produce carboxylic-functionalized semiconducting polymer NPs (Fig. 1C). 79 It is important to point out that the degree of hydrophilic functionalization plays a critical role in determining the stability and fluorescence performance of the NPs, and that the density of hydrophilic groups should be kept at a low level for biological applications. In another study, a cross-linking strategy was developed to synthesize functionalized semiconducting polymer NPs. 89 PFBT polymer with side-chain amine groups was first prepared and then cross-linked by a functional polymer, poly(isobutylene- alt -maleic anhydride) (PIMA) or PSMA, to form covalent crosslinks with the semiconducting polymer while simultaneously providing carboxyl groups for bioconjugation. The strategy generated stable carboxy-functionalized PFBT NPs that had a small particle diameter of 10 nm or less.
With the improved reprecipitation methods, many dye-doped semiconducting polymer NPs were prepared for biological imaging. The small molecules were either physically trapped into particles by co-precipitation with semiconducting polymers,90-92 or chemically incorporated into polymers by conjugation prior to precipitation. 38, 39, 88 The high light-harvesting capability of semiconducting polymer NPs was maintained, while their fluorescent properties were tuned by the doped dyes. These features are highly favorable for the development of fluorescent nano-sized imaging agents.
Besides semiconducting polymers, many amphiphilic polymers, such as poly(acrylic acid) (PAA), 93 PLGA, 8394 poly(gamma-benzyl-l-glutamate) (PBLG) polypeptide 95 and polyurethane, 96 have been used in reprecipitation methods to trap small-molecule fluorophores to yield dye-doped polymer NPs. In most cases, PEG-modified polymers were needed to form an encapsulation shell, which involves a self-assembly process after precipitation. The encapsulation is important for the stability of the dye-doped amphiphilic polymer NPs because the hydrophobic interaction between small molecules is rather weak. Based on the reprecipitation-self-assembly method, bright fluorescent small-molecule organic NPs were prepared from AIE-typed fluorogens 41 or spirobifluorene derivatives that could avoid self-quenching, 97 respectively, with the assistance of DSPE-PEG or PEG-grafted poly(maleic anhydridealt-1-octadecene) (C18PMH-PEG).
As another variation of the reprecipitation method, a one-step reprecipitation-encapsulation method was developed to produce dye-doped biocompatible core-shell NPs. 98, 99 In this method (Fig. 2), a hydrophobic polymer (PS) and a alkoxy silane (DTS) were coprecipitated with selected fluorophores into a basic water containing positively charged poly-L-lysine (PLL). The PS functioned as the particle matrix and the DTS was the silica-based encapsulation agent. Base-catalyzed hydrolysis and condensation of alkoxysilane led to the silica-based encapsulation of particles; PLL molecules were subsequently absorbed onto particle surface by electrostatic attractive force between amino and silanol groups to create the surface modification. This simple strategy not only can be generalized to most hydrophobic dyes to produce fluorescent NPs, but also afford surface amino groups for further conjugation with a wide range of biomolecules. 100
Figure 2.
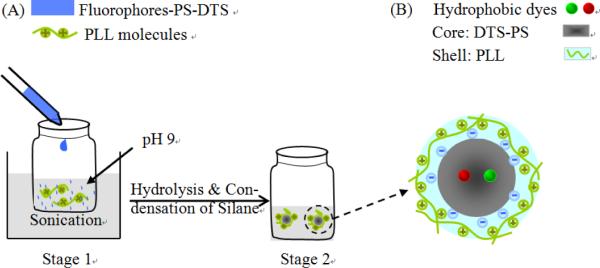
Schematic representation of the encapsulation-reprecipitation process. Electrostatic attraction between amino and silanol groups enables the PLL modification (Adapted with permission from ref. 99, Copyright 2012, Royal Society of Chemistry).
2.3 Self-assembly method
In the self-assembly method, the starting materials aggregate to form NPs via electrostatic attraction and/or hydrophobic interaction. Since the self-assembling process takes place in aqueous solution, stability and hydrophilicity of the resulting organic NPs are high. The fluorophores can be introduced into the particles either by direct co-assembling them with the starting materials or first conjugating them to the starting materials. The self-assembly approach is suitable for synthesis of fluorescent organic NPs for biological applications. Based on the nature of the starting materials, artificial or natural, the preparation methods are grouped into synthetic polymer-based self-assembly or biomaterial-based self-assembly (Table 4).
Table 4.
Fluorescent soft nano-sized imaging agents prepared by self-assembly and other methods*
| Particle type | Structure Properties |
Optical properties | Applications | ||
|---|---|---|---|---|---|
| size (nm) | composition | surface + ligands | |||
| Conjugated polymer NPs | 50 | cationic PFO; poly(L-glutamic acid)-doxorubicin (Dox) | COOH | λex = 380 nm; λem = 454 nm (turn on with the release of Dox) | in vitro imaging of drug delivery ( A549 cells)104 |
| 40-232 | amphiphilic fluorene oligomers | NH2 | λex = 335 nm, 430 nm; λem =545 nm (0.35-0.75) | nonspecific in vitro imaging (HeLa cells)106 | |
| 61 | star-shaped glycosylated conjugated oligomer | glycose (sugar) | λex = 388 nm λem = 553 nm (0.1) |
specific in vitro imaging (HeLa cells)107 | |
| Small-molecule organic NPs | ca.20 | AIE dye (BLSA); peroxalate (CPPO) | Pluronic F-127 (surfactant) | AIE flu. λex = 412 nm; λem = 578 nm (0.67) | in vitro ( RAW264.7 macrophages) and in vivo imaging of H2O2 (mice ankle)125 |
| 70-90 | AIE dye (C18-R) - PEG-polymer | PEG | AIE flu. λex = 365 nm; λem = 593 nm | nonspecific in vitro imaging (A549 cells)126 | |
| 109 | Amphiphilc perylene diimide derivative | PEG; NH2 | DIE multicolour flu. λem = 525 nm, = 630nm (susceptible to photobleaching) | nonspecific in vitro imaging (yeast cells)108 | |
| Helical nano-architectures | evolved shape | cyanostilbene-naphthalimide dyad | NH2 | phototunable flu. λem = 460-570 nm | nonspecific in vitro imaging (HeLa cells)109 |
| Polymeric micelles | 39-45 | Dox; PEG-b-P(S-co-AIE dye (PPSEMA)) | PEG | λex = 383 nm; λem = 503 nm (quenched by Dox) | in vitro imaging of drug delivery (HT-29 cells)111 |
| 15 | AIE dye (DSA)-PCL-b-PEG-folate | PEG; folate | λex = 410 nm; λem = 520 nm (27%) | specific in vitro imaging (HeLa cells) | |
| spiropyran-based hyperbranched star | hydrophilic shell (PDMAEMA | photoswitchable Flu. λex = 490 nm; | nonspecific in vitro imaging (HeLa cells)113 | ||
| 120 | copolymer ; hydropboic dye (NBD) | -co-PSPMA) | λem = 610 nm (upon UV irradiation) λem = 530 nm (upon visible-light irradiation) |
||
| 38±15 | Paclitaxel; porphyin (TAPP)-PCL-mPEO | PEO | λex = 423 nm; λem = 655 nm | in vitro (LoVo cells) and specific in vivo imaging of tumor (mice)114 | |
| 150 | ICG-copolymer-PEG-folate | PEG; folate | λex = 765 nm; λem = 820 nm | specific in vivo imaging of tumor (mice)115 | |
| Polymeric vesicles | No | Nile Blue-biomimetic block (PMPC) -pH responsive block (PDPA) | Non specific | pH-sensitive absorption and emission | nonspecific in vitro imaging (lysosomes, early endosomes, interstitial hypoxic/acidic regions)116 |
| ca.110 | NIR dye DiD (or DiR); PEO-b-PBD; | PEO; streptavidin + biotinylated antibody | λex=650 nm (or 754 nm); λem=683 nm (or 787 nm) | specific in vivo imaging of tumor (mice)117 | |
| Nanogels | 170 | PS-alt-Man-NIPAm-methylcoumarin | negatively charged | λex= 370 nm; λem= 450 nm (decreased with temperature) | nonspecific in vitro imaging of temperature (MDCK cells)118 |
| 73± 21 | Hyaluronic acid (HA)- poly(p-amino ester) -ICG | HA (CD44-receptor-targeting) | λex= 765 nm; λem = 820 nm (at pH 7.4 is off, at pH 5.5 on) | specific in vitro imaging (MDA-MB-231 cells)119 | |
| Biomaterials NPs | 7 | IR783-peptide; human serum albumin-Cy5 | peptides (targeting αvβ3-integrins) | λex = 627 nm; λem = 672 nm (Cy5, FRET to IR783) = 811 nm (IR783) | specific in vitro (MDA-MB-231 cells) and in vivo imaging of sentinel lymph node (mice)121 |
| 40 | fluorescent proteins (DsRed or eGFP); hepatitis B virus | Non specific | enhanced flu. (160-170 fold) | nonspecific in vivo imaging (mouse)123 | |
| 151±53 | hydrophobic macrocycle (C-SNAF-cycl) | Non specific | λex= 673 nm; λem= 707 nm | specific in vivo imaging caspase activity (mouse)124 | |
| Dye doped polymer NPs | 500 | oxygen probe (PtTF20PP) and naphthalimide derivative; PS NPs | NH2 | Ratiometric flu. λex = 380 nm λem = 490 nm (reference) λem = 650 nm (probe) |
nonspecific in vitro oxygen imaging (NRK cell) |
| 25 (100) | NIR dye Sq730 (or Itrybe); PS NPs | COOH | λex= 640 nm; λem = 740 nm (25nm-Itrybe, lifetime= 0.9 ns ; 100 nm-Sq730, 2.8 ns) | in vitro lifetime multiplexed discrimination of cells (fibroblasts vs. macrophages)128 a | |
| 7 | DiO; high-density lipoprotein | Non specific | λex = 490 nm λem = 507 nm |
nonspecific in vitro imaging (macrophages)129 b | |
Notes: The NPs listed in the table are prepared by self-assembly method, except for that labeled with (a) are by post staining and (b) by microfluidic technology.
2.3.1 Synthetic polymer-based self-assembly
Amphiphilic molecules are the foundation for the self-assembly method. The molecules consist of (i) hydrophobic segments that are chromophores or used to encapsulate chromophores and (ii) hydrophilic portions that are responsible for both their solubility and biofunctionability. The two parts work synergistically to determine the structure, size and morphology of the self-assembled NPs. When amphiphilic molecules with small percentage of hydrophilic segments are used, the resulting fluorescent NPs usually take on a rather compact structure comprised of a hydrophobic particle core and a hydrophilic surface. For instance, the cationic semiconducting polymer Polyfluorene (PFO) can be self-assembled into solid NPs (50 nm) with the aid of the anionic hydrophilic poly(L-glutamic acid) by electrostatic attraction. 104 Hydrophobic chromophores are more often covalently linked with hydrophilic groups, in the form of amphiphilic oligomers, to self-assemble into compact particles. In this case, the hydrophobic interaction is responsible for the self-assembly process. Schenning and co-workers synthesized a type of π-conjugated amphiphilic oligomer by decorating a hydrophobic chromophore with gallic acid derivatives bearing alkyl tails at one end and ethylene glycol chains at the other; functional groups were next introduced at the periphery of the ethylene glycol side chains. By co-assembling the differently functionalized oligomers, fluorescent NPs with tuned ligand composition and density were made. 105 Furthermore, the investigators functionalized the ethylene glycol side chains with amine groups. By controlling the density of surface amino groups, i.e. the ratio of amine-functionalized oligomer to the primary one, size-adjustable amino-functionalized NPs were created (40-232 nm). 106 In another study reported by Liu and coworkers, star-shaped glycosylated conjugated oligomers were synthesized to prepare fluorescent organic NPs. With the high density of hydrophilic sugar side groups, the nonionic oligomers self-assembled into NPs with an average diameter of 61 nm. 107
Similarly, small hydrophilic dyes can aggregate into solid NPs by the self-assembly method with the assistance of amphiphilic polymers. In order to avoid the common ACQ effect, AIE-typed 107, 108 fluorophores were used to ensure strong fluorescence for biological imaging. 104, 105 Even without a surfactant, certain hydrophilic dyes with special structure can self-assemble into stable NPs. One example is a perylene diimide derivative. 108 The nano-sized assemblies were non-fluorescent in water but recovered fluorescence when absorbed by cells due to a dissociation-induced-emission (DIE) effect. Zhao and co-workers reported a type of helical nano-sized assembly based on a hierarchical self-assembly of an amino-functionalized cyanostilbene-naphthalimide dyad. 109 Tunable fluorescence (from blue to yellow) was obtained by the self-assembly disorder and the dual fluorescent characteristic that comes from the Z-to-E photoisomerization of the cyanostilbene unit.
