

Abstract
In vitro and in vivo experimental models are frequently used to assess a new chemical entity’s (NCE) biopharmaceutical performance risk for food effect (FE) in humans. Their ability to predict human FE hinges on replicating key features of clinical FE studies and building an in vitro-in vivo relationship (IVIVR). In this study, 22 compounds that span a wide range of physicochemical properties, Biopharmaceutics Classification System (BCS) classes, and food sensitivity were evaluated for biorelevant dissolution in fasted- and fed-state intestinal media and the dog fed/fasted-state pharmacokinetic model. Using the area under the curve (AUC) as a performance measure, the ratio of the fed-to-fasted AUC (FE ratio) was used to correlate each experimental model to FE ratio in humans. A linear correlation was observed for the in vitro dissolution-human IVIVR (R2 = 0.66, % mean square error 20.7%). Similarly, the dog FE ratio correlated linearly with the FE ratio in humans (R2 = 0.74, % mean square error 16.25%) for 15 compounds. Data points near the correlation line indicate dissolution-driven mechanism for food effect, while deviations from the correlation line shed light on unique mechanisms that can come into play such as GI physiology or unusual physicochemical properties. In summary, fed/fasted dissolution studies and dog PK studies show a reasonable correlation to human FE, hence are useful tools to flag high-risk NCEs entering clinical development. Combining kinetic dissolution, dog FE model and in silico modeling one can study FE mechanism and formulation strategies to mitigate the FE risk.
KEY WORDS: biorelevant dissolution, dog food effect model, food effect, in vitro-in vivo relationship
INTRODUCTION
The FDA guidance on food-effect (FE) bioavailability and fed bioequivalence studies highlights the need to understand the effect of food on a new drug’s rate and extent of absorption in human pharmacokinetic studies (1). The change in pharmacokinetics induced by a meal can have significant repercussion for narrow therapeutic-index drugs, compromising safety if exposure increases to a large extent, or compromising efficacy for drugs with a steep dose-response. Food can insert large pharmacokinetic variability through varied mechanisms such as delayed gastric emptying, bile solubilization, change in pH in the gastrointestinal tract, physicochemical interaction with food components, a change in absorption mechanism from transcellular diffusion to lymphatic absorption, change in luminal metabolism, or increased splanchnic blood flow (2,3). In early clinical development, as knowledge on a new chemical entity (NCE) emerges, getting an early read on the impact of food can go a long way in recognizing and/or defining the pharmacokinetic variability to be anticipated, mitigating the effect through formulation technologies, and making appropriate recommendations in the drug approval label as to the dose administration with regard to food.
To study the risk of FE in early formulation development, various in vitro and in vivo methods have been described in literature including recent reviews of methodologies (2,4,5). The experimental approaches can broadly be classified as statistical or mechanistic in nature. Statistical approaches examine a large number of compounds to provide a qualitative assessment of the risk and recommend threshold physicochemical parameters that influence the correlation (6–8). They pull-out trends in large data sets, but don’t provide information on the mechanism behind the rate-limiting steps for a compound of interest. Mechanistic approaches on the other hand examine the in vitro dissolution and in vivo pharmacokinetics (PK) for an individual compound and may utilize physiologically based pharmacokinetic (PBPK) modeling to draw conclusions on the primary mechanisms causing the FE (9–11). In vitro dissolution in fed- and fasted-state intestinal media has been shown to correlate to the human FE for lipophilic positive FE compounds (4,12,13) with permeability playing a significant role for negative, or no FE drugs (13). In the in vivo realm, the canine FE model has been reported to be predictive of human FEs when the PK profiles are compared for dogs administered drug in the fed and fasted state (4,14,15). More recently, an integrated approach using in vitro dissolution, in vivo PK, and in silico modeling has be shown to shed light on the primary mechanism (or hypothesis) behind an observed positive or negative FE (9–11). Whether these are retrospective, post hoc analyses with clinical FE data, or prospective analyses prior to clinical trial FE data, in vitro dissolution and preclinical FE models are essential to gauge the food-related risk.
