

3D bioprinting shows significant promise for creating complex tissue and organ mimics to solve transplant needs and to provide platforms for drug testing and studying tissue morphogenesis[1][2][3]. However, the lack of 3D printable and cell-compatible bioinks as well as the limited ability to tune bioink material properties are cited as significant inhibitors to the growth of bioprinting[4][5]. Pioneers in the field of tissue engineering and biomaterials have established and validated that changing materials properties such as stiffness[6], bioactive moieties[7][8], and degradation[7][9] significantly impacts cell behavior and tissue formation. Thus, developing novel, versatile and tunable bioink methods that will facilitate advanced material and construct design will have important implications in the field of bioprinting and biofabrication. Versatile bioink synthesis techniques, ones that can be used with many materials, will improve both printability of existing bioinks and most importantly, can add completely new biomaterials to the 3D bioprinting material palette. Furthermore, development of tunable bioink methods will provide additional means to customize mechanical, chemical, physical, and biological properties of printed structures towards creating compositionally and structurally complex structures and functional tissues beyond the rudimentary tissue structures presented thus far.
To date, nearly all bioink methods (particularly for robotic dispensing) require printing a polymer solution while initiating subsequent gelation after extrusion. Due to the inability of a solution to be self-supporting for layer-by-layer fabrication, the solution must either be made very viscous or gelled rapidly on the printing substrate. High polymer fraction solutions (> 5 wt%) provide necessary viscosity for printing definition[10][11][12], yet are not ideal for tissue engineering. The use of dense polymer matrices can inhibit matrix remodeling and vascularization in vivo[13][14]. As well, for cell-encapsulating bioinks, high polymer fractions can be debilitating to cells, preventing spreading, migration, and proliferation, and therefore, are not ideal candidates for cell-laden constructs[15][16]. Although necessary gelation kinetics are provided, gelation layer-by-layer during printing mostly relies on cross-linking that is inherent to the material, such as materials that are thermally[17][18] or ionically[19][20] gelled (e.g. gelatin, poly(N-isopropylacrylamide), alginate). Furthermore, many high resolution structures printed with gelation layer-by-layer have also used high polymer fraction solutions[18][19][20][21]. Developing a bioink synthesis technique compatible with low polymer fractions as well as many cross-linking chemistries could significantly expand the number of 3D printable, cell-compatible soft materials. Therefore, the goal of this work was to establish a versatile method to create hydrogel bioinks of varying materials and permit the ability to tune mechanical, chemical, physical, and biological properties of the resulting structures.
In this work, we present a single bioink method capable of producing extrudable, gel phase bioinks from a variety of materials, both synthetic and natural. A few studies have reported gel phase bioinks, yet have not reported such versatility and tunability[22][23][24][25]. We demonstrated with 35 formulations that bioinks can be customized with regard to composition (additives, composites), degree of cross-linking, and polymer concentration in order to optimize structural and biological performance while maintaining printability. Additionally, we start to uncover specific properties of these gels that make them printable through rheological studies. In this method, polymer solutions were lightly cross-linked with a long length (5000 g/mol) chemical cross-linker, a homobifunctional polyethylene glycol (PEG) ending in two reactive groups (PEGX). Polymers can be linear or branched, as well as have multi-functional groups for primary (bioink synthesis) and secondary (post-printing) cross-linking (Scheme 1a). Mixtures of different polymers presenting the same functional group for cross-linking may also be used. PEG is an ideal cross-linker since it is commercially available in many physical (linear, multi-arm, molecular weight variation) and chemical variants (Scheme 1b,f)[26][27]. Furthermore, PEG is an established biomaterial with FDA-approved uses, and therefore, is a suitable additive to other biocompatible biomaterials. Since we explored ink synthesis with several protein-based materials in these studies, bioinks were synthesized by amine-carboxylic acid coupling (X in PEGX= succinimidyl valerate: SVA) to make use of a common functional group (amines) without need for modification. We envision that other cross-linking chemistries, such as click chemistries and Michael-type additions, would also be possible with this PEGX method to further expand eligible materials beyond those that are amine-presenting[26][27][28,29] (Scheme 1f). For example, thiol-containing polymers could be cross-linked with PEG acrylates, PEG maleimides, or PEG vinyl sulfones and would be an excellent system for cell-encapsulating bioinks.
Scheme 1.

a) Polymer or polymer mixtures can be linear (e.g. gelatin), branched (e.g. 4 arm PEG amine) or multi-functional (e.g. gelatin methacrylate). In example, the red circles may represent amines, blue triangles methacrylate groups, and the yellows stars SVA groups of PEGX, as demonstrated in this paper. b) PEGX can be linear or multi-arm and can be various chain lengths. c) Cells can be optionally incorporated by d) mixing with polymers and PEGX to form the bioink. e) Optional, secondary cross-linking to increase mechanical robustness may be performed post-printing. f) By changing the reactive groups of PEGX, polymers of other functional groups may be cross-linked. For example, purple polygons could represent thiols, cross-linkable with maleimide PEGX (pink squares) and green ellipses could represent alkynes, cross-linkable with azide PEGX (orange hexagon). g) Printing process of PEGX bioink method and corresponding phase: PEGX with or without cells are mixed within the polymer solution and loaded into the printing cartridge. After gel formation and stable mechanical properties are achieved, gels can be 3D printed into multi-layer structures.