When amphiphilic block copolymers are used, the self-assembled particles normally take the form of micelles, which are a loose nanostructure consisting of a hydrophobic core and a hydrophilic shell. 110 Several fluorescent polymeric micelles have been prepared from dye-conjugated amphiphilic block copolymers111, 112 or hyperbranched star copolymers.113, 114 In a typical study, PEGylated copolymers were conjugated with the NIR dye ICG and the ligand folate; the self-assembled polymeric micelles were about 150 nm in hydrodynamic size and 40-60 nm in the solid state. 115 During the self-assembly process, additional small hydrophobic species can be simultaneously encapsulated into the particle core to create multi-colored fluorescent imaging agents. 111, 113, 114
Self-assembly of amphiphilic block copolymers can also result in another loose nanostructure, the polymer vesicle. Armes et al. reported a polymer vesicle prepared from block copolymers comprised of the biomimetic PMPC block copolymer and the pH-responsive PDPA block copolymer labeled with Nile Blue. 116 Deprotonation of the PDPA block, as just above pH 6, led to in situ self-assembled vesicles. The PDPA-based vesicle membranes provided a highly hydrophobic environment that blue-shifted the absorption wavelength of Nile Blue label and increased its quantum yield. In another work, fluorescent polymer vesicles were prepared from the amphiphilic block copolymer (PEO-b-PBD) by a sequential process of mixing, drying, ultrasonication, and dialysis. 117 The hydrophobic membrane can encapsulate hydrophobic dyes and also be easily modified with ligands. Unfortunately, the whole preparation process requires four separate operations and two more days of reaction time, which makes this approach somewhat tedious.
For amphiphilic polymers with a high content of hydrophilic ingredients, the nano-sized assemblies resemble nanogels. A temperature-sensitive fluorescent nanogel was prepared from a well-known thermoresponsive polymer called NIPAm- combined with a water-sensitive fluorophore and a hydrophilic unit. 118 The fluorophores were quenched by neighboring water molecules in hydrated nanogels. But higher temperatures shrank the nanogel because of the hydrophobic interaction, resulting in the exclusion of water molecules out from nanogel and the emission of strong fluorescence. pH-activable fluorescent nanogels were also reported by self-assembly of a copolymer consisting of a targeting ligand HA, a pH-sensitive unit PBAE and an ICG copolymer. 119 ICG dyes are in quenched state when encapsulated by nanogels, but acidic pH can solubilize the PBAE units and recover the fluorescence of the ICG by releasing them from nanogels. 66
The synthetic polymer-based self-assembly method is similar to the post-polymerization method because they both rely on pre-synthesized materials to form particles via noncovalent forces. But the latter method needs a foreign force to disperse the precursor solution into tiny droplets. Specifically, the miniemulsion method needs water-immiscible solvents to produce an emulsion, and the reprecipitation method requires a quick dispersion time to render a sudden decrease in solubility. In comparison, the self-assembly process is rather mild. A longer time is needed to form thermodynamically stable particles by self-assembly. In addition, the particle size of self-assembled fluorescent NPs is generally bigger than those formed by reprecipitation, because the self-assembled fluorescent NPs are composed of loosely aggregated polymer chains of amphiphilic molecules.
2.3.2 Biomaterial-based self-assembly
Some biomaterials are of the nanoscale size so they can be converted into fluorescent NPs by complexing them with fluorescent molecules via self-assembly. Human serum albumin (HSA) is a good biological nano-sized carrier, which was used to noncovalently self-assemble complexes with the NIR dye ICG. 120 When co-assembled with the peptide-modified cationic dye IR783, the self-assembled particles led to peptide-coated fluorescent NPs for targeted imaging. 121 Bacteria are another type of nano-sized carriers that were used to assemble with conjugated polymer nanoparticles (CPNs). 122 Multicolour microparticles were developed by tuning fluorescence resonance energy transfer (FRET) efficiencies among CPNs under single excitation wavelength based on the self-assembly between bacteria and CPNs.
The HSA and bacterial fluorescent NPs are hybrids of biological matter and synthetic fluorophores. Genetically encoded fluorophores, such as a fluorescent protein, have been used to prepare purely biological NPs. Lee and coworkers synthesized fluorescent capsid nanoparticles (FCNPs) by genetically inserting enhanced green fluorescent protein (eGFP) (or red fluorescent protein from Discosoma, DsRed) into each of 240 surface spike tips of hepatitis B virus (HBV) capsid particles. The result was spherical NPs with an uniform diameter of about 40 nm because of the self-assembly function of the HBV core protein when expressed in E. Coil (Fig. 3). By adding flexible linker peptides in between DsRed (or eGFP) and the capsid, the fluorescence intensity of FCNPs was significantly amplified by 160-170 folds because fluorescence quenching was reduced. 123
Figure 3.
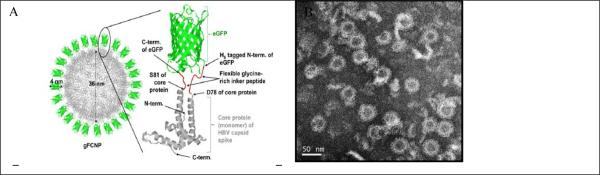
(A) Schematics of green fluorescent capsid nanoparticles (FCNPs). (B) TEM images of purified gFCNPs (Adapted with permission from ref. 123, Copyright 2012, Elsevier).
Although the self-assembly of biomaterials in aqueous solution can be troublesome, controlled self-assembly in living animals is even more challenging because of the complex and dynamic in vivo physiological environment. Very recently, Rao and coworkers reported an optimized first-order bioorthogonal cyclization reaction to control the self-assembly of a fluorescent small molecule in both apoptotic cells and tumor tissues. 124 A caspase-sensitive nano-sized aggregation fluorescent probe (C-SNAF) was designed to be biocompatible, possess NIR spectral properties and undergo triggered self-assembly through condensation chemistry in vivo (Fig. 4). In the presence of caspase-3/7, the L-DEVD capping peptide was cleaved, which triggered the intramolecular condensation of C-SNAF into the macrocycle C-SNAF-cycl. C-SNAF-cycl, which was rigid, hydrophobic and susceptible to intermolecular interactions (i.e. hydrophobic π-π stacking), self-assembled into nano-sized aggregates in situ.
Figure 4.
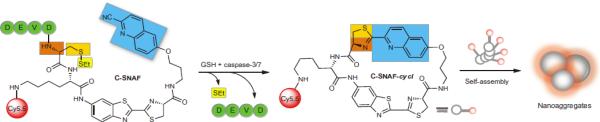
Proposed caspase-3/7 and reduction-controlled conversion of C-SNAF into C-SNAF-cycl through the bio-orthogonal intra-molecular cyclization reaction, followed by self-assembly into nano-aggregates in situ. Blue, the CHQ group; dark and light orange, amino and thiol groups of D-cysteine, respectively; yellow, thioethyl masking group; green, the capping peptide residues; red, NIR fluorophore Cy5.5 (reprinted with permission from ref. 124, Copyright 2014, Nature Publishing Group)
The biomaterial-based self-assembly method provides more biocompatible and even endogenous fluorescent organic NPs. In this regard, it is very favorable for the preparation of in vivo imaging agents. A disadvantage is that the available fluorophores that match the biomaterials or in vivo environments are limited.
2.4 Other methods
Post staining
In this method, fluorescent nano-sized imaging agents are prepared from NP precursors (commercially available or pre-fabricated) to function as the host for organic fluorophores (Table 4). Polystyrene NPs are popular because they are not very toxic to cells and available with different surface groups for bio-functionalization. 130-132 In a typical synthesis, amino-functionalized PS NPs were swollen in a water/organic solvent mixture under sonication. Two hydrophobic dyes (PtTF20PP and naphthalimide derivatives) then were added to allow for encapsulation into the NPs. 127 The swelling and sonication during the preparation process did not affect the size (and size distribution) and surface chemistry of the original monodispersed particles. Hydrophobic dyes (asymmetric cyanine Itrybe and squaraine dye Sq730) were also encapsulated by carboxyl-functionalized PS NPs via the staining procedure to produce NIR fluorescent NPs. 128 A straightforward strategy such as this one facilitates the creation of fluorescent organic NPs as well as the tuning of fluorescent properties by encapsulation with different dyes. However, the loaded dyes are highly likely to leak out of the NPs.
Microfluidic technology
The methods mentioned so far are capable of producing various biocompatible nano-sized imaging agents, but the NPs suffer from low yield and batch-to-batch variation. Recently, microfluidic-based approaches have emerged as a production method to yield reproducible and homogeneous batches of organic NPs in simple and efficient ways (Table 4). A variety of biocompatible NPs have been prepared by microfluidics, including liposome NPs, 133 polymeric NPs 134, 135 and lipid-polymer hybrid NPs. 136 Biologically active high-density lipoprotein-mimicking NPs (μHDL) also were prepared by microfluidics, which were loaded with either hydrophobic molecules (3,3-dioctadecyoxacarbocyanine perchlorate, DiO) or inorganic NPs (quantum dots). 129 The fluorescent μHDLs were reconstituted using a single-step, self-assembly method in a single layer, 3-inlet microfluidic device (Fig. 5). This microfluidic device generated tunable, dual microvortices and a focusing pattern at Reynolds number (Re) ~150 (Figure 1b-d). The setup allowed for rapid and effective mixing of precursors in the central inlet and the two outer inlets. As a result, the transition of the phospholipids (DMPC and MHPC)/dopants (e.g. DiO) from an organic solution into an aqueous one initiated the formation of lipid aggregates, while the microvortices caused apolipoprotein A-I (apoA-I) to swiftly incorporate in the nascent aggregates. The result was self-assembled small-sized μHDL NPs. The particle size of the μHDLs could be tuned by changing the Re value or the ratio of lipid to apoA-I. For the fluorescent DiO-μHDL, the mean size of the NPs was about 7 nm with a polydispersity of less than 0.1. The microfluidic approach resulted in a yield that was a continuous production of μHDL at a rate of 420 mg/h.
Figure 5.

Microfluidic reconstitution of HDL-derived NPs (μHDL). (A) A schematic depiction of a microfluidic platform that allows single-step and large-scale production of the HDL: reconstituted HDL by microfluidics (μHDL); DiO-μHDL for fluorescence; [S]-μHDL for a therapeutic platform; Au-μHDL for CT imaging; FeO-μHDL for MRI; and QD-μHDL for fluorescence. (B) Photograph of the microfluidic device. (C) Microscope image of dual microvortices at Re = 150. Scale bar is 500 μm. (D) Computational fluid dynamics simulation showing the microvortices at Re = 150 (reprinted with permission from ref. 129. Copyright 2013, American Chemical Society).