The correlation of in vitro dissolution to in vivo PK in the fed and fasted state for a given drug is heavily linked to the assumptions with the in vitro model and the rate-limiting step that controls the overall FE observation. Careful scrutiny of the assumptions is prudent to understand scenarios that make the in vitro models predictive of human FE and/or recognize scenarios when the rate-limiting steps are not captured (e.g., physiology-related factors such as delayed gastric emptying for narrow window of absorption, transporter/enzyme competition at the cell membrane, etc.). Published case studies tend to highlight successful in vitro-in vivo mechanistic correlations for individual drugs, focusing on the drug tested in in vitro and in vivo systems. Examples that exhibit unusual behavior are less frequently reported as they invoke confounding mechanisms that cannot be easily explained. To better understand such scenarios, it is necessary to focus on the experimental model and test many compounds to extract general trends (IVIVRs) and knowledge about situations when compounds deviate from the trend. This practice increases our confidence in experimental models and provides a holistic picture that connects experimental model data to clinical PK performance.
In this study, we adopt a hybrid approach that merges elements of statistical and mechanistic methods to get a bigger picture of experimental models bioperformance data. First, we generated dissolution profiles in biorelevant intestinal fasted- and fed-state media for a diverse set of 22 distinct chemotypes, spanning a range of physicochemical properties and Biopharmaceutics Classification System (BCS) classes using consistent dissolution methodology. Next, an in vitro dissolution in vivo human FE relationship (IVIVR) analysis was conducted to test if a quantitative relationship can be obtained that correlates the in vitro dissolution FE ratio to the human FE ratio but importantly, closely analyze the data points that stray from the correlation to investigate the underlying mechanism. As verification of in vitro dissolution risk in animal models is required, the third objective was to explore the correlation of the dog FE model to human FE risk for a subset of 15 compounds. Trends are discussed, and examples of distinctive FE mechanisms are highlighted for both IVIVRs. The primary intent of these trend analyses was to enhance our confidence in the risk-indicating aspects of these experimental models, so they can be applied to new NCEs before Phase 1 studies are conducted.
This retrospective correlation analysis includes clinical and dog PK data for 17 proprietary compounds generated over a span of two decades. Study protocols, dose selection criteria, and formulation aspects may differ between drugs that are intended for different molecular targets across therapeutic areas. However, the fact that these are conducted as crossover studies in dogs and humans using the same active pharmaceutical ingredient (API) or dosage form, it is possible to use the fed:fasted ratio comparison to gain a wider perspective of food-effect knowledge across drugs.
MATERIALS AND METHODS
Simulated intestinal fluid (SIF) powder used to make fasted-state simulated intestinal fluid (FaSSIF) and fed-state simulated intestinal fluid (FeSSIF) was purchased from Biorelevant.com (Croydon, Surrey, United Kingdom). Sodium phosphate dibasic was purchased from EM Science (Gibbstown, NJ). Hydrochloric acid was purchased from J.T. Baker (Phillipsburg, NJ). Methocel A4M was purchased from Dow Chemical Company (Midland, MI). Water was purified by Milli-Q UV plus systems (Millipore Co., Bedford, MA). All other chemicals were of analytical grade and purchased from commercial vendors.
Test Compounds and Physicochemical Properties
All BMS compounds were synthesized by Bristol-Myers Squibb Company (New Brunswick, NJ). The API was used for in vitro dissolution studies. API suspension or conventional immediate release (IR) dosage forms were used for the dog and clinical studies. The formulation compositions contained no exotic excipients that would influence bioavailability. Enabled formulation approaches (lipid-based formulations, amorphous dispersions, or modified-release formulations) that can mitigate food effects were excluded from this analysis. Commercially available drugs: ketoconazole, ribavirin, triclabendazole, and danazol were purchased from Sigma–Aldrich (St. Louis, MO).
The BMS compounds are 17 distinct chemotypes for unique molecular targets across therapeutic areas. The molecular weights range from 129 to 748 g/mole. Physicochemical characterization studies were conducted using standard pharmaceutical industry techniques. Equilibrium solubility was measured with excess solid phase in the fluid media for 24–48 h. The octanol water partition coefficient (LogP) was measured using the octanol/pH 6.5 buffer shake flask method. Permeability was measured by Caco-2 and PAMPA membranes permeability assays (16). The Caco-2 permeability values were validated against the human jejunal permeability for several compounds reported by Lennernas (17). The BCS class was assigned based on the solubilization of the dose across the physiological pH range 1–7.5 and the permeability measurements.