Although, in general, gels behave as solids and thus do not flow, we demonstrated that when chemical cross-linking is carefully controlled, gel bioinks can be extruded through fine diameter nozzles (200 μm) while maintaining integrity (Figure 1a). To make these printable gel-phase bioinks, polymer and PEGX, with or without cells, were mixed together and allowed to gel within a printing cartridge (Scheme 1d,g). Successful bioinks were generated from polymer solutions of natural proteins (gelatin and fibrinogen), modified proteins (gelatin methacrylate) and synthetic polymers (4 arm PEG amine), as well as protein mixtures (gelatin-atelocollagen, gelatin-fibrinogen) and synthetic-natural mixtures (4 arm PEG amine-gelatin, gelatin-peptide amphiphiles) (Table S1). When gel properties stabilized, formulations were extruded through a syringe by hand through a 200 μm nozzle to confirm continuous, shape-maintaining gel filaments, and selected bioinks were printed layer-by-layer on an EnvisionTEC 3D-Bioplotter® to build defined, self-supporting (i.e. subsequent layer is supported without collapse) structures (Figure 1b–d, Supplementary Movie 3). We define gels possessing these properties as 3D “printable”. In contrast, when a warmed gelatin solution, without PEGX, was printed onto a 5°C substrate (to induce rapid thermal gelation), filaments became flattened, and subsequent layers collapsed (Figure 1e). Poor z-layer definition compromises printing definition with multi-layer constructs and may also interfere with the ability to pattern different materials due to material spreading. Extrudable, self-supporting bioink gels from the PEGX method yielded optimal layer-by-layer definition that enabled the ability to print thick, self-supporting constructs and different materials with ease.
Figure 1.

3D bioprinting PEGX-gelatin a) extrusion through 200 μm tip b) 15×15 mm square printed, 4 layers c) 20 layer porous hexagon shape ~ 5 mm thick d) inner structure of 4 layer printed structure, scale bar 500 μm, struts ~350–450 μm diameter e) warm gelatin (no PEGX) printed on 5 °C substrate with 200 μm tip, 2 layers, scale bar 500 μm, struts ~1.1–1.3 mm diameter.
The amount of PEGX needed for printability is dependent on both polymer and PEGX properties, such as molecular weight, concentration, and amount and display of functional groups. To identify printable formulations, phase plots were created by screening various polymer concentrations against varying PEGX:polymer mass to mass (m:m) ratios (Figure 2a, Figure 3a–c, Figure S5a–b). Formulations were mixed and incubated at 37 °C for 120 minutes. Material “phase” (solution or gel behavior) was determined by tube inversion. Gels were subsequently designated as either “soft” if they were spreadable or “robust” if they retained shape when manipulated with a spatula (Figure S1). Robust gels were either unable to be extruded or required significant pressure, producing inconsistent strands at very slow mass flow rates, both of which are not ideal for 3D printing. In extrusion tests, soft gels ejected as smooth, continuous strands of gel desirable for printing, and therefore, soft gels became candidate bioinks in later experiments (Figure 1a). Expectedly, soft gels were found at PEGX concentrations at or near the gel point. As polymer fraction decreased, a larger PEGX to polymer ratio was required for gelation to occur and therefore, degree of cross-linking associated with a soft gel is concentration dependent. Importantly, the ability to produce printable gels at multiple polymer concentrations (including concentrations <5 wt%) illustrates the capacity to tune material properties (i.e. modulus, degradation) and subsequently, cell response. As mentioned, many previously reported bioink methods have used high polymer fraction (>5 wt%) inks that yield viscous solutions good for printability, but not ideal for cell-encapsulation. This PEGX bioink method overcomes this limitation by allowing low polymer fraction inks to be printable.
Figure 2.
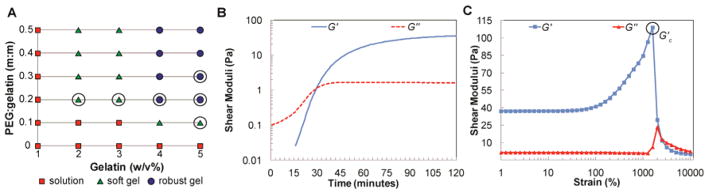
a) Phase plot of varying gelatin concentrations with varying PEGX:gelatin (m:m) ratios, circled formulas were rheologically tested. b) Gelation profile of 5 w/v% gelatin and 0.1 PEGX:gelatin (m:m) after addition of PEG cross-linker and c) response to increasing strains, failure (G′c) at 1590% strain.
Figure 3.

a) Phase plot of fibrinogen b) phase plot of 4 arm PEG amine (10,000 g/mol) c) phase plot of gelatin cross-linked with physical PEGX variant, PEG-SVA (1000 g/mol). Scanning electron micrographs of d) PEGX-gelatin (5 w/v%) at 0.1 PEGX:polymer mass ratio e) PEGX-gelatin (3 w/v%)-atelocollagen (0.06 w/v%) at 0.2 PEGX:polymer mass ratio f) PEGX-gelatin (2.25 w/v%)-peptide amphiphiles (0.75 w/v%) at 0.4 PEGX:polymer mass ratio, Ca2+-treated, scale bars 50 nm.