3. Biological and biomedical applications
3.1 In vitro imaging
3.1.1 Nonspecific imaging
Soft fluorescent NPs exhibit relatively low cytotoxicity compared with hard NPs because of the biocompatible nature of most organic materials. The NPs can be easily internalized by living cells via the process of endocytosis, regardless of whether the particle surface is bare or modified. In addition, the encapsulation of large quantity of fluorophores per NP ensures sufficient brightness for fluorescence imaging, even with common fluorescence microscopy under low staining concentrations. Therefore, nonspecific in vitro imaging is an elementary step for showing cellular images labeled with soft fluorescent NPs. A lot of fluorescent organic NPs have been used to label cells, including fluorescent conjugated polymer NPs, 53, 75, 106 dye-doped polymeric NPs, 58, 66, 78, 129 AIE- 126 and DIE- 108 type small-molecule NPs, self-fluorescent biodegradable NPs 69 and phototunable NPs 110, 114.
Aside from the simple lighting up of cells, nonspecific soft fluorescent NPs have been used to study the interactions between cells and NPs. For example, the mechanism of cellular uptake was studied with bare semiconducting polymer NPs (PFBT, 18 ± 5 nm). 137 Intracellular fluorescence colocalization of bare NPs and Texas Red dextran indicated that cellular uptake occurred through a common endocytic mechanism. The endocytic process was more vividly imaged by using of a dual-factor triggered NIR nanoprobe, boron dipyrromethene (fgBODIPY)-based biodegradable NPs (fg-nanoprobe). 103 The fg-nanoprobes showed an enhanced signal-to-noise ratio because of the synergistic effects of increased fluorogenicity triggered by cellular compartmentalization and the pH-tunable fluorescence on/off characteristic. The increased signal-to-noise ratio of fg-nanoprobes made them eligible for ultrahigh contrast NIR fluorescence imaging and super-resolution subdiffraction imaging. The time-dependent fluorogenic process during endocytosis was investigated with time-lapse confocal microscopy (Fig. 6). A low probe concentration (50 nM) incubated with cells (HUVEC) were used to illustrate the subcellular localization of fg-nanoprobe via endocytosis and activation (Fig. 6A). At the beginning, the fg-nanoprobe was approaching the cell, as seen by the released fg-BODIPY dyes. The dyes were activated and randomly anchored on cell membrane because the burst/release effect of biodegradable NPs caused by the microporous channels on the particle surfaces (Fig. 6B). When the fg-nanoprobe was trapped into the endo/lysosome compartments in the cytosol, the acidic environment and associated lipids accelerated the release of fg-BODIPY followed by fast protonation that gave rise to fluorogenicity. At later time points, the released dyes accumulated in the micellar-like compartments, such as clathrin (Fig. 6C). The series of activation processes of fg-nanoprobes from membrane lipid association to pH change in cytosolic membrane compartments clearly demonstrated the dynamic behavior of the cellular uptake of NPs. 103
Figure 6.

Fluorogenic progress of fg-nanoprobe during cellular uptake. (A) Time-dependent HUVEC uptake of fg-nanoprobe and subcellular distribution analysis (tracked via clathrin) by time-lapse confocal microscopy (TLCM). Inset in each picture shows the colocalization of activated fg-nanoprobe with clathrin. Blue: nucleus; green: clathrin; orange/yellow: co-localization of activated fg-BODIPY and clathrin; red: activated fg-BODIPY from fg-nanoprobe; (B) Early time point (4 h) subcellular distribution of fg-nanoprobe in HUVEC cells. Blue: nucleus; cyan: actin; green: tubulin; red: fg-nanoparticle; (C) Late time point (24 h) subcellular distribution of fg-nanoprobe in HUVEC cells (reprinted with permission from ref. 103, Copyright 2012, Elsevier).
The efficiency of cellular uptake of NPs was also studied with nonspecific fluorescent organic NPs. 59 In a recent work, in vitro quantitative evaluation of cellular uptake was performed using dye-doped polymeric NPs, of different sizes and varied surface charge, which were stabilized by the surfactant Tween 80. 57 By combining flow cytometry and plate fluorimetry, the number of particles internalized in each cell was evaluated. The results indicated that the uptake process reached a maximum in 2–5 h, and then stabilized within 24 h at a somewhat lower plateau value. Larger NPs with positive charge had higher cellular uptake and were better retained inside the cells than negatively charged NPs of similar size. The higher uptake of positively charged NPs makes sense because of the attraction between the NPs and the negatively charged cytoplasmic membrane of cells Smaller NPs were less dependent on surface charge for their uptake by cells..
As cellular uptake of NPs depends on size of NPs, the size of the particle may in turn provide a simple means to discriminate cell types. Hence fluorescent NPs of different sizes may have the ability to target certain cell types without being conjugated to any targeting ligands. Resch and coworkers demonstrated the strategy by using differently sized dye-loaded polystyrene NPs. 128 The experiment was designed under the following considerations: (i) macrophage cells are capable of an efficient internalization of particles with sizes up to 3 μm while fibroblast cells only internalize NPs less than 100 nm in diameter and (ii) the NIR dye Itrybe and Sq730 display a high degree of spectral overlap in emission but have different fluorescent lifetimes. In the experiment, 25-nm PS-NPs-Itrybe and 100-nm PS-NP-Sq730 were used to simultaneously incubate in a co-culture of 3T3 fibroblast and J774 macrophage cells. Both cell types competed for the uptake of PS-NPs. The macrophage and the fibroblast cells could not be spectrally distinguished by a confocal laser scanning microscropy (CLSM) image when detected in the same spectral window (Fig. 7A). But the fluorescence decay curves recorded on the two cell types revealed significantly different decay characteristics (Fig. 7B). Macrophage cells (red squares) exhibit longer decay times than fibroblast cells (black circles), suggesting a more efficient uptake of PS-NPs with longer lifetimes by the macrophage. 128
Figure 7.

(A) CLSM intensity image of a macrophage cell (1) and a fibroblast cell (2) in a co-culture. (B) Measured (symbols) and three-exponentially fitted (thick solid lines) fluorescence decay curves of the individual cells (macrophage: red squares, fibroblast: black circles) exposed to PS-NP-Itrybe (25 nm) and PS-NP-Sq730 (100 nm) in a mass ratio of 1:1. The residual traces in the bottom panel demonstrate the quality of the fits (reprinted with permission from ref. 128. Copyright 2013, American Chemical Society).
3.1.2. Specific imaging
In comparison to nonspecific imaging, the requirements for fluorescent organic NPs aimed at specificin vitro imaging applications are more stringent. Firstly, the fluorescent organic NPs should have high brightness so that the targeted subcellular structures can be distinguished from other parts of the cell. In this respect, many fluorescent organic NPs can fulfill this demand, such as semiconducting polymer NPs, dye-doped polymer NPs with high loading efficiency, and AIE-typed NPs. Secondly, the NPs need surface modification with hydrophilic surface groups (e.g. PEG) to reduce nonspecific absorption. Lastly, but most importantly, ligands for targeting must be conjugated to particles so that the modified particles can specifically bind cells via ligand-receptor interactions.
Antibodies
Among the various targeting ligands, antibodies are most widely used because they bind antigens, which are unique to each cell line, in a highly selective manner. Antibodies can be directly conjugated onto the surface of fluorescent soft NPs to selectively label cells. For example, self-fluorescent polyacrylonitrile NPs were conjugated with the anti-ErbB2 antibody via EDC/NHS activation. These antibody-conjugated NPs were then used for the specific targeting and imaging of human breast cancer cells (SKBR-3). 55 In another study, the monoclonal antibody trastuzumab (which has the trade name Herceptin) was coupled with conjugated-polymer-loaded NPs to selectively image SKBR-3 cells (against MCF-7 and NIH/3T3 cells), which overexpress the human epidermal growth factor receptor 2 (HER2), the target of trastuzumab (Figure 8). 76 The strong and regular green fluorescent profile of SKBR-3 breast cancer cells (Fig. 8A) indicated that the cell membrane was successfully stained by the conjugated-polymer-loaded NPs. In contrast, only weak and randomly distributed fluorescent signals were detected from MCF-7 and NIH/3T3 cell membrane (Fig. 8B-C). It is apparent that the specific interaction of trastuzumab-NPs conjugates with HER2 in the cell membrane allows clear discrimination of HER2-positive cancer cells from others. 76
Figure 8.
CLSM images of fixed SKBR-3 breast cancer cells (A), MCF-7 breast cancer cells (B) and NIH/3T3 fibroblast cells (C) stained with conjugated-polymer-loaded NPs. The cell nuclei are stained by propidium iodide (PI) (reprinted with permission from ref. 76, Copyright 2011, Wiley-VCH).
Alternatively, fluorescent organic NPs can be first conjugated to IgG or streptavidin and then used to specifically label cells based on the anti-IgG/IgG or biotin/streptavidin immunoreaction. Our group demonstrated this approach by using fluorescent PFBT NPs to label a specific cellular target, EpCAM, an epithelial cell-surface marker currently used to detect circulating tumor cells. 84 As seen in Figure 9A, EpCAM receptors on the surface of live MCF-7 human breast cancer cells were successfully labeled by the NP-IgG conjugates when they were co-incubated with a monoclonal primary anti-EpCAM antibody. In contrast, we did not detect a signal from the NPs when the cells were incubated without the primary antibody (Figure 9B). The NP-streptavidin conjugates also proved to specifically label EpCAM on the surface of live MCF-7 cells, together with the primary anti-EpCAM antibody and a biotinylated goat anti-mouse-IgG secondary antibody. 84 Furthermore, we upgraded the NP-streptavidin probes by using small-sized CN-PPV (cyano-polyphenylene vinylene) NPs (~10 nm) with high quantum yield (60%). With these probes, we achieved specific subcellular imaging of microtubule structures in HeLa cells 138 where the cells were fixed, permeabilized and incubated with a biotinylated monoclonal anti-α-tubulin antibody. In the confocal microcopy images (Figure 9C-D), bright fluorescence from well-resolved tubular structures was observed in the positively labeled cells, while the control samples showed no detectable nonspecific binding, demonstrating the sensitivity and selectivity of the CN-PPV NP-streptavidin bioconjugates. Although the NP-IgG and NP-streptavidin conjugates are both highly specific for cellular imaging, the streptavidin/biotin approach seems to be more attractive because most antibodies can be easily derivatized with biotin. Therefore, many fluorescent polymer NPs have been developed to specifically image live cells using this strategy. 86-88, 101, 102
Figure 9.

Specific cellular imaging with semiconducting polymer NPs-bioconjugates. (A) Fluorescence imaging of the cell-surface marker EpCAM on MCF-7 cells incubated sequentially with the anti-EpCAM primary antibody and NPs-IgG conjugates. (B) Fluorescence imaging of the control sample in which the cells were incubated with NPs-IgG alone (no primary antibody) (reprinted with permission from ref. 84, Copyright 2010, American Chemical Society). (C) Fluorescence imaging of microtubule structures in HeLa cells incubated sequentially with biotinylated anti-α-tubulin antibody and CN-PPV-streptavidin probes. (D) Fluorescence imaging of the control sample in which the cells were incubated with the probe conjugates only (no primary antibody). The scale bar in (A) and (C) is 10 μm, and in (D) is 50 μm (reprinted with permission from ref. 138, Copyright 2012, Royal Society of Chemistry).