The drugs studied include mostly BCS-II drugs and few BCS-III and BCS-IV drugs (Table I). The unbalanced distribution is reflective of the typical development pipeline with more than 2/3rd being BCS-II drugs (18,19), that have a higher FE risk probability.
Table I.
Compound Table with Physicochemical Properties, BCS Class, In Vitro Dissolution, Dog and Clinical Food-Effect Ratio
| Drug | Mol. type | BCS class | Log P | In vitro AUC FE ratio | Dog dose (mg) | Dog AUC FE ratio | Human dose (mg) | Human AUC FE ratio | Reference |
|---|---|---|---|---|---|---|---|---|---|
| BMS-A | Base | 2 | 2.36 | 1.04 | 30 | 0.9 | 40 | 1.17 | BMS |
| BMS-B | Base | 4 | 3.87 | 3.29 | – | – | 300 | 3.17 | BMS |
| BMS-C | Acid | 2 | 5.09 | 2.05 | 100 | 2.1 | 200 | 2.1 | BMS |
| BMS-D | Base | 3 | 2.8 | 1.52 | 400 | 2.65 | 400 | 1.7 | BMS |
| BMS-E | Base | 2 | 1.5 | 1.17 | 200 | 2.5 | 800 | 2.5 | BMS |
| BMS-F | Acid | 3 | 0.59 | 0.7 | 20 | 0.7 | 20 | 0.69 | BMS |
| BMS-G | Acid | 2 | 3.73 | 1.1 | 150 | 5.2 | 600 | 1.2 | BMS |
| BMS-H | Base | 3 | −3.37 | 0.7 | 500 | 0.75 | 850 | 0.75 | BMS |
| BMS-I | Base | 3 | 2.3 | 0.94 | 25 | 0.86 | 25 | 0.61 | BMS |
| BMS-J | Base | 2 | 5.3 | 1.38 | 15 | 1.5 | 15 | 1.2 | BMS |
| BMS-K | Base | 2 | 2.08 | 1.25 | – | – | 300 | 1.09 | BMS |
| BMS-L | NI | 4 | 3.26 | 1.08 | – | – | 400 | 0.91 | BMS |
| BMS-M | NI | 2 | 4.6 | 2.26 | 75 | 2.3 | 75 | 1.87 | BMS |
| BMS-N | Base | 2 | 3.1 | 0.91 | 50 | 1.16 | 300 | 2.3 | BMS |
| BMS-O | Base | 2 | 4.9 | 2.3 | 50 | 2.3 | 40 | 1.2 | BMS |
| BMS-P | Base | 2 | 2.3 | 4.23 | 150 | 5.5 | 800 | 3.7 | BMS |
| BMS-Q | Acid | 2 | 1 | – | 300 | 0.83 | 300 | 1.08 | BMS |
| Ketoconazole | Base | 2 | 3.54 | 2.46 | – | – | 600 | 1.45 | (39) |
| Ribavirin | Base | 3 | −2.6 | 1.2 | – | – | 200 | 1.42 | (40) |
| Triclabendazole | Base | 2 | 6.34 | 10.01 | – | – | 600 | 3.7 | (26) |
| Danazol | NI | 2 | 4.7 | 3.56 | – | – | 100 | 3 | (41) |
| Celecoxib | Acid | 2 | 3 | – | 100 | 2.2 | 200 | 1.2 | (42) |
No value denotes the drug was not tested in that model. Molecule type: acid, base, or non-ionizable (NI)
In Vitro Microdissolution Assay
Dissolution studies were conducted in the pION μDiss Profiler (pION Inc., Billerica, MA) fitted with UV-fiber optic probes for real-time analysis of drug concentration in solution. APIs were dosed directly in FaSSIF and FeSSIF fluids that were prepared as per the Dressman original recipe (20).
All fluids were pre-heated to 37°C and degassed for 30 min prior to use. Test compounds were dosed into 15 mL of FaSSIF or FeSSIF and dissolution followed up to 180 min. The dissolution media were mixed throughout the experiment using small magnetic stirrer bars at 150 rpm. Prior to running the dissolution experiment, a standard curve was established for each experiment using drug stock solution in organic solvent diluted in FaSSIF or FeSSIF. Typically, <2% v/v organic solvent was used for standard solutions with minimal interference to the measured UV spectral signal. The detection wavelength selected was specific to each drug. The path length of the UV-fiber optic probes used was typically 5 mm. The 2nd derivative of the UV spectra was used to prevent signal interference from undissolved material.