The use of phase plots for initial screening of potential inks served as a very useful tool to narrow down and identify 3D printable gel formulations using PEGX. To relate these observations of printability with more quantitative characteristics of the bioinks, we determined the degree of cross-linking and rheological properties of the PEGX-gelatin system. The degree of cross-linking was quantified by the 2,4,6-trinitrobenzenesulfonic acid (TNBS) assay, which determines the amount of free amines after gelation[30][31]. Soft gels ranged from 25–45% reacted amines confirming that these gels were loosely cross-linked (Figure S2). To probe mechanical properties that may be associated with the printability of soft gels, several PEGX-gelatin formulations (circled in Figure 2a) were tested rheologically. Comprehensive rheological studies that correlate with printing observations are greatly needed in order to advance bioink development[4]. Most rheological characterizations of bioinks have focused on reporting solution viscosity[20][32][33]; however, since these bioinks were gels, other types of analysis were necessary. After loading a freshly prepared formulation into the rheometer, an oscillatory time sweep (1% strain, 10 rad/s angular frequency) was performed for 120 minutes at 37 °C to monitor gelation, followed by a frequency sweep at 1% strain to confirm expected gel viscoelasticity (G′ ~ independent of frequency and G′ ≫ G″; see Figure S3a), and two strain sweeps at 10 rad/s. Key results are summarized in Table 1. Gelation (G′-G″ cross-over; Figure 2b) occurred between 15 and 30 minutes. Storage moduli were mostly stable by 120 min, but displayed modest growth over longer times. During 3D printing of these soft gel formulations over the course of several hours, however, drastic changes in printing parameters (e.g. extrusion pressure) were not needed. It is important to note that the few studies on near-gel (G′ ≈ G″) or gel phase bioinks are lacking adequate rheology that validates that they are, in fact, gels and not viscous solutions just before the gel point. Additionally, these studies do not discuss if there is a specified time frame for printing (i.e. over extended periods of time, do inks cross-link too much to the point when are no longer printable?)[22][23][24][25]. Our presented method overcomes time-dependent printing since the degree of cross-linking is controlled by small additions of cross-linker, and printing occurs when cross-linking, and therefore G′, have stabilized. After 120 minutes, soft gel formulations possessed mean storage moduli (G′2 hrs) ranging from ~1–100 Pa and robust gels were over 150 Pa. In the first strain sweep, gels exhibited a linear response at strains up to ~50%; after 50%, they exhibited strain-hardening until catastrophic yielding. Interestingly, soft gels yielded at remarkably high strains (up to 2200%; Figure 2c) while robust gels yielded at lower strains, less than 800%. Storage modulus (G′2 hrs), critical storage modulus (G′c), and critical stress (σc) increased while critical strain (γc) decreased when the polymer concentration increased at a fixed PEG ratio or when the PEG ratio increased at a fixed polymer concentration (Table 1). Repeated amplitude sweeps showed that samples underwent catastrophic failure at the critical yielding point (Figure S3b–c). Such extensive damage, however, was not observed following extrusion. In extrusion, shear stress is maximized at the nozzle walls, in contrast to rheological testing where stress is experienced homogeneously throughout the sample. We hypothesize that extrusion is facilitated by localized yielding/rupture at the nozzle surface when the wall shear stress (σwall) exceeds the critical stress (σc) necessary to induce yielding. An estimate of the magnitude of wall shear stress may be made based on a highly simplified assumption that the full pressure drop driving ink printing (ΔP) is applied only over the capillary extrusion nozzle of radius R and length L (Equation 1):
| Equation 1 |
Table 1.
Rheology summary of formulations (indicated in Figure 2a). Mean and standard deviation displayed, N=3. Values taken from time and first amplitude sweeps.
| Increasing Gelatin Concentration | Increasing PEGX:Gelatin (m:m) | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Formulation | 2% gelatin 0.2 PEGX:gelatin |
3% gelatin 0.2 PEGX:gelatin |
4% gelatin 0.2 PEGX:gelatin |
5% gelatin 0.2 PEGX:gelatin |
5% gelatin 0.1 PEGX:gelatin |
5% gelatin 0.2 PEGX:gelatin |
5% gelatin 0.3 PEGX:gelatin |
| G′-G″cross-over [minutes] | 84 ± 22 | 23 ± 9 | 28 ± 6 | 15 ± 8 | 30 ± 6 | 15 ± 8 | 22 ± 4 |
| G′2 hrs [Pa] | 1.21 ± 1.01 | 62.7 ± 7.14 | 157 ± 41.1 | 554 ± 258 | 37.4 ± 1.96 | 554 ± 258 | 589 ± 219 |
| δ2hrs [°] | 29.8 ± 11.9 | 0.72 ± 0.14 | 0.73 ± 0.17 | 0.28 ± 0.12 | 2.73 ± 0.10 | 0.28 ± 0.12 | 1.13 ± 1.35 |
| γc [%] | 2200 ± 537 | 1000 ± 0 | 794 ± 0 | 461 ± 193 | 1587 ± 5.77 | 461 ± 193 | 378 ± 107 |
| G′c [Pa] | 7.53 ± 6.08 | 213 ± 29.5 | 403 ± 89.1 | 791 ± 277 | 115 ± 16.3 | 791 ± 277 | 868 ± 50.3 |
| σc [Pa] | 144 ± 78.6 | 2130 ± 295 | 3197 ± 708 | 3407 ± 1329 | 1815 ± 254 | 3407 ± 1329 | 3295 ± 1014 |
Using parameters relevant for the printing conditions used here (ΔP = 1.5 × 105 Pa, R = 0.1 mm, L = 2 mm), σwall is estimated to be 3750 Pa, which is of the same order of magnitude as the stresses at which yielding occurs in these soft gels in rheological testing (Table 1). With higher critical stresses than soft gels, robust gels may not be capable of yielding under such printing conditions. Localized yielding at the walls, however, cannot be the sole explanation of how these gels extrude for ideal 3D printing since other weak hydrogels may fragment into discrete gel pieces and water when extruded (unpublished observations). Conversely, PEGX-polymer bioinks remained intact and extruded as continuous, cohesive filaments, an important requirement for 3D printing, even when subjected to the enormous deformation associated with extruding the gel from the much larger printing cartridge barrel through the tiny capillary nozzle (Supplementary Movie 2). Furthermore, the printed gel filaments displayed elastic behavior when uniaxially stretched, which is consistent with the ability to retain cohesiveness under severe deformation (Figure S4, Supplementary Movie 3). Future testing of more sophisticated experimental set-ups is needed to quantitatively assess this elastic behavior that may help produce a more complete picture of material requirements for 3D printing gels beyond the critical stress argument discussed above.