Small-molecule targeting ligands
In addition to large-molecule antibodies that are expensive and potentially immunogenic, smaller targeting moieties have been used to conjugate to fluorescent NPs for specific cellular imaging applications. Examples include peptides (F3-Cys), 67 vitamins, sugars (e.g. glucose 107 and hyaluronic acid 119) and aptamers. 139 Folic acid also is widely adopted for this purpose because it selectively binds to folate receptors that are over-expressed in tumor cells. Self-fluorescent abietane NPs, 70 dye-doped PLA NPs 79 and small-molecule NPs 97, 112 were all conjugated to folic acid and used to specifically label live cells.
Bioorthogonal labeling based on click chemistry
The copper (I)-catalyzed Huisgen 1, 3-dipolar cycloaddition between azides and terminal alkynes is one of the most useful and reliable click reaction. 140 It has been applied to numerous NPs-related research fields. 141-145 In terms of specific imaging, both azide and alkyne groups can be considered to be bioorthogonal chemical reporters because they do not interact with any native biological functional groups. If one bioorthogonal reporter was incorporated into a target biomolecule using the biosynthetic machinery of the cell, and the other reporter is conjugated to fluorescent NPs, then it is possible to achieve specific labeling in situ via a click reaction. We demonstrated this approach by visualizing newly synthesized proteins which were modified by bioorthogonal noncanonical amino-acid tagging. 85 Newly synthesized proteins in MCF-7cells were metabolically labeled with an azide- (or alkyne-) bearing artificial amino acid, such as azidohomoalanine or homopropargylglycine, followed by tagging with alkyne- (or azide-) functionalized fluorescent semiconducting polymer NPs by click chemistry. Similarly, the clickable fluorescent NPs were used to selectively target glycoproteins. The highly efficient, specific, and bright protein labeling based on clickable polymer NPs suggest this bioorthogonal labeling is a promising strategy for visualizing various cellular processes.
3.2 In vivo imaging
The translation of fluorescence imaging from in vitro to in vivo remains a challenging task. There are two main issues that require serious consideration. One consideration is the penetration depth of optical signals, which is greatly attenuated by the photon absorption, scattering and autofluorescence in biological substances. To tackle this problem, fluorescent organic NPs with longer-wavelength emission, particularly in NIR range, are much preferred because of the reduced interaction within the “tissue optical window” (600-1300 nm). 146 Optical penetration depths around 1 mm in the NIR-I window (700 - 900 nm), and up to several millimeters in the NIR-II window (1-1.4 μm) have been explored with whole animals. 147
The other consideration is the biological barriers which the body uses to attack and remove exogenous materials (e.g., bacteria, viruses, medical implants, and drugs). The barriers imposed by the body can be broadly classified as either physiological or cellular. Physiological barriers include the blood, liver, spleen, kidneys, immune system, and the guards that prevent extravasation of foreign substances from the blood. Therefore, the particle surface of any nano-sized imaging agent should be carefully designed so it can bypass the biological barriers and reach the target sites before being removed from the body. 5 Cellular barriers include the cell membrane, the endosome/lysosome trafficking system, and intracellular trafficking.
3.2.1 Nonspecific imaging
Many fluorescent NPs with red or NIR emission have been prepared for nonspecific in vivo imaging. The particles are stabilized by either surfactants, 43, 56 amphiphilic polymers 78 or PEG 41, 93. By tracking the fluorescent trail of organic NPs, objects of interest can be imaged in vivo. For example, human amniotic fluid cells labeled with RhB-doped polymer NPs were grafted in the brain ventricles of healthy mice, and the longitudinal bio-distribution of the human amniotic fluid cells in different brain areas were determined by fluorescence imaging. 56
Blood vessels are ideal for nonspecific in vivo imaging because they are the main place for particle transportation after intravenous injection. In one study, ultra-bright AIE-type NPs were used for real-time intravital two-photon imaging of blood vessels. 41 By using of a two-photon fluorescence imaging technique, real-time vasculature imaging in live mice was investigated in three models (the brain, bone marrow and ear) with deep-tissue penetration and high contrast. Klymchenko and coworkers devised a type of bright fluorescent nano-sized droplets (termed as DiI-TPB) to perform in vivo microangiography imaging on live zebrafish embryos. 43 The nano-sized droplets were injected in the sinus venosus of the embryos. The confocal microscopy images showed that the nano-sized droplets remained in the blood circulation without interacting with the vessel wall and without dye leaking from them even 60 minutes after injection (Figure 10). Moreover, due to the extreme brightness of the nano-sized droplets (100-fold brighter than quantum dots of equivalent size), blood flow was measured by single-particle tracking of the nano-droplets in vivo, revealing both the slow and fast phases of the cardiac cycle.
Figure 10.
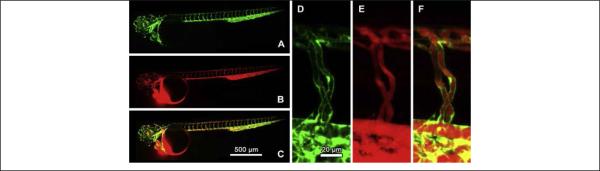
Zebrafish microangiography using 90-nm nano-sized droplets containing 8 wt% of DiI-TPB injected in Tg(fli1:eGFP)y1 embryons 3 days post fertilization. Images present the global view (A-C) and a zoom in of the trunk vasculature (D-F). Images in green (A and D) present endothelial cells expressing eGFP, while images in red present the fluorescence of DiI-TPB (590 nm), 30 min after injection of the nano-emulsion. The merged images (C and F) show no colocalization of DiI-TPB with the endothelial cells (reprinted with permission from ref. 43, Copyright 2014, Elsevier).
Particle size influences the in vivo trafficking of NPs so as a consequence, certain organs can be imaged. For instance, Kim and co-workers took advantage of the mechanism of lymphatic drainage and lymph node retention of NPs of a certain size to successfully image the sentinel lymph node with NIR semiconducting polymer NPs (termed as NIR-cvPDs, 60 nm in diameter). 54 When injected intradermally into the forepaw pad of a mouse, the fast movement of the fluorescence signal along lymphatic vessels was clearly detected (Fig. 11A). The results indicated that NIR-cvPDs drained rapidly from the interstitial site of injection into the lymphatics and accumulated at the regional lymph node without any flow toward the next tier nodes. Recently, fluorescent organic NPs with longer emission wavelength were developed for in vivo imaging. 78 In a typical work, NIR-II NPs (termed as IR-PEG) with emission wavelength longer than 1000 nm were used. 93 The whole-body imaging showed that, after intravenous injection, the IR-PEG NPs sequentially passed through lungs, kidneys and to other parts of the mouse with the direction of the blood flow (Fig. 11B1-3). Based on the time-dependent variation of the NIR-II fluorescence throughout the body, principal component analysis was applied to convert the time-dependent variation of the fluorescence intensity at various locations into spatially resolved components. 148 The principal component analysis results (pseudo color) vividly delineated the different organs inside the mouse, including the lungs, the kidneys, and the skin (Fig. 14B4). 93 Obviously, the NIR-II NPs enable in vivo imaging with deeper tissue penetration, higher spatial resolution, and better image fidelity than traditional fluorescence imaging in the <900 nm NIR region. To get deeper penetration, fluorescence-based multiple modality imaging may tackle this problem. For example, fluorescence properties can be incorporated into magnetic NPs, which can be used as magnetic resonance imaging (MRI) contrast agent because of their deep tissue penetration. 149
Figure 11.

(A) Pseudo-color NIR fluorescence images of a mouse injected intradermally with NIR-cvPDs into the right paw. Imaging time points after injection are shown. Arrows and arrowheads indicate axillary lymph node and lymphatic vessels, respectively (reprinted with permission from ref. 54, Copyright 2010, Royal Society of Chemistry). (B) 1-3) In vivo NIR-II imaging of a nude mouse with intravenously injected IR-PEG NPs. NIR-II fluorescence images of the entire body of the mouse were recorded at various times after injection of the IR-PEG NPs into the mouse tail vein. 4) Image with overlaid principal component analysis data, based on the first 200 consecutive images of video-rate NIR-II imaging (reprinted with permission from ref. 93, Copyright 2013, Wiley-VCH).
Figure 14.
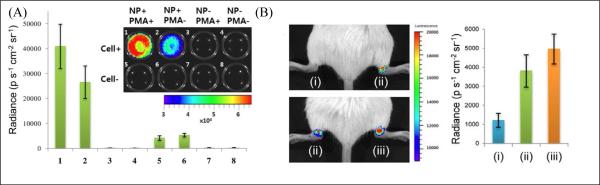
(A) In vivo imaging of PMA-stimulated H2O2 production in RAW264.7 macrophages labeled with CL NPs. Representative image (inset) and quantification of CL emission intensity from experimental groups 1-8 (n = 4) with or without CL NPs, cells, and phorbol myristate acetate (PMA) stimulation, as indicated in the inset. (B) In vivo CL imaging of the LPS-induced arthritis model with undoped (green luminescent) or Nile Red-doped (red luminescent) CL NPs. Spectrally unfiltered images (left) and quantification (right) of CL emission intensity from experimental groups (n = 4): (i) preinjected PBS + green CL NPs, (ii) preinjected LPS + green CL NPs, and (iii) preinjected LPS + red CL NPs (reprinted with permission from ref. 125, Copyright 2012, American Chemical Society).
3.2.2 Specific imaging
For specific imaging, the ligands described in Section 3.1.2 (specific in vitro imaging) are still valid for in vivo active targeting. The enhanced permeability and retention (EPR) effect is effective for fluorescent NPs to passively target tumors without ligands. Based on the EPR effect, fluorescent PEO-stabilized drug-loaded micelles were used to image drug delivery via the blood circulation in tumor-bearing mice. 114 The real-time fluorescent imaging revealed that the micelles accumulated at tumor site until they reach a maximum level at 24h post-injection; they then were slowly cleared from the tumor. Although EPR-based passive targeting is easy to carry out, active targeting exhibits better specificity with the established ligand-receptor interactions. Most of the specific in vivo imaging studies performed with targeting fluorescent NPs, such as NIR polymer vesicles modified with antibodies, 117 NIR polymer NPs conjugated with folate groups 77, 115 and peptide-coated NIR NPs, 121 give significantly better detection specificity and accuracy because of the selectivity between receptors and ligands.
In addition to ligands that bind specifically to the target, other ligands may also be needed to help traverse special biological barriers. For example, in vivo fluorescent imaging of brain is difficult at present, because of the presence of the blood-brain barrier and the complex dependence of the probe's penetration of the barrier on size and surface properties. Recently, several attempts have been made to specifically image brains with NP-ligand bioconjugates. Tosi et al. reported a type of PLGA NP engineered with both a NIR dye and a brain-specific peptide (simil-opioid glycopeptides) for brain targeting. 94 After intravenous injection of the NIR NPs into a mouse, in vivo time-domain optical imaging showed brain localization of the NIR NPs. Fluorescent and confocal microscopy studies on the brain tissues confirmed the penetration of the NIR NPs across blood-brain barrier and distribution within the cells of the cerebral tissue. Our group developed another kind of bioconjugate for in vivo brain tumor imaging called Pdot-CTX. The probes were based on 15-nm semiconducting polymer NPs (emitting at 650 nm) conjugated with a tumor-specific peptide ligand (Chlorotoxin). 35 The Pdot-CTX conjugates were designed to traverse the blood-brain barrier and specifically target brain tumors in a transgenic mouse model called ND2:SmoA1. The Pdot-CTX conjugates were injected into either ND2:SmoA1 or wild-type control mice. Fluorescence imaging showed that Pdot-CTX conjugates preferentially accumulated in the brain tumor regions of the ND2:SmoA1 mice (Figure 12A). Quantitative evaluation of NP accumulation and histological analysis further confirmed the selective accumulation of the Pdot-CTX conjugates in the malignant brain tumors (Figure 12B-C).
Figure 12.