The target amount for the dissolution test was based on the clinical dose administered with a glass of water, i.e., clinical dose/250 mL, scaled to the microdissolution test volume. In general, dissolution studies were conducted at the target dose except on the occasion where there was major interference in UV signal from undissolved material; a lower target amount was studied. Dose can influence dissolution results. However, the lower doses tested did not impact the overall result as excess undissolved drug (at least twice that of the peak solubility) was always present to ensure maximal dissolution. The concentration-time profile was analyzed for area under the curve (AUC) and peak concentration. Each condition was conducted in replicates of n = 3–4.
The in vitro food effect was calculated using the following equation:
where AUC is the area under the curve for the concentration-time profiles for FaSSIF and FeSSIF over 180 min, calculated using the trapezoidal rule on Microsoft Excel.
Test compounds with poor wetting properties (that tend to float) were pre-suspended in 0.5% Methocel A4M to enhance wetting and dosed within 2 min of preparation. Weak base drugs that show strong pH-dependent solubility were studied at a common pH in both fluids. The FeSSIF pH was raised to 6.5 to deconvolute the pH dependency of the API from bile-mediated solubilization. This strategy was applied to ketoconazole, BMS-C, BMS-D, BMS-I, BMS-K, and BMS-N.
Dog PK Model for Food Effect
A total of 15 BMS and commercially available compounds were studied in the dog FE model under conditions that mimic the human FE studies. These were conducted in a crossover design, two treatment (fasted and fed), in 4 animals. Male Beagle dogs (~10 kg) were fasted overnight with no intake of water 1 h before and after dosing. Pentagastrin (6 μg/kg, intramuscular) was administered 30 min prior to drug dose to control gastric pH in the range pH 1–3 (15). The fasted-state gastric pH has been reported to span a wide range between 1.6 and 8.5 and can vary between dog colonies (15,21,22). To reduce this inherent gastric pH variability and control it in a range more typical of human stomach pH, pentagastrin, a synthetic peptide that simulates the secretion of gastric acid secretion in a dose and time-dependent manner in dog and man was used (15,23). To maintain consistency of study design, mimic the human gastric pH, and improve overall predictability of the dog FE model as suggested by Lentz, 2008 (14), dogs were pretreated with pentagastrin in the fed state too. The oral drug dose was followed immediately by oral gavage with up to 50 mL water (fasted state) or 50 mL of a high-fat meal supplement (fed state). The high-fat meal supplement contains Boost® drink supplemented with Coffeemate® powder at neutral pH. The meal provides 100 calories/dog and represents the same caloric breakdown of a high-fat American breakfast (roughly 60% calories from fat, 25% from carbohydrate, and 15% from protein). This high-fat meal supplement provides a commercially sourced, practical, consistent meal type, in an easy to dose form. The composition of the meal being the primary difference between this study and that by Lentz et al. (14) that used homogenized American breakfast. Blood samples (2 mL) were withdrawn from the cephalic vein at predetermined time points over 24 h and placed in EDTA-containing Vacutainer blood collection tubes. Samples were centrifuged at 5000 rpm for 10 min at 4°C. The supernatant plasma was harvested and stored at −80°C until HPLC-LC/MS analysis. Studies were performed in accordance with the standards recommended by the Guide for Care and Use for Laboratory Animals (Institute of Animal Laboratory Resources, 1995) and were approved by the institutional animal care use committee with full consideration to experimental refinement, reduction in animal use, and replacement with in vitro methods.
Dose selection for dog FE studies and human studies may differ. A dog FE study is typically conducted well before a clinical FE study. In an ideal scenario, the dog dose would be scaled to the human FE dose, or the same dose/dosage form is used that is tested in the human. However, this is rarely the case in drug development practice. Human FE studies are conducted based on Phase 1 trial PK data which may be different from the dose originally projected, leading to a dose discrepancy between dog and human. High dose in dogs usually represents the use of the human dosage form, i.e., FE risk assessment for the worst case scenario. Low dog doses are used in instances when emesis (especially for oncology drugs) or other adverse events can confound PK data interpretation.