By exploiting a prevalent functional group (amines) for cross-linking, we proved that the PEGX bioink method can create printable bioinks from multiple materials. A list of all printable formulations can be found in Table S1. Soft, printable gels of fibrinogen were produced at identical formulations as gelatin (Figure 3a), and printable mixtures of gelatin and fibrinogen were synthesized similarly (Figure S5a). Atelocollagen was a targeted material; however, commercially available atelocollagen solutions are only offered at very low polymer fractions (0.3 w/v%). In PEGX-gelatin and PEGX-fibrinogen, printable formulations were not obtained at polymer concentrations under 1 w/v%, and therefore, the concentration of atelocollagen remained a challenge. However, we successfully mixed atelocollagen with gelatin to create a printable formulation. Incorporation of atelocollagen resulted in a more fibrous structure (Figure 3e) compared to PEGX-gelatin (Figure 3d), thus indicating assembly of collagen molecules into triple helices. Not only do these fibers instill a nanostructure that cells may more readily recognize, but they can also enhance the robustness and alter construct degradation. For example, addition of fibrinogen to PEGX-gelatin prolonged the degradation time in media to more than 2 weeks compared to the 2 day degradation of the PEGX-gelatin alone. The PEGX method could be extended to include incorporation of other isolated extracellular matrix (ECM) such as fibronectin and laminin or even tissue-specific ECM (e.g. decellularized ECM). Synthetic self-assembling oligopeptides such as peptide amphiphiles (PAs) can also add nanostructural features and provide tailorable bioactivity (i.e. inclusion of ECM-mimetic, growth factor-mimetic, or growth factor-binding peptides)[34][35]. PAs were successfully incorporated into PEGX-gelatin bioinks at multiple concentrations (Figure S5b). Since PAs can assemble into nanofibrous structures through ionic triggers, post-printing treatment of calcium chloride solution led to a more fibrous nanostructure within PEGX-gelatin-PA constructs (using negatively charged PAs) (Figure 3f), which further increased robustness and prolonged scaffold degradation. Customizing the PEGX bioink formulation of natural biopolymers to amplify bioactivity, as well as match tissue-specific compositions and physical properties may help lead to more mimetic biofabricated tissues[36][37][38].
As mentioned previously, PEG can be synthesized in many physical and chemical variations. We showed that a lower molecular weight (1000g/mol) homobifunctional linear PEG-SVA (1000 g/mol) also produced printable gels (Figure 3c). Compared to the 1000 g/mol PEGX, the higher molecular weight PEGX (5,000g/mol), however, was able to produce printable inks at lower polymer concentrations, indicating that the molecular weight of PEGX might be altered to include a broader range of polymer concentrations within these bioinks. Synthetic materials like PEG have been heavily used by tissue engineers for synthesis of hydrogels with easily tailorable biological and mechanical properties. In order to take advantage of the versatility of synthetic polymers, we aimed to create 3D printable formulations that were solely composed of synthetic polymer using 4-arm PEG-amine (10,000 g/mol), that is both physically and chemically drastically different from natural polymers. We were able to identify multiple printable formulations of PEGX-PEG amine at varying PEG amine concentrations (Figure 3b). Furthermore, since PEG has been added to proteins for enhancing mechanical robustness and slowing degradation[20][21][22], we cross-linked mixtures of PEG amine and gelatin and also successfully produced printable formulations. These printable blends can be alternatively viewed as an enhancement to the synthetic PEG, as gelatin inherently provides cell adhesion sites. We envision that the PEGX method will easily be extended to mixtures of synthetic peptides and PEG for controllable cell adhesion and degradation properties.
A secondary, post-printing cross-linking step can be used to manipulate modulus and degradation properties of the resulting printed constructs[12][22]. Since the PEGX method only partially cross-links the polymer bioinks, the degradation of printed structures for some materials (such as gelatin) may be too fast for long-term cell culture and tissue engineering applications. EDC (N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide)/NHS (N-Hydroxysuccinimide) is a ubiquitous cross-linking agent that couples amines and carboxylic acid groups or carboxylic acid groups together, and has been used in many natural, unmodified materials, including collagen[39], gelatin[31], hyaluronic acid[40], as well as mixtures[41][42]. When PEGX-gelatin was cross-linked with EDC/NHS, the modulus increased from Pa to kPa, covering the range of mechanical properties relevant for soft tissue engineering (Table 1, Figure S6)[43]. Degradation of these cross-linked scaffolds in serum-containing media was also significantly extended to greater than four weeks when compared to those without secondary cross-linking, which degraded within two days (Figure S7). Using fibrinogen within the PEGX bioinks afforded another type of post-printing stabilization and modification method. Fibrinogen-containing formulations were secondary cross-linked by treating with a thrombin-Ca2+ solution, after which gels became opaque, were significantly more robust with longer degradation times, and exhibited a fibrous structure indicative of fibrin assembly (Figure S5c–d). Finally, biopolymers can be modified to provide heterogeneous functionality for orthogonal primary (bioink fabrication) and secondary (post-printing) cross-linking (Scheme 1e–g)[15][23]. Gelatin was modified with methacrylate groups through reaction of amines. The degree of methacrylate functionalization was ~35%, according to the TNBS assay. Printable gels were produced using a similar formulation for unmodified gelatin, indicating that remaining, unreacted amines were present in adequate quantities (after methacrylation) for effective primary PEGX cross-linking. Secondary cross-linking between methacrylate groups was then initiated by UV light leading to increased robustness and stability of constructs within serum-containing media for prolonged times (Figure S8). PEGX-gelatin methacrylate verifies that modified biopolymers[44][45][46], or other multi-functional polymers, can be used within the PEGX method to introduce greater tailorability of the resulting 3D printed constructs. This may also lead to the use of heterogeneous multi-arm PEGX that can be used for both primary and subsequent, secondary polymer-PEGX or PEGX-PEGX (interpenetrating network) cross-linking, which would most likely lead to structures having very different physical properties.