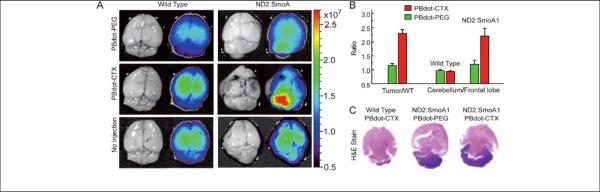
Pdot-CTX conjugates for in vivo tumor targeting. A) Fluorescence imaging of healthy brains in wild-type mice (left) and medulloblastoma tumors in ND2:SmoA1 mice (right). Each mouse received injection through the tail vein with control Pdot-PEG conjugates (top, the 15-nm semiconducting polymer NPs conjugated with PEG), targeting Pdot-CTX (middle), or no injection (bottom; control). B) Tumor-targeting efficiency by quantifying fluorescence signals in the cerebellum and frontal lobe regions of the brain of ND2:SmoA1 and wild-type mice. C) Histological examination of the mouse brains in A). The dark purple regions in the H&E-stained cerebellum of ND2:SmoA1 mice confirmed the presence of tumor (reprinted with permission from ref. 35, Copyright 2011, Wiley-VCH).
Bone has also been targeted for in vivo imaging with fluorescent NPs as well. In one study, FITC-doped PBLG NPs conjugated with alendronate, a targeting moiety that has a strong affinity for bone, were used to specifically target bone in rats. 95 The NPs showed bright fluorescence in femur tissue, indicating the specificity of the conjugates. In order to obtain better imaging quality, NIR crosslinked bisphosphonate NPs were exploited for bone targeting. 68 The bisphosphonate groups have high affinity for the bone mineral hydroxyapatite. In vivo imaging on a whole chicken embryo clearly indicated the affinity of the bisphosphonate NPs for the chicken embryo bones.
3.3 Imaging of biological processes
Cell division, growth, metabolism, apoptosis and many other cellular processes involve various biochemical and physical parameters, such as electrolytes, temperature, dissolved oxygen, and reactive species. Sensing and imaging of these factors of biological processes helps us to better understand cellular events and also develop novel diagnostic and therapeutic techniques. However, fluorescent soft NPs for imaging biological processes are sensitive to their surrounding environment, which affects their imaging stability.
Temperature
Some dyes are very sensitive to temperature, as represented by Rhodamine B (RhB) and europium (Eu) beta-diketonate chelate. They usually exhibit decreased fluorescence intensity/lifetime at higher temperature. Doping those temperature indictors into organic NPs can give them the ability to work as thermometers. One example is Eu-tris(dinaphthoylmethane)-bis-(trioctylphosphine oxide) doped PMMA NPs. 98 In the physiological temperatures range (25-45 °C), the fluorescence intensity of Eu-NPs decreased by −3.07 % per °C as the temperature increased, corresponding to a resolution of 0.3 °C. Our group also constructed RhB-doped semiconducting polymer NPs for ratiometric fluorescence sensing and temperature-based imaging. 38 The RhB-NPs showed a linear fluorescence response in the temperature range of 10-70 °C and efficient cellular uptake; the quantitative determinations of intracellular temperature based on cellular imaging were in good agreement with the results measured by a thermocouple.
Another type of fluorescent thermometer is the fluorophore-doped PNIPAm nanogel. 64, 65, 118 The gel usually exhibits enhanced fluorescence with a temperature increase. Seiichi and coworkers developed a fluorescent polymeric thermometer for intracellular temperature mapping, where the fluorescence lifetime of the fluorescent polymeric thermometer was adopted as a temperature-dependent variable. By using time-correlated single-photon counting system-based fluorescence lifetime imaging microcopy, high spatial (200 nm) and temperature resolution (0.18-0.58 °C) were achieved. Based on the fluorescent polymeric thermometer, the intracellular temperatures were successfully mapped to demonstrate that (i) the temperatures of nucleus and centrosome were both higher than the cytoplasm; (ii) the temperature gap between the nucleus and the cytoplasm differed depending on the cell cycle; (iii) the temperature near the mitochondria was higher than the rest of the space in the cytosol because of the heat generated by the organelle.
Dissolved oxygen
Fluorescent oxygen nanosensors can be similarly constructed by incorporating oxygen probes into polymer NPs. Based on oxygen-quenched phosphorescence, many oxygen nanosensors have been developed for cellular oxygen sensing and imaging, including Pt(II) octaethylporphine-doped semiconducting polymer NPs 37 and PS NPs, 99 Pd-tetra-(4-carboxyphenyl) tetrabenzoporphyrin dendrimer (G2)-doped NIR nanogels, 71 platinum(II) meso-tetrakis-(pentafluorophenyl)porphyrinato-doped PS NPs, 127 Pt(II)-meso-tetra(pentafluorophenyl)porphine (Pt-TFPP)-doped cationic hydrogel NPs150 and Ru-complex doped PS NPs. 151 Furthermore, assisted by the accurate fluorescence lifetime imaging microcopy technique, the intracellular oxygen distribution in neurospheres was mapped with PtTFPP-doped semiconducting polymer NPs. 152 When oxygen nanosensors with different targeting abilities were used, the pericellular, extracellular, intracellular and even intramitochondrial oxygen concentrations were detected and discriminated. 153, 154 Accurate determination of cellular oxygen concentrations can find applications in enzymatic assays, analysis of cell respiration, and potential cancer cell screening. For more detailed biological applications of oxygen sensing, please refer to a recent review. 155
In comparison to cellular oxygen imaging, in vivo imaging of oxygen based on fluorescence methods is rarely studied. Recently, Vinogradov and coworkers reported direct in vivo measurements of local oxygen tension (pO2) in the bone marrow of live mice by using of a macromolecular platinum porphyrin nanoprobe (PtP-C343). 156 By two-photon phosphorescence lifetime microscopy, the absolute pO2 of the bone marrow was determined to be quite low (<32 mm Hg) despite very high vascular density, and heterogeneities in local pO2 were detected. These pO2 values were found to change markedly after radiation and chemotherapy, pointing to the role of stress in altering the cell metabolic microenvironment.
pH
The fluorescent organic NPs for pH imaging usually are designed by conjugating pH-indicator dyes onto the surface of fluorescent NPs. Our group described a ratiometric methodology for pH imaging, in which the pH-sensitive FITC dye was covalently attached to fluorescent semiconducting polymer NPs. 39 The efficient energy transfer from the donor NPs to the acceptor FITC molecules enabled a rapid and robust sensing of pH in the range of 5-8. We used the probes quantitatively to determine the intracellular pH of HeLa cells. In another study, FITC dye molecules were conjugated onto oxygen-sensitive fluorescent NPs with a core shell to simultaneously measure the intracellular pH and oxygen in the cytosol. 157 The fluorescent NPs showed a large signal change between pH 5 and 8 (with a pKa value of 6.4). After being internalized into the cellular cytosol via electroporation, the intracellular pH values were sensed and imaged. Another type of a pH nanosensor was also developed by incorporating a pH indicator into a soft nanogel. 96 The pH nanogels were prepared from an inert, but biocompatible, polyurethane polymer, which was made pH-sensitive by loading it with the pH indicator bromothymol blue. The fluorescence was rendered ratiometric by the addition of two standard fluorophores that underwent efficient FRET inside the nanogel. The ratiometric fluorescent nano-sized gels were easily taken up by cells and capable of sensing intracellular pH values in the physiological range of 6-8 (with a pKa value of 7.64). In addition, polymeric vesicles were developed to image pH based on Nile Blue-labeled PMPC-PDPA diblock copolymers. The biomimetic PMPC block was responsible for cell uptake; the PDPA block was the pH-responsive component that enabled easy vesicle self-assembly in aqueous solution; and the Nile Blue dye was sensitive to pH. 116 These biocompatible vesicles were used to image pH gradients in a tumor model and to probe intracellular microenvironments by selective intracellular staining of lysosomes and early endosomes via subtle changes in fluorescence emission.
Caspase activity
The effector caspases (for example, caspase-3 and -7) are ideal targets for tumor apoptosis imaging because their activation commits the cell to programmed death. Taking advantage of a caspase-sensitive self-assembled fluorescent probe (C-SNAF), which was controlled by a first-order bioorthogonal cyclization reaction, the activity of caspase-3/7 during chemotherapy in mouse models xenografted with human tumors was successfully imaged in vivo. 124 Female nude mice bearing subcutaneous HeLa tumors received either intravenous chemotherapy of doxorubicin (DOX) or saline (Fig. 13). Then C-SNAF or either of the control probes (L-ctrl and D-ctrl) was administered intravenously to the tumor-bearing mice. In healthy nude mice, C-SNAF was cleared rapidly from the circulation with a blood half-life of ~1 hour, and completely eliminated from the body at 12 hours after administration. The whole body imaging clearly showed that C-SNAF was significantly brighter in DOX-treated than in saline-treated tumors, with the maximum fluorescence one hour after administration (Fig. 13A). In 4 hours, the fluorescence signal was increased by 1.6-fold in DOX-treated tumors compared to that in saline-treated mice. Neither L-ctrl nor D-ctrl showed negligible increases (Fig. 13B). The serial tumor response to chemotherapeutic intervention in the same mouse could also be imaged by C-SNAF (Fig. 13C). One dose of DOX (×1 DOX) resulted in a moderate increase in tumor fluorescence, and third round of DOX therapy gave a significant signal enhancement compared to both saline and ×1 DOX treatment. The results indicated that the caspase-3/7-triggered bioorthogonal macrocyclization and fluorescent nanoaggregation of C-SNAF are effective to monitor the tumor-therapy response in vivo.
Figure 13.

Non-invasive imaging of apoptosis in tumor-bearing mice treated with DOX. (A) Longitudinal fluorescence imaging with C-SNAF of ×3 DOX-treated (top) and saline-treated (bottom) tumor-bearing mice. Anatomical locations of the tumors and kidneys are indicated by white arrows. Whole-body fluorescence was monitored longitudinally using a Maestro fluorescence imager. (B) The percent difference in tumor fluorescence intensity between ×3 DOX and saline treatment groups over the course of imaging for C-SNAF (blue), L-ctrl (black) and D-ctrl (red) (C) A comparison of the average tumor fluorescence intensity at 2 h after C-SNAF administration in saline-treated mice, or after a ×1 DOX or ×3 DOX treatments in the same animals (Reprinted by permission from ref. 124, Copyright 2014, Nature Publishing Group).
H2O2
Hydrogen peroxide (H2O2) is an endogenous molecule that plays diverse physiological and pathological roles in living systems. Chemiluminescence (CL) can be used to sensitively image H2O2 because there isn't any autofluorescence from the background. In a recent study, CL NPs comprised of the green emitting AIE-active dye (BLSA) and the H2O2-responsive peroxalate (CPPO) were prepared for in vitro and in vivo imaging of endogenous H2O2. 125 CPPO can convert chemical reaction energy into electronic excitation. BLSA allows for high-density loading of dyes into the particle matrix without fluorescence quenching but it also enables efficient intraparticle CL energy transfer to a low-energy dopant (e.g. Nile Red) to tune the spectrum toward the longer-wavelength window that is more biologically transparent. Based on the CL NPs, endogenous H2O2 produced during an immune response was imaged in macrophages (strong signal) and in untreated control cells (weak signal) (Figure 14A). In vivo visualization of H2O2 associated with early stage inflammation as seen in arthritis was performed on mouse ankle joints by using Nile Red-doped or undoped CL NPs (Figure 14B). H2O2 was only detected from the lipopolysaccharide (LPS)-treated inflamed joints with the two CL NPs. Importantly, the Nile Red-doped CL NPs exhibited improved visualization of H2O2 than the undoped NPs, indicating the deeper penetration depth of long-wavelength emission.