Reporting of Clinical Food-Effect Results
Human FE is represented as a ratio of mean pharmacokinetic parameters (AUC or Cmax) between the fed state and fasted state.
A ratio of 1 signifies no food effect, values >1 indicating a trend towards a positive FE, and value <1 indicating a trend towards a negative FE. Typically, these are conducted as crossover studies where subjects get the test drug under fasted and fed conditions (e.g., a high-fat American breakfast meal), as per FDA guidance recommendations. Variations to the meal are sometimes considered based on the caloric content (standard or light meal) and/or breakdown of the food groups. However, in this study, these parameters were not explored. Food can influence the rate of absorption (through altered Cmax and Tmax) and extent of absorption (AUC). As AUC is the most commonly used PK parameter of overall FE (7,8,14), and the fact that food induced changes to the rate of absorption may not reflect a change in extent of absorption (2,24), AUC is used as the primary parameter in this article.
The correlation of in vitro dissolution to human FE assumes dissolution-precipitation behavior of drugs under the fed- and fasted-state conditions is the predominant driver for the food effect. Other mechanisms such as physiological states altered by food (blood flow, gastric motility/residence time, competition of cytochrome-mediated metabolism, influx or efflux by food components, etc., are possible but are difficult to prove from this correlation analysis. Certain food components can affect metabolizing enzyme pathways and transporters in the gut surface through competition with API (2,24). Additionally, food co-administration can influence hepatic first-pass (18,25). Such interactions are not the focus of this article.
Statistics and Trend Analysis
The linear regression analysis was conducted on Microsoft Excel. The regression was represented as the correlation coefficient (R2) for the line equation y = ax + b.
The root mean square error (RMSE) was estimated as follows:
where Xobs is observed values and Xmodel is modeled values for a particular value of i.
The normalized RMSE value in the IVIVR is estimated as follows:
where the denominator, Xobs,max − Xobs,min indicates the range of the observed data.
Data tables were visualized for correlations using Spotfire software (Tibco Spotfire, Somerville, MA). Graphs were plotted using SigmaPlot (Systat Software Inc, San Jose, CA).
RESULTS
In Vitro Dissolution Studies to Predict Food Effect in Humans
The in vitro dissolution model is a convenient way to obtain real-time kinetic dissolution behavior for an API in FaSSIF and FeSSIF. Figure 1, illustrates the concentration-time profile of a model drug that shows a strong positive FE. The ratio of the area under the curve for FeSSIF:FaSSIF dissolution (the in vitro FE ratio = 3.7) reflects the severity of the risk. The concentration-time profiles themselves provide useful insights into the kinetics of dissolution/precipitation behavior in the GI lumen. For example in Fig. 2, BMS-C dissolution in FaSSIF has a rate of dissolution (4 μg/mL/min) that is about half that observed in FeSSIF. While the dissolution curves merge achieving the same plateau concentration, the significantly slower initial dissolution rate may be attributed to poor wetting of BMS-C in FaSSIF that could impact it’s absorption over 0–3 h.
Fig. 1.
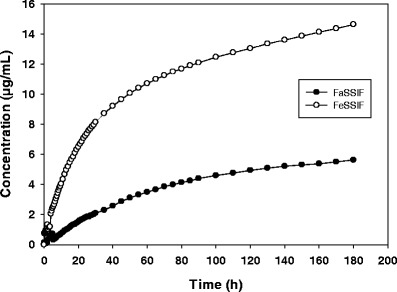
In vitro dissolution profile of a model positive FE compound BMS-P in FaSSIF and FeSSIF. Data shown is the mean profile, n = 3–4
Fig. 2.
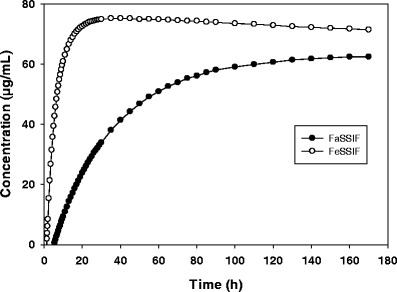
In vitro dissolution profile of BMS-C in FaSSIF and FeSSIF. Data shown is the mean dissolution profile, n = 3–4
The FE ratio for in vitro FE and human FE for 20 compounds is summarized in Table I. The in vitro dissolution FE ratio correlated with the human FE ratio with an apparent linear relationship (Fig. 3a, b). Triclabendazole (the point furthest to the right marked in red) has an unusually high in vitro FE ratio of 10, significantly extending the upper range on the x-axis (Fig. 3a). With triclabendazole included, the correlation equation is: y = 0.35x + 1.04, with a correlation coefficient R2 = 0.53; root mean square error RMSE = 0.6, and %NRMSE = 19.2%. When triclabendazole is excluded as an outlier (Fig. 3b), the correlation improves significantly, R2 = 0.66, RMSE = 0.53, and %NRMSE = 20.7%. The regression equation is: y = 0.74x + 0.42
Fig. 3.