To investigate the utility of these inks in biofabrication and tissue engineering, multi-material printing and cell studies were performed. PEGX-fibrinogen and PEGX-gelatin were successfully co-printed to demonstrate the ability to spatially organize multiple types of extracellular matrix within one 3D construct (Figure 4a–b). Furthermore, PEGX-PEG and PEGX-gelatin were also successfully co-printed into one construct. This resulted in seeded human dermal fibroblasts (HDFs) preferentially adhering to the gelatin struts, signifying the ability to direct cell adhesion to only certain portions of a scaffold (Figure 4d). The ability to incorporate cells into the inks is of great importance for complex tissue and organ engineering through controlled 3D spatial organization of multiple cell types. Cells (HDFs) were mixed within bioink formulations (2E6 cells/mL) of PEGX-gelatin and incubated for 2 hours to achieve a printable consistency. To ensure that cells do not settle within the printing cartridge during the incubation time, sequential aliquots of the entire cell/PEGX-gelatin mixture were dispensed and analyzed for DNA content. The number of cells per aliquot throughout the full ink volume was consistent, indicating that the cells did not settle and remained homogeneous throughout the ink during the incubation time (Figure S9). Furthermore, the number of cells per mg ink was near the calculated value. As seen in our rheological analysis, although gelation of this formulation occurs at ~30 minutes, the viscosity of the solution rapidly increases immediately upon addition of the cross-linker until it approaches infinity at the gel point. This increase in viscosity helps the cells remain homogeneously suspended until the gel point is reached.
Figure 4.
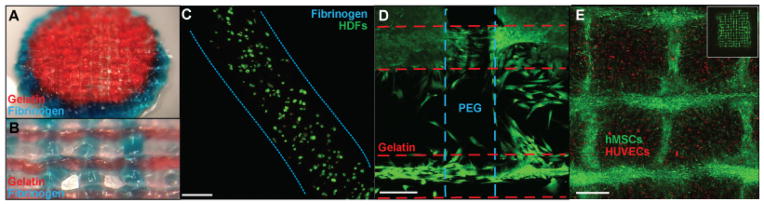
a) PEGX-gelatin (red) and PEGX-fibrinogen (blue) co-printed cylinder, 15 mm diameter b) PEGX-gelatin (red) and PEGX-fibrinogen (blue) co-printed construct with inner structure patterned, struts ~650 μm diameter c) Cell viability using Live/Dead assay at one day post-printing using 3 w/v% fibrinogen at a 0.2 PEG ratio, scale bars 200 μm d) PEGX-PEG and PEGX-gelatin co-printed structure seeded with HDFs (green, calcein AM). Cells preferentially adhere to PEGX-gelatin e) XY projection of 3D reconstruction. HUVECs (CellTracker™ Red) printed in 5 w/v% gelatin and 0.1 PEGX:gelatin (m:m) seeded with hMSCs (CellTracker™ Green) into internal porous structure. hMSCs fill empty spaces of inner structure, inset: whole construct, scale bar 400 μm. hMSCs spread into open spaces of construct and onto printed bioink strands at Day 4.
Cell viability after printing was qualitatively assessed with the Live/Dead assay in PEGX-gelatin and PEGX-fibrinogen. This bioink method successfully supported viability of HDFs and human umbilical vein endothelial cells (HUVECs) after printing (Figure 4c, Figure S10). Additionally, we investigated spatially organizing two cell types within the same printed construct as a technique for engineering more mimetic tissues. HUVECs stained with CellTracker™ Red were encapsulated in PEGX-gelatin bioink and printed into 15×15 mm, 4 layer structures. Human mesenchymal stem cells (hMSCs) stained with CellTracker™ Green were subsequently seeded onto printed cell-laden constructs to fill the open spaces of the internal structure (Figure S10g–h). At four days, hMSCs began to migrate and completely fill the open spaces of the construct and were spread onto the printed filaments, clearly following the 3D grid pattern of the printed construct (Figure 4e). The addition of hMSCs also slowed the degradation of the gel by two days compared to HUVEC-laden constructs without hMSCs seeded within the pores. Although the gel degraded, hMSCs maintained a grid-like pattern after two weeks and surprisingly, the cells deposited sufficient matrix that led to a robust and opaque skin-like tissue that was lifted and handled with forceps. This emphasizes the importance of optimizing bioink degradation with matrix deposition and remodeling by cells for engineering more natural tissues.
In conclusion, we present a versatile and cell-compatible bioink method of creating soft, printable gels from a variety of amine-containing polymers and polymer mixtures, both synthetic and natural. We observe that the critical strain and critical stress of these gel phase bioinks are new parameters that may be correlated with 3D printability; however, gel cohesiveness is also an important requirement that needs further quantification. In future studies, we expect that other PEGX (chemical and physical variants) will permit additional cross-linking chemistries for bioink synthesis that can further expand the number of 3D printable bioinks available. Furthermore, polymer type and concentration, degree of cross-linking, as well as post-printing cross-linking can be tailored with ease to tune material properties of the bioinks and 3D printed structures. We envision that the PEGX bioink method can be used towards developing tailorable platforms for studying cell-cell signaling and tissue morphogenesis in 3D, as well as creating more customized and biomimetic 3D printed tissue constructs that are optimized for complex tissue and organ engineering.