4. Conclusion and outlook
We have summarized recent progress in the development of soft fluorescent NPs for biological and biomedical imaging. In comparison to solid fluorescent NPs, they are more competent for biological and biomedical imaging. Various fluorescent organic NPs have been developed as imaging agents, including dye-doped polymer NPs, semiconducting polymer NPs, small-molecule organic NPs, nanogels, polymeric micelles and vesicles. These organic NPs can be made by direct polymerization, post-polymerization (miniemulsion and reprecipitation) processes or self-assembly methods. The methods take into account the nature of starting materials, which determines the interaction forces between molecules and the stability of particles, and the aggregation states of fluorophores inside the NPs, which influence the fluorescence.
The soft fluorescent NPs exhibit diverse optical properties and imaging capabilities. Comparatively, fluorescent semiconducting polymer NPs and AIE-type small-molecule organic NPs are known for ultra-high brightness, which is desirable for high-contrast imaging. Dye-doped polymeric NPs may have the weak point of low brightness, but a large library of dyes is available for constructing dye-doped NPs with versatile imaging capabilities. Moreover, dye-doped polymeric NPs based on biodegradable molecules (or biomaterials) are suitable for in vivo imaging because of their negligible toxicity. For nanogels, polymer micelles and vesicles, the loose nanostructures enable the bio-analytes/stimuli to affect particle matrix and/or inner fluorophores, and cellular processes can be imaged accordingly. These nano-sized imaging agents have been widely demonstrated for in vitro imaging; however, there is still much room for improvement, such as tailored particle size and surface chemistry to specifically target and image organelles. Unlike cell-surface labeling, which generally rely on specific ligands or antibodies conjugated to soft fluorescent NPs, organelle labeling places more stringent requirements on the imaging agents - besides ligands, surface charge and particle size are also critical in controlling their destination within the cell.
With regard to in vivo imaging, soft fluorescent NPs based on long-wavelength fluorophores are commonly adopted. Unfortunately, fluorescence imaging is limited to small animals or tissues not buried deep in the body, even though NIR-II organic NPs have been developed to increase the penetration depth. 93 In order to tackle this shortcoming, one possible strategy is to develop multimodality nanoprobes in which other contrast agents (with deeper penetration) are combined with the fluorescent NPs. For example, 64Cu-DOTA (64Cu(II)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) can be adopted to construct PET (Positron emission tomography)/optical dual-modality imaging agents, and Gd-DTPA (Gd(III)-diethylenetriaminepentacetate) MRI/optical agents. 158 Alternatively, the nano-sized soft fluorescent imaging agents may find more practical applications in surgical operation, where deep penetration is no longer necessary as the body covering would be opened.
Acknowledgement
We are grateful to the National Institutes of Health (R21CA186798 and R01CA175215) for support of this work.
References
- 1.Hell SW. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 2.Heintzmann R, Gustafsson MGL. Nat Photonics. 2009;3:362–364. [Google Scholar]
- 3.Huang B, Bates M, Zhuang XW. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunstein M, Wicker K, Herault K, Heintzmann R, Oheim M. Opt Express. 2013;21:26162–26173. doi: 10.1364/OE.21.026162. [DOI] [PubMed] [Google Scholar]
- 5.Kievit FM, Zhang MQ. Adv Mater. 2011;23:H217–H247. doi: 10.1002/adma.201102313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nalwa HS, editor. Soft nanomaterials. American Scientific Publishers; 2009. [Google Scholar]
- 7.Roy M, Niu CJ, Chen YH, McVeigh PZ, Shuhendler AJ, Leung MK, Mariampillai A, DaCosta RS, Wilson BC. Small. 2012;8:1780–1792. doi: 10.1002/smll.201102105. [DOI] [PubMed] [Google Scholar]
- 8.So MK, Xu CJ, Loening AM, Gambhir SS, Rao JH. Nat Biotechnol. 2006;24:339–343. doi: 10.1038/nbt1188. [DOI] [PubMed] [Google Scholar]
- 9.Warnement MR, Tomlinson ID, Rosenthal SJ. Curr Nanosci. 2007;3:273–284. [Google Scholar]
- 10.He XX, Wang YS, Wang KM, Chen M, Chen SY. Anal Chem. 2012;84:9056–9064. doi: 10.1021/ac301461s. [DOI] [PubMed] [Google Scholar]
- 11.Wu XM, Chang S, Sun XR, Guo ZQ, Li YS, Tang JB, Shen YQ, Shi JL, Tian H, Zhu WH. Chem Sci. 2013;4:1221–1228. [Google Scholar]
- 12.Hemmer E, Venkatachalam N, Hyodo H, Hattori A, Ebina Y, Kishimoto H, Soga K. Nanoscale. 2013;5:11339–11361. doi: 10.1039/c3nr02286b. [DOI] [PubMed] [Google Scholar]
- 13.Li JL, Tang B, Yuan B, Sun L, Wang XG. Biomaterials. 2013;34:9519–9534. doi: 10.1016/j.biomaterials.2013.08.066. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Guo BD, Rao ZY, Zhang BH, Gong JR. Nano Lett. 2013;13:2436–2441. doi: 10.1021/nl400368v. [DOI] [PubMed] [Google Scholar]
- 15.Sharma J, Yeh HC, Yoo H, Werner JH, Martinez JS. Chem Commun. 2011;47:2294–2296. doi: 10.1039/c0cc03711g. [DOI] [PubMed] [Google Scholar]
- 16.Yu JH, Choi SM, Richards CI, Antoku Y, Dickson RM. Photochem Photobiol. 2008;84:1435–1439. doi: 10.1111/j.1751-1097.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tantra R, Knight A. Nanotoxicology. 2011;5:381–392. doi: 10.3109/17435390.2010.512987. [DOI] [PubMed] [Google Scholar]
- 18.Swierczewska M, Lee S, Chen XY. Mol Imaging. 2011;10:3–16. [PMC free article] [PubMed] [Google Scholar]
- 19.Cho EC, Glaus C, Chen JY, Welch MJ, Xia YN. Trends Mol Med. 2010;16:561–573. doi: 10.1016/j.molmed.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng L. T. a. J. Nano Today. 2013;8:290–312. doi: 10.1016/j.nantod.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benya R QJ, Brundage B. Cathet Cardiovasc Diagn. 1989;17:231–233. doi: 10.1002/ccd.1810170410. [DOI] [PubMed] [Google Scholar]
- 22.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Annu Rev Chem Biomol. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maldonado CR, Salassa L, Gomez-Blanco N, Mareque-Rivas JC. Coordin Chem Rev. 2013;257:2668–2688. [Google Scholar]
- 24.Vollrath A, Schubert S, Schubert US. J Mater Chem B. 2013;1:1994–2007. doi: 10.1039/c3tb20089b. [DOI] [PubMed] [Google Scholar]
- 25.Wu CF, Chiu DT. Angew Chem Int Edit. 2013;52:3086–3109. doi: 10.1002/anie.201205133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng LH, Zhu CL, Yuan HX, Liu LB, Lv FT, Wang S. Chem Soc Rev. 2013;42:6620–6633. doi: 10.1039/c3cs60036j. [DOI] [PubMed] [Google Scholar]
- 27.Hong YN, Lam JWY, Tang BZ. Chem Soc Rev. 2011;40:5361–5388. doi: 10.1039/c1cs15113d. [DOI] [PubMed] [Google Scholar]
- 28.Li K, Liu B. Chem Soc Rev. 2014;43:6570–6597. doi: 10.1039/c4cs00014e. [DOI] [PubMed] [Google Scholar]
- 29.Gong J, Shen QM, Fan QL, Huang W. Prog Chem. 2013;25:1928–1941. [Google Scholar]
- 30.Pecher J, Mecking S. Chem Rev. 2010;110:6260–6279. doi: 10.1021/cr100132y. [DOI] [PubMed] [Google Scholar]
- 31.Ong BS, Wu YL, Liu P, Gardner S. Adv Mater. 2005;17:1141–+. [Google Scholar]
- 32.Wang F, Han MY, Mya KY, Wang YB, Lai YH. J Am Chem Soc. 2005;127:10350–10355. doi: 10.1021/ja0521730. [DOI] [PubMed] [Google Scholar]
- 33.Szymanski C, Wu CF, Hooper J, Salazar MA, Perdomo A, Dukes A, McNeill J. J Phys Chem B. 2005;109:8543–8546. doi: 10.1021/jp051062k. [DOI] [PubMed] [Google Scholar]
- 34.Wu C, Bull B, Szymanski C, Christensen K, McNeill J. Acs Nano. 2008;2:2415–2423. doi: 10.1021/nn800590n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu CF, Hansen SJ, Hou QO, Yu JB, Zeigler M, Jin YH, Burnham DR, McNeill JD, Olson JM, Chiu DT. Angew Chem Int Edit. 2011;50:3430–3434. doi: 10.1002/anie.201007461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birks JB. Photophysics of Aromatic Molecules. Wiley; London: 1970. [Google Scholar]
- 37.Wu CF, Bull B, Christensen K, McNeill J. Angew Chem Int Edit. 2009;48:2741–2745. doi: 10.1002/anie.200805894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye FM, Wu CF, Jin YH, Chan YH, Zhang XJ, Chiu DT. J Am Chem Soc. 2011;133:8146–8149. doi: 10.1021/ja202945g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan YH, Wu CF, Ye FM, Jin YH, Smith PB, Chiu DT. Anal Chem. 2011;83:1448–1455. doi: 10.1021/ac103140x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin W, Ding D, Liu JZ, Yuan WZ, Hu Y, Liu B, Tang BZ. Adv Funct Mater. 2012;22:771–779. [Google Scholar]
- 41.Ding D, Goh CC, Feng GX, Zhao ZJ, Liu J, Liu RR, Tomczak N, Geng JL, Tang BZ, Ng LG, Liu B. Adv Mater. 2013;25:6083–6088. doi: 10.1002/adma.201301938. [DOI] [PubMed] [Google Scholar]
- 42.Goutayer M, Dufort S, Josserand V, Royere A, Heinrich E, Vinet F, Bibette J, Coll JL, Texier I. Eur J Pharm Biopharm. 2010;75:137–147. doi: 10.1016/j.ejpb.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 43.Kilin VN, Anton H, Anton N, Steed E, Vermot J, Vandamme TE, Mely Y, Klymchenko AS. Biomaterials. 2014;35:4950–4957. doi: 10.1016/j.biomaterials.2014.02.053. [DOI] [PubMed] [Google Scholar]
- 44.Albanese A, Tang PS, Chan WCW. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 45.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharmaceut. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang JG, Leshuk T, Gu FX. Nano Today. 2011;6:478–492. [Google Scholar]
- 47.Garnett MC, Kallinteri P. Occup Med-Oxford. 2006;56:307–311. doi: 10.1093/occmed/kql052. [DOI] [PubMed] [Google Scholar]
- 48.Cu Y, Saltzman WM. Nat Mater. 2009;8:11–13. doi: 10.1038/nmat2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain RK, Stylianopoulos T. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schadlich A, Caysa H, Mueller T, Tenambergen F, Rose C, Gopferich A, Kuntsche J, Mader K. Acs Nano. 2011;5:8710–8720. doi: 10.1021/nn2026353. [DOI] [PubMed] [Google Scholar]
- 51.Wang M, Thanou M. Pharmacol Res. 2010;62:90–99. doi: 10.1016/j.phrs.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Tuncel D, Demir HV. Nanoscale. 2010;2:484–494. doi: 10.1039/b9nr00374f. [DOI] [PubMed] [Google Scholar]
- 53.Pecher J, Huber J, Winterhalder M, Zumbusch A, Mecking S. Biomacromolecules. 2010;11:2776–2780. doi: 10.1021/bm100854a. [DOI] [PubMed] [Google Scholar]
- 54.Kim S, Lim CK, Na J, Lee YD, Kim K, Choi K, Leary JF, Kwon IC. Chem Commun. 2010;46:1617–1619. doi: 10.1039/b923309a. [DOI] [PubMed] [Google Scholar]
- 55.Lee KJ, Oh WK, Song J, Kim S, Lee J, Jang J. Chem Commun. 2010;46:5229–5231. doi: 10.1039/c002533j. [DOI] [PubMed] [Google Scholar]
- 56.Cova L, Bigini P, Diana V, Sitia L, Ferrari R, Pesce RM, Khalaf R, Bossolasco P, Ubezio P, Lupi M, Tortarolo M, Colombo L, Giardino D, Silani V, Morbidelli M, Salmona M, Moscatelli D. Nanotechnology. 2013;24 doi: 10.1088/0957-4484/24/24/245603. [DOI] [PubMed] [Google Scholar]
- 57.Ferrari R, Lupi M, Falcetta F, Bigini P, Paolella K, Fiordaliso F, Bisighini C, Salmona M, D'Incalci M, Morbidelli M, Moscatelli D, Ubezio P. Nanotechnology. 2014;25 doi: 10.1088/0957-4484/25/4/045102. [DOI] [PubMed] [Google Scholar]
- 58.Thielbeer F, Chankeshwara SV, Bradley M. Biomacromolecules. 2011;12:4386–4391. doi: 10.1021/bm201394k. [DOI] [PubMed] [Google Scholar]
- 59.Zupke O, Distler E, Baumann D, Strand D, Meyer RG, Landfester K, Herr W, Mailander V. Biomaterials. 2010;31:7086–7095. doi: 10.1016/j.biomaterials.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 60.Averick SE, Magenau AJD, Simakova A, Woodman BF, Seong A, Mehl RA, Matyjaszewski K. Polym Chem-Uk. 2011;2:1476–1478. [Google Scholar]
- 61.Fu GD, Jiang H, Yao F, Xu LQ, Ling J, Kang ET. Macromol Rapid Comm. 2012;33:1523–1527. doi: 10.1002/marc.201200337. [DOI] [PubMed] [Google Scholar]
- 62.Goncalves M, Maciel D, Capelo D, Xiao SL, Sun WJ, Shi XY, Rodrigues J, Tomas H, Li YL. Biomacromolecules. 2014;15:492–499. doi: 10.1021/bm401400r. [DOI] [PubMed] [Google Scholar]
- 63.Sun GR, Berezin MY, Fan JD, Lee H, Ma J, Zhang K, Wooley KL, Achilefu S. Nanoscale. 2010;2:548–558. doi: 10.1039/b9nr00304e. [DOI] [PubMed] [Google Scholar]
- 64.Chen CY, Chen CT. Chem Commun. 2011;47:994–996. doi: 10.1039/c0cc04450d. [DOI] [PubMed] [Google Scholar]
- 65.Okabe K, Inada N, Gota C, Harada Y, Funatsu T, Uchiyama S. Nat Commun. 2012;3 doi: 10.1038/ncomms1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou SY, Dou HJ, Zhang ZF, Sun K, Jin YQ, Dai TT, Zhou GD, Shen Z. Polym Chem-Uk. 2013;4:4103–4112. [Google Scholar]
- 67.Wang SY, Kim G, Lee YEK, Hah HJ, Ethirajan M, Pandey RK, Kopelman R. Acs Nano. 2012;6:6843–6851. doi: 10.1021/nn301633m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gluz E, Grinberg I, Corem-Salkmon E, Mizrahi D, Margel S. J Polym Sci Pol Chem. 2013;51:4282–4291. [Google Scholar]
- 69.Zhang XY, Wang SQ, Xu LX, Feng L, Ji Y, Tao L, Li SX, Wei Y. Nanoscale. 2012;4:5581–5584. doi: 10.1039/c2nr31281f. [DOI] [PubMed] [Google Scholar]
- 70.Chen Y, Wilbon PA, Zhou JH, Nagarkatti M, Wang CP, Chu FX, Tang CB. Chem Commun. 2013;49:297–299. doi: 10.1039/c2cc37386f. [DOI] [PubMed] [Google Scholar]
- 71.Lee YEK, Ulbrich EE, Kim G, Hah H, Strollo C, Fan WZ, Gurjar R, Koo SM, Kopelman R. Anal Chem. 2010;82:8446–8455. doi: 10.1021/ac1015358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Landfester K, Montenegro R, Scherf U, Guntner R, Asawapirom U, Patil S, Neher D, Kietzke T. Adv Mater. 2002;14:651–655. [Google Scholar]
- 73.Kietzke T, Neher D, Landfester K, Montenegro R, Guntner R, Scherf U. Nat Mater. 2003;2:408–U407. doi: 10.1038/nmat889. [DOI] [PubMed] [Google Scholar]
- 74.Howes P, Thorogate R, Green M, Jickells S, Daniel B. Chem Commun. 2009:2490–2492. doi: 10.1039/b903405f. [DOI] [PubMed] [Google Scholar]
- 75.Howes P, Green M, Levitt J, Suhling K, Hughes M. J Am Chem Soc. 2010;132:3989–3996. doi: 10.1021/ja1002179. [DOI] [PubMed] [Google Scholar]
- 76.Li K, Liu YT, Pu KY, Feng SS, Zhan RY, Liu B. Adv Funct Mater. 2011;21:287–294. [Google Scholar]
- 77.Ding D, Liu J, Feng GX, Li K, Hu Y, Liu B. Small. 2013;9:3093–3102. doi: 10.1002/smll.201300171. [DOI] [PubMed] [Google Scholar]
- 78.Reul R, Tsapis N, Hillaireau H, Sancey L, Mura S, Recher M, Nicolas J, Coll JL, Fattal E. Polym Chem-Uk. 2012;3:694–702. [Google Scholar]
- 79.Mackiewicz N, Nicolas J, Handke N, Noiray M, Mougin J, Daveu C, Lakkireddy HR, Bazile D, Couvreur P. Chem Mater. 2014;26:1834–1847. [Google Scholar]
- 80.Fryd MM, Mason TG. Annu Rev Phys Chem. 2012;63:493–518. doi: 10.1146/annurev-physchem-032210-103436. [DOI] [PubMed] [Google Scholar]
- 81.Kurokawa N, Yoshikawa H, Hirota N, Hyodo K, Masuhara H. Chemphyschem. 2004;5:1609–1615. doi: 10.1002/cphc.200400117. [DOI] [PubMed] [Google Scholar]
- 82.Wu CF, Szymanski C, McNeill J. Langmuir. 2006;22:2956–2960. doi: 10.1021/la060188l. [DOI] [PubMed] [Google Scholar]
- 83.Ma CP, Ling QQ, Xu SD, Zhu HN, Zhang G, Zhou X, Chi ZG, Liu SW, Zhang Y, Xu JR. Macromol Biosci. 2014;14:235–243. doi: 10.1002/mabi.201300259. [DOI] [PubMed] [Google Scholar]
- 84.Wu CF, Schneider T, Zeigler M, Yu JB, Schiro PG, Burnham DR, McNeill JD, Chiu DT. J Am Chem Soc. 2010;132:15410–15417. doi: 10.1021/ja107196s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu CF, Jin YH, Schneider T, Burnham DR, Smith PB, Chiu DT. Angew Chem Int Edit. 2010;49:9436–9440. doi: 10.1002/anie.201004260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rong Y, Wu CF, Yu JB, Zhang XJ, Ye FM, Zeigler M, Gallina ME, Wu IC, Zhang Y, Chan YH, Sun W, Uvdal K, Chiu DT. Acs Nano. 2013;7:376–384. doi: 10.1021/nn304376z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Yu JB, Gallina ME, Sun W, Rong Y, Chiu DT. Chem Commun. 2013;49:8256–8258. doi: 10.1039/c3cc44048f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chan YH, Gallina ME, Zhang XJ, Wu IC, Jin YH, Sun W, Chiu DT. Anal Chem. 2012;84:9431–9438. doi: 10.1021/ac302245t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu JB, Wu CF, Zhang XJ, Ye FM, Gallina ME, Rong Y, Wu IC, Sun W, Chan YH, Chiu DT. Adv Mater. 2012;24:3498–3504. doi: 10.1002/adma.201201245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davis CM, Childress ES, Harbron EJ. J Phys Chem C. 2011;115:19065–19073. [Google Scholar]
- 91.Jin YH, Ye FM, Zeigler M, Wu CF, Chiu DT. Acs Nano. 2011;5:1468–1475. doi: 10.1021/nn103304m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun W, Yu JB, Deng RP, Rong Y, Fujimoto B, Wu CF, Zhang HJ, Chiu DT. Angew Chem Int Edit. 2013;52:11294–11297. doi: 10.1002/anie.201304822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tao ZM, Hong GS, Shinji C, Chen CX, Diao S, Antaris AL, Zhang B, Zou YP, Dai HJ. Angew Chem Int Edit. 2013;52:13002–13006. doi: 10.1002/anie.201307346. [DOI] [PubMed] [Google Scholar]
- 94.Tosi G, Bondioli L, Ruozi B, Badiali L, Severini GM, Biffi S, De Vita A, Bortot B, Dolcetta D, Forni F, Vandelli MA. J Neural Transm. 2011;118:145–153. doi: 10.1007/s00702-010-0497-1. [DOI] [PubMed] [Google Scholar]
- 95.Ozcan I, Bouchemal K, Segura-Sanchez F, Ozer O, Guneri T, Ponchel G. J Pharm Sci-Us. 2011;100:4877–4887. doi: 10.1002/jps.22678. [DOI] [PubMed] [Google Scholar]
- 96.Peng HS, Stolwijk JA, Sun LN, Wegener J, Wolfbeis OS. Angew Chem Int Edit. 2010;49:4246–4249. doi: 10.1002/anie.200906926. [DOI] [PubMed] [Google Scholar]
- 97.An FF, Ye J, Zhang JF, Yang YL, Zheng CJ, Zhang XJ, Liu Z, Lee CS, Zhang XH. J Mater Chem B. 2013;1:3144–3151. doi: 10.1039/c3tb20271b. [DOI] [PubMed] [Google Scholar]
- 98.Peng HS, Stich MIJ, Yu JB, Sun LN, Fischer LH, Wolfbeis OS. Adv Mater. 2010;22:716–+. doi: 10.1002/adma.200901614. [DOI] [PubMed] [Google Scholar]
- 99.Wang XH, Peng HS, Ding H, You FT, Huang SH, Teng F, Dong B, Song HW. J Mater Chem. 2012;22:16066–16071. [Google Scholar]
- 100.Wang XH, Peng HS, Yang L, You FT, Teng F, Tang AW, Zhang FJ, Li XH. J Mater Chem B. 2013;1:5143–5152. doi: 10.1039/c3tb20884b. [DOI] [PubMed] [Google Scholar]