Experimental Section
Bioink synthesis
Homobifunctional PEG SVA (5000 g/mol or 1000 g/mol, Laysan Bio) was dissolved concentrated in pre-warmed phosphate-buffered saline solution (PBS) just prior to ink preparation. Concentrated solutions of hydrogel precursor were prepared as follows: 10 w/v% gelatin type A (Sigma) in PBS; 10 w/v% gelatin methacrylate in PBS; 10 w/v % fibrinogen (Sigma) in 0.9% NaCl; 20 w/v% 4 arm PEG amine (10,000 g/mol, Laysan Bio) in PBS; and 5 w/v% transforming growth factor beta binding (TGFβ) PA in deionized water at 37°C. TGFβ-binding PA having a sequence of HSNGLPLGGGSEEEAAAVVV(K)-CO(CH2)10CH3 was synthesized and purified as previously described[47] using standard solid phase peptide synthesis and HPLC methods, respectively. Atelocollagen solution (Advanced Biomatrix) at 4 °C was mixed into warm gelatin after neutralizing with 1 M NaOH and room temperature PBS and PEGX. Gelatin methacrylate was synthesized as previously published[15]. Briefly, methacrylic anhydride was added dropwise to a 10% gelatin at 40 °C. After 1 hour, gelatin was diluted to 2% and dialyzed at 40 °C for 1 week. Solution was then lyophilized and stored dry at −20 °C. Gelatin methacrylate inks also included 1w/v% photoinitiator [2-Hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Sigma) dissolved in 1-Vinyl-2-pyrrolidinone (Sigma)]. Cells were concentrated in PBS. Hydrogel precursor polymer solution, PEG cross-linker (PEGX), PBS (as needed for dilution), and cells (optional) were thoroughly mixed at various amounts to produce different bioink formulations, immediately transferred to a glass vial, syringe, or printing cartridge, and held at 37 °C for 1–2 hours. Unless otherwise stated, 5000 g/mol PEGX was used.
Phase plots
Bioinks were prepared in glass vials. At 120 minutes, vials were inverted to determine if it was a solution or gel phase. Gels were manipulated with a spatula and if it could be spread on the glass vial wall, the gel was designated “soft”. If the gel retained its shape, it was designated “robust”. Soft gels were extruded through a syringe and nozzle by hand.
TNBS assay
TNBS assay to determine percent reacted amines was performed according to previously published studies[30][31]. Briefly, after 120 minutes, gels were incubated with TNBS solution (0.01 M in sodium bicarbonate buffer) for 2 hours at 40 °C. The gels were then treated HCl (1 M) and SDS (10%) to stop TNBS reaction and dissolve gel overnight at 40 °C. Solutions were diluted and absorbance at 340 nm was read on a SpectraMax M5 microplate reader.
Rheology
Testing was performed using an Anton-Paar MCR 302 rheometer with a cone-plate fixture. Temperature was controlled at 37 °C during testing. Formulations were prepared as above immediately prior to testing, loaded on the warmed plate and the measuring cone was lowered into position. After applying mineral oil to the edges of the fixture to prevent dehydration, a time sweep was performed for 120 min, followed by a frequency sweep and then two amplitude sweeps, using testing parameters described in the text.
Printing
Bioinks were prepared in a conical tube and quickly transferred to EnvisionTEC printer cartridges. The cartridges were stored in a cell culture incubator or within the printing magazine to maintain 37 °C. At 1–2 hours, a printing nozzle (200 μm) was fixed onto the cartridge, and gels were printed on the EnvisionTEC 3D-Bioplotter®. Printing pressures and speeds were altered depending on ink flow properties. Gels were typically printed by applying a pressure of 1–2.5 bar at a printing speed of 5 mm/s onto autoclaved glass slides. Gelatin and fibrinogen co-printed inks were mixed with red and blue food coloring respectively for visualization.
Degradation Studies
Gelatin scaffolds were cross-linked with 15 mM EDC (Sigma) and 6 mM NHS (Sigma) in deionized water for up to 60 minutes. UV cross-linking of printed gelatin methacrylate was performed with B-100AP lamp (UVP) lamp at wavelength 365 nm and intensity 15–20 mW/cm2 for up to ten minutes. Fibrinogen-containing samples were treated post-printing with 10 U/mL thrombin (Sigma) in 40 mM CaCl2 for ~30 minutes. PA-containing samples were treated with 40 mM CaCl2 solution to induce self-assembly. Structures were prepared sterile and incubated in a cell culture incubator at 37 °C in either PBS or DMEM supplemented with penicillin-streptomycin and 10% fetal bovine serum. Degradation was assessed by visual inspection and pictures. Degradation was indicated by dissolution of printed strands or structures.
Mechanical Testing
Compression testing was performed on a LF Plus mechanical tester at 0.5 mm/s (Lloyd instruments, 50N load cell). Gels (200 μL) were prepared between glass coverslips and cross-linked with 15 mM EDC (Sigma) and 6 mM NHS (Sigma) in deionized water for 1 hour to yield flat gel cylinders of 7 mm diameter and 1 mm height. Modulus was taken over 0–20% strain.