- 101.Zhang XJ, Yu JB, Wu CF, Jin YH, Rang Y, Ye FM, Chiu DT. Acs Nano. 2012;6:5429–5439. doi: 10.1021/nn301308w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu PJ, Kuo SY, Huang YC, Chen CP, Chan YH. Anal Chem. 2014;86:4831–4839. doi: 10.1021/ac404237q. [DOI] [PubMed] [Google Scholar]
- 103.Wang Z, Zhang XX, Huang P, Zhao WD, Liu DB, Nie LM, Yue XY, Wang SJ, Ma Y, Kiesewetter D, Niu G, Chen XY. Biomaterials. 2013;34:6194–6201. doi: 10.1016/j.biomaterials.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feng XL, Lv FT, Liu LB, Tang HW, Xing CF, Yang QO, Wang S. Acs Appl Mater Inter. 2010;2:2429–2435. doi: 10.1021/am100435k. [DOI] [PubMed] [Google Scholar]
- 105.Petkau K, Kaeser A, Fischer I, Brunsveld L, Schenning APHJ. J Am Chem Soc. 2011;133:17063–17071. doi: 10.1021/ja2075345. [DOI] [PubMed] [Google Scholar]
- 106.Fischer I, Petkau-Milroy K, Dorland YL, Schenning APHJ, Brunsveld L. Chem-Eur J. 2013;19:16646–16650. doi: 10.1002/chem.201302647. [DOI] [PubMed] [Google Scholar]
- 107.Wang G, Pu KY, Zhang XH, Li K, Wang L, Cai LP, Ding D, Lai YH, Liu B. Chem Mater. 2011;23:4428–4434. [Google Scholar]
- 108.Montalti M, Battistelli G, Cantelli A, Genovese D. Chem Commun. 2014;50:5326–5329. doi: 10.1039/c3cc48464e. [DOI] [PubMed] [Google Scholar]
- 109.Zhu LL, Li X, Zhang Q, Ma X, Li MH, Zhang HC, Luo Z, Agren H, Zhao YL. J Am Chem Soc. 2013;135:5175–5182. doi: 10.1021/ja400456h. [DOI] [PubMed] [Google Scholar]
- 110.Miyata K, Christie RJ, Kataoka K. React Funct Polym. 2011;71:227–234. [Google Scholar]
- 111.Chen JI, Wu WC. Macromol Biosci. 2013;13:623–632. doi: 10.1002/mabi.201200396. [DOI] [PubMed] [Google Scholar]
- 112.Zhang Y, Chen YJ, Li X, Zhang JB, Chen JL, Xu B, Fu XQ, Tian WJ. Polym Chem-Uk. 2014;5:3824–3830. [Google Scholar]
- 113.Wang Y, Hong CY, Pan CY. Biomacromolecules. 2012;13:2585–2593. doi: 10.1021/bm3008346. [DOI] [PubMed] [Google Scholar]
- 114.Zhang LZ, Lin Y, Zhang YJ, Chen R, Zhu ZS, Wu W, Jiang XQ. Macromol Biosci. 2012;12:83–92. doi: 10.1002/mabi.201100197. [DOI] [PubMed] [Google Scholar]
- 115.Miki K, Oride K, Inoue S, Kuramochi Y, Nayak RR, Matsuoka H, Harada H, Hiraoka M, Ohe K. Biomaterials. 2010;31:934–942. doi: 10.1016/j.biomaterials.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 116.Madsen J, Canton I, Warren NJ, Themistou E, Blanazs A, Ustbas B, Tian XH, Pearson R, Battaglia G, Lewis AL, Armes SP. J Am Chem Soc. 2013;135:14863–14870. doi: 10.1021/ja407380t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li ZH, Wu LY, Hu PR, Han SH, Zhang T, Fan HL, Jin W, Jin QH, Mu Y. Nanoscale. 2012;4:7097–7105. doi: 10.1039/c2nr32047a. [DOI] [PubMed] [Google Scholar]
- 118.Qiao J, Qi L, Shen Y, Zhao LZ, Qi C, Shangguan DH, Mao LQ, Chen Y. J Mater Chem. 2012;22:11543–11549. [Google Scholar]
- 119.Park HS, Lee JE, Cho MY, Hong JH, Cho SH, Lim YT. Macromol Rapid Comm. 2012;33:1549–1555. doi: 10.1002/marc.201200246. [DOI] [PubMed] [Google Scholar]
- 120.Buckle T, van Leeuwen AC, Chin PTK, Janssen H, Muller SH, Jonkers J, van Leeuwen FWB. Nanotechnology. 2010;21 doi: 10.1088/0957-4484/21/35/355101. [DOI] [PubMed] [Google Scholar]
- 121.Bunschoten A, Buckle T, Kuil J, Luker GD, Luker KE, Nieweg OE, van Leeuwen FWB. Biomaterials. 2012;33:867–875. doi: 10.1016/j.biomaterials.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Feng XL, Yang GM, Liu LB, Lv FT, Yang Q, Wang S, Zhu DB. Adv Mater. 2012;24:637–+. doi: 10.1002/adma.201102026. [DOI] [PubMed] [Google Scholar]
- 123.Yoo L, Park JS, Kwon KC, Kim SE, Jin X, Kim H, Lee J. Biomaterials. 2012;33:6194–6200. doi: 10.1016/j.biomaterials.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 124.Ye DJ, Shuhendler AJ, Cui LN, Tong L, Tee SS, Tikhomirov G, Felsher DW, Rao JH. Nat Chem. 2014;6:519–526. doi: 10.1038/nchem.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee YD, Lim CK, Singh A, Koh J, Kim J, Kwon IC, Kim S. Acs Nano. 2012;6:6759–6766. doi: 10.1021/nn3014905. [DOI] [PubMed] [Google Scholar]
- 126.Zhang XY, Zhang XQ, Yang B, Zhang YL, Liu MY, Liu WY, Chen YW, Wei Y. Colloid Surface B. 2014;113:435–441. doi: 10.1016/j.colsurfb.2013.09.031. [DOI] [PubMed] [Google Scholar]
- 127.Wang XD, Gorris HH, Stolwijk JA, Meier RJ, Groegel DBM, Wegener J, Wolfbeis OS. Chem Sci. 2011;2:901–906. [Google Scholar]
- 128.Hoffmann K, Behnke T, Drescher D, Kneipp J, Resch-Genger U. Acs Nano. 2013;7:6674–6684. doi: 10.1021/nn4029458. [DOI] [PubMed] [Google Scholar]
- 129.Kim Y, Fay F, Cormode DP, Sanchez-Gaytan BL, Tang J, Hennessy EJ, Ma MM, Moore K, Farokhzad OC, Fisher EA, Mulder WJM, Langer R, Fayad ZA. Acs Nano. 2013;7:9975–9983. doi: 10.1021/nn4039063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Behnke T, Wurth C, Laux EM, Hoffmann K, Resch-Genger U. Dyes Pigments. 2012;94:247–257. [Google Scholar]
- 131.Laux EM, Behnke T, Hoffmann K, Resch-Genger U. Anal Methods-Uk. 2012;4:1759–1768. [Google Scholar]
- 132.Behnke T, Mathejczyk JE, Brehm R, Wurth C, Gomes FR, Dullin C, Napp J, Alves F, Resch-Genger U. Biomaterials. 2013;34:160–170. doi: 10.1016/j.biomaterials.2012.09.028. [DOI] [PubMed] [Google Scholar]
- 133.Jahn A, Stavis SM, Hong JS, Vreeland WN, Devoe DL, Gaitan M. Acs Nano. 2010;4:2077–2087. doi: 10.1021/nn901676x. [DOI] [PubMed] [Google Scholar]
- 134.Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC. Nano Lett. 2008;8:2906–2912. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 135.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. P Natl Acad Sci USA. 2010;107:17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim YT, Chung BL, Ma MM, Mulder WJM, Fayad ZA, Farokhzad OC, Langer R. Nano Lett. 2012;12:3587–3591. doi: 10.1021/nl301253v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fernando LP, Kandel PK, Yu JB, McNeill J, Ackroyd PC, Christensen KA. Biomacromolecules. 2010;11:2675–2682. doi: 10.1021/bm1007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ye FM, Wu CF, Jin YH, Wang M, Chan YH, Yu JB, Sun W, Hayden S, Chiu DT. Chem Commun. 2012;48:1778–1780. doi: 10.1039/c2cc16486h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu CC, Chen T, Han D, You MX, Peng L, Cansiz S, Zhu GZ, Li CM, Xiong XL, Jimenez E, Yang CJ, Tan WH. Acs Nano. 2013;7:5724–5731. doi: 10.1021/nn402517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Edit. 2001;40:2004–+. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 141.Li JT, Wang L, Benicewicz BC. Langmuir. 2013;29:11547–11553. doi: 10.1021/la401990d. [DOI] [PubMed] [Google Scholar]
- 142.Demay-Drouhard P, Nehlig E, Hardouin J, Motte L, Guenin E. Chem-Eur J. 2013;19:8388–8392. doi: 10.1002/chem.201300903. [DOI] [PubMed] [Google Scholar]
- 143.Amici J, Kahveci MU, Allia P, Tiberto P, Yagci Y, Sangermano M. J Mater Sci. 2012;47:412–419. [Google Scholar]
- 144.Mahtab F, Lam JWY, Yu Y, Liu JZ, Yuan WZ, Lu P, Tang BZ. Small. 2011;7:1448–1455. doi: 10.1002/smll.201002195. [DOI] [PubMed] [Google Scholar]
- 145.Hayashi K, Ono K, Suzuki H, Sawada M, Moriya M, Sakamoto W, Yogo T. Chem Mater. 2010;22:3768–3772. doi: 10.1021/am100237p. [DOI] [PubMed] [Google Scholar]
- 146.Pansare VJ, Hejazi S, Faenza WJ, Prud'homme RK. Chem Mater. 2012;24:812–827. doi: 10.1021/cm2028367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hong GS, Lee JC, Robinson JT, Raaz U, Xie LM, Huang NF, Cooke JP, Dai HJ. Nat Med. 2012;18:1841–+. doi: 10.1038/nm.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Welsher K, Sherlock SP, Dai HJ. P Natl Acad Sci USA. 2011;108:8943–8948. doi: 10.1073/pnas.1014501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mahmoudi M, Hosseinkhani H, Hosseinkhani M, Boutry S, Simchi A, Journeay WS, Subramani K, Laurent S. Chem Rev. 2011;111:253–280. doi: 10.1021/cr1001832. [DOI] [PubMed] [Google Scholar]
- 150.Fercher A, Borisov SM, Zhdanov AV, Klimant I, Papkovsky DB. Acs Nano. 2011;5:5499–5508. doi: 10.1021/nn200807g. [DOI] [PubMed] [Google Scholar]
- 151.Wang XD, Achatz DE, Hupf C, Sperber M, Wegener J, Bange S, Lupton JM, Wolfbeis OS. Sensor Actuat B-Chem. 2013;188:257–262. [Google Scholar]
- 152.Kondrashina AV, Dmitriev RI, Borisov SM, Klimant I, O'Brien I, Nolan YM, Zhdanov AV, Papkovsky DB. Adv Funct Mater. 2012;22:4931–4939. [Google Scholar]
- 153.Dmitriev RI, Zhdanov AV, Jasionek G, Papkovsky DB. Anal Chem. 2012;84:2930–2938. doi: 10.1021/ac3000144. [DOI] [PubMed] [Google Scholar]
- 154.Wang XH, Peng HS, Yang L, You FT, Teng F, Hou LL, Wolfbeis OS. Angew Chem Int Edit. 2014;53:12471–12475. doi: 10.1002/anie.201405048. [DOI] [PubMed] [Google Scholar]
- 155.Papkovsky DB, Dmitriev RI. Chem Soc Rev. 2013;42:8700–8732. doi: 10.1039/c3cs60131e. [DOI] [PubMed] [Google Scholar]
- 156.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu JW, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, Yusuf R, Cote D, Vinogradov SA, Scadden DT, Lin CP. Nature. 2014;508:269–+. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang XD, Stolwijk JA, Lang T, Sperber M, Meier RJ, Wegener J, Wolfbeis OS. J Am Chem Soc. 2012;134:17011–17014. doi: 10.1021/ja308830e. [DOI] [PubMed] [Google Scholar]
- 158.Louie A. Chem Rev. 2010;110:3146–3195. doi: 10.1021/cr9003538. [DOI] [PMC free article] [PubMed] [Google Scholar]