Cell Studies
P4-P6 human dermal fibroblasts (Cell Applications, Inc.) and human umbilical vein endothelial cells (Lonza Inc.) were incorporated into 5 w/v% gelatin or 3 w/v% fibrinogen at PEGX ratios of 0.1 and 0.2 respectively. Cell homogeneity throughout the bioink was assessed by sequentially collecting extruded bioink into tared microtubes. Wet weight was collected, gels were degraded with proteinase K (Sigma), and the solutions were analyzed for DNA content by the Picogreen assay (Life Technologies) using a microplate reader. Cell viability was assessed with the Live/Dead® assay (Life Technologies) according to manufacturer’s instructions. P4-P6 bone marrow-derived human mesenchymal stem cells (Lonza, Inc.) were seeded onto HUVEC encapsulated PEG-gelatin printed constructs. HUVECs and hMSCs were labeled with CellTracker™ Red (Molecular Probes®) and CellTracker™ Green, respectively according to manufacturer’s instructions. Cells were imaged with a Nikon C2+ confocal and Nikon AZ 100 fluorescent stereoscope.
Imaging
Printed constructs were photographed with a Canon camera or cell phone camera. Photojojo macro lens was also used for pictures and movies taken with cell phone camera. Printed constructs were also imaged with a Leica M205 C stereoscope. Confocal stacks were analyzed with ImageJ software. SEM analysis was performed with a LEO Gemini 1525 after glutaraldehyde fixing, critical point drying, and osmium coating of samples.
Supplementary Material
Acknowledgments
The authors thank Sumanas W. Jordan and Phillip L. Lewis for assistance with printing cell encapsulating bioinks. The authors would also like to thank the Woodruff Lab (PI: Teresa Woodruff, Ph.D.) at Northwestern University for gracious use of their Leica stereoscope. Authors acknowledge use of the following facilities: Northwestern University Cell Imaging Facility generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, EPIC facility (NUANCE Center – Northwestern University) which has received support from NSF DMR-0520513 and Engineering Center EEC-0118025|003, and the Peptide and Equipment Core Facilities at the Simpson Querrey Institute for BioNanotechnology at Northwestern University developed by support from The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University. This work was supported by the National Institutes of Health (1K01DK099454-01) and National Science Foundation (Graduate Research Fellowship for A.L. Rutz).
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Alexandra L. Rutz, Institute for BioNanotechnology in Medicine, Chicago, IL 60611, USA. Department of Biomedical Engineering, Evanston, IL 60208, USA
Kelly E. Hyland, Department of Materials Science and Engineering, Evanston, IL 60208, USA. Institute for BioNanotechnology in Medicine, Chicago, IL 60611, USA
Adam E. Jakus, Department of Materials Science and Engineering, Evanston, IL 60208, USA. Institute for BioNanotechnology in Medicine, Chicago, IL 60611, USA
Prof. Wesley R. Burghardt, Department of Materials Science and Engineering, Evanston, IL 60208, USA. Department of Chemical and Biological Engineering Evanston, IL 60208, USA
Prof. Ramille N. Shah, Email: ramille-shah@northwestern.edu, Department of Materials Science and Engineering, Evanston, IL 60208, USA. Department of Surgery - Organ Transplantation, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. Institute for BioNanotechnology in Medicine, Chicago, IL 60611, USA
References
- 1.Derby B. Science (80- ) 2012;338:921. doi: 10.1126/science.1226340. [DOI] [PubMed] [Google Scholar]
- 2.Mironov V, Kasyanov V, Markwald RR. Curr Opin Biotechnol. 2011;22:667. doi: 10.1016/j.copbio.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Ozbolat IT, Yu Y. IEEE Trans Biomed Eng. 2013;60:691. doi: 10.1109/TBME.2013.2243912. [DOI] [PubMed] [Google Scholar]
- 4.Malda J, Visser J, Melchels FP, Jüngst T, Hennink WE, Dhert WJA, Groll J, Hutmacher DW. Adv Mater. 2013;25:5011. doi: 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- 5.Ringeisen BR, Pirlo RK, Wu PK, Boland T, Huang Y, Sun W, Hamid Q, Chrisey DB. MRS Bull. 2013;38:834. [Google Scholar]
- 6.Discher DE, Janmey P, Wang YL. Science (80- ) 2005;310:1139. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 7.Lutolf MP, Weber FE, Schmoekel HG, Schense JC, Kohler T, Müller R, Hubbell Ja. Nat Biotechnol. 2003;21:513. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 8.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotechnol. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 9.Nair LS, Laurencin CT. Prog Polym Sci. 2007;32:762. [Google Scholar]
- 10.Gaetani R, Doevendans PA, Metz CHG, Alblas J, Messina E, Giacomello A, Sluijter JPG. Biomaterials. 2012;33:1782. doi: 10.1016/j.biomaterials.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Chang CC, Boland ED, Williams SK, Hoying JB. J Biomed Mater Res B Appl Biomater. 2011;98:160. doi: 10.1002/jbm.b.31831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuurman W, Levett Pa, Pot MW, van Weeren PR, Dhert WJa, Hutmacher DW, Melchels FPW, Klein TJ, Malda J. Macromol Biosci. 2013;13:551. doi: 10.1002/mabi.201200471. [DOI] [PubMed] [Google Scholar]
- 13.Sung HJ, Meredith C, Johnson C, Galis ZS. Biomaterials. 2004;25:5735. doi: 10.1016/j.biomaterials.2004.01.066. [DOI] [PubMed] [Google Scholar]
- 14.Brandl F, Sommer F, Goepferich A. Biomaterials. 2007;28:134. doi: 10.1016/j.biomaterials.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Biomaterials. 2010;31:5536. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phelps Ea, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek Ta, Barker TH, García AJ. Adv Mater. 2012;24:64. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Yan Y, Pan Y, Xiong Z, Liu H, Cheng J, Liu F, Lin F, Wu R, Zhang R, Lu Q. Tissue Eng. 2006;12:83. doi: 10.1089/ten.2006.12.83. [DOI] [PubMed] [Google Scholar]
- 18.Fedorovich NE, Swennen I, Girones J, Moroni L, van Blitterswijk Ca, Schacht E, Alblas J, Dhert WJa. Biomacromolecules. 2009;10:1689. doi: 10.1021/bm801463q. [DOI] [PubMed] [Google Scholar]
- 19.Khalil S, Sun W. J Biomech Eng. 2009;131:1. doi: 10.1115/1.3128729. [DOI] [PubMed] [Google Scholar]
- 20.Fedorovich NE, Schuurman W, Wijnberg HM, Prins HJ, van Weeren PR, Malda J, Alblas J, Dhert WJA. Tissue Eng Part C Methods. 2012;18:33. doi: 10.1089/ten.tec.2011.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Censi R, Schuurman W, Malda J, di Dato G, Burgisser PE, Dhert WJa, van Nostrum CF, di Martino P, Vermonden T, Hennink WE. Adv Funct Mater. 2011;21:1833. [Google Scholar]
- 22.Skardal A, Zhang J, McCoard L, Xu X, Oottamasathien S, Prestwich GD. Tissue Eng Part A. 2010;16:2675. doi: 10.1089/ten.tea.2009.0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skardal A, Zhang J, Prestwich GD. Biomaterials. 2010;31:6173. doi: 10.1016/j.biomaterials.2010.04.045. [DOI] [PubMed] [Google Scholar]
- 24.Cohen DL, Malone E, Lipson H, Bonassar LJ. Tissue Eng. 2006;12:1325. doi: 10.1089/ten.2006.12.1325. [DOI] [PubMed] [Google Scholar]
- 25.Loozen LD, Wegman F, Öner FC, Dhert WJa, Alblas J. J Mater Chem B. 2013;1:6619. doi: 10.1039/c3tb21093f. [DOI] [PubMed] [Google Scholar]
- 26.Roberts MJ, Bentley MD, Harris JM. Adv Drug Deliv Rev. 2002;54:459. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 27.Zhu J. Biomaterials. 2010;31:4639. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nimmo CM, Shoichet MS. Bioconjug Chem. 2011;22:2199. doi: 10.1021/bc200281k. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z. Biomaterials. 2014;35:4969. doi: 10.1016/j.biomaterials.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Habeeb AFSA. Anal Biochem. 1966;14:328. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- 31.Kuijpers AJ, Engbers HM, Krijgsveld J, Zaat SAJ, Dankert J, Feijen J. J Biomater Sci Polym Ed. 2000;11:225. doi: 10.1163/156856200743670. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh S, Parker ST, Wang X, Kaplan DL, Lewis JA. Adv Funct Mater. 2008;18:1883. [Google Scholar]
- 33.Hanson Shepherd JN, Parker ST, Shepherd RF, Gillette MU, Lewis Ja, Nuzzo RG. Adv Funct Mater. 2011;21:47. doi: 10.1002/adfm.201001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui H, Webber MJ, Stupp SI. Biopolymers. 2010;94:1. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matson JB, Zha RH, Stupp SI. Curr Opin Solid State Mater Sci. 2011;15:225. doi: 10.1016/j.cossms.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skardal A, Smith L, Bharadwaj S, Atala A, Soker S, Zhang Y. Biomaterials. 2012;33:4565. doi: 10.1016/j.biomaterials.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pati F, Jang J, Ha D, Kim SW, Rhie J, Shim J, Kim D, Cho D. Nat Commun. 2014;5:1. doi: 10.1038/ncomms4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badylak SF, Taylor D, Uygun K. n.d doi: 10.1146/annurev-bioeng-071910-124743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capito RM, Spector M. Gene Ther. 2007;14:721. doi: 10.1038/sj.gt.3302918. [DOI] [PubMed] [Google Scholar]
- 40.Tomihata K, Ikada Y. J Biomed Mater Res. 1997;37:243. doi: 10.1002/(sici)1097-4636(199711)37:2<243::aid-jbm14>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 41.Rafat M, Li F, Fagerholm P, Lagali NS, Watsky Ma, Munger R, Matsuura T, Griffith M. Biomaterials. 2008;29:3960. doi: 10.1016/j.biomaterials.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 42.Gorgieva S, Kokol V. J Biomed Mater Res A. 2012;100:1655. doi: 10.1002/jbm.a.34106. [DOI] [PubMed] [Google Scholar]
- 43.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park YD, Tirelli N, Hubbell Ja. Biomaterials. 2003;24:893. doi: 10.1016/s0142-9612(02)00420-9. [DOI] [PubMed] [Google Scholar]
- 45.Fu Y, Xu K, Zheng X, Giacomin AJ, Mix AW, Kao WJ. Biomaterials. 2012;33:48. doi: 10.1016/j.biomaterials.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandler EM, Berglund CM, Lee JS, Polacheck WJ, Gleghorn JP, Kirby BJ, Fischbach C. Biotechnol Bioeng. 2011;108:1683. doi: 10.1002/bit.23079. [DOI] [PubMed] [Google Scholar]
- 47.Shah RN, Shah Na, Del Rosario Lim MM, Hsieh C, Nuber G, Stupp SI. Proc Natl Acad Sci U S A. 2010;107:3293. doi: 10.1073/pnas.0906501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.