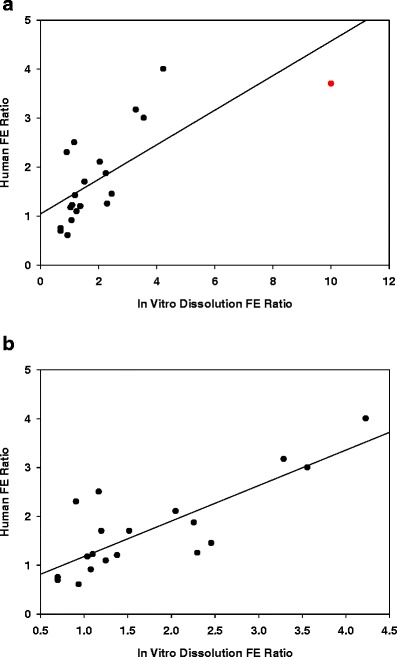
In vitro-in vivo relationship for predicting human FE for 20 compounds (Fig. 3a) and excluding triclabendazole (Fig. 3b)
This IVIVR assumes that API dissolution in FaSSIF and FeSSIF is the primary mechanism driving the observed human FE. As dissolution has the greatest impact on Cmax, we also examined the correlation of in vitro FE ratio with the human Cmax ratio. Curiously, the correlation coefficient was reduced R2 = 0.53, the RMSE increased to 0.91 and the % NRMSE to 27.4% (excluding triclabendazole). Correlating the in vitro dissolution peak dissolution ratio with the clinical Cmax FE ratio, the correlation coefficient was 0.48 (data not shown). Since total exposure was the most pertinent parameter in driving pharmacokinetic-pharmacodynamic relationships for most of the drugs studied, the AUC-based correlation was considered the most representative for further discussion in this article.
Table II lists the equilibrium FaSSIF and FeSSIF solubility along with the peak solubility from the kinetic dissolution for 9 compounds (7 positive FE and 2 no FE). The comparison between the equilibrium and kinetic solubility values highlights the supersaturation potential within the 3 h duration of the dissolution experiment. Using the same qualitative FE risk categories for human FE ratio (described in Table III): positive FE >1.2; no FE 0.8–1.2; negative FE <0.8; the propensity of equilibrium solubility to correctly predict the clinical FE ratio was 4 out of 9 compounds, while using kinetic dissolution peak values, the score was 8 out of 9 compounds.
Table II.
Equilibrium and Peak Kinetic Solubility (mg/mL) in FaSSIF and FeSSIF
| Drug | Eq. FaSSIF sol. | Eq. FeSSIF sol. | Eq. FE ratio | Kinetic FaSSIF sol. | Kinetic FeSSIF sol. | Kinetic FE ratio | Human FE ratio | Correlates to human FE ratio | |
|---|---|---|---|---|---|---|---|---|---|
| Eq. sol. ratio | Kinetic sol. ratio | ||||||||
| BMS-B | 0.013 | 0.018 | 1.38 | 0.0115 | 0.0378 | 3.29 | 3.17 | Yes | Yes |
| BMS-C | 0.11 | 0.07 | 0.64 | 0.05 | 0.075 | 1.5 | 2.1 | No | Yes |
| BMS-D | 0.037 | 0.05 | 1.35 | 0.079 | 0.109 | 1.38 | 1.7 | Yes | Yes |
| BMS-E | 0.031 | 0.033 | 1.06 | 0.0479 | 0.0589 | 1.23 | 2.5 | No | Yes |
| BMS-M | 0.007 | 0.022 | 2.8 | 0.009 | 0.024 | 2.67 | 1.87 | Yes | Yes |
| BMS-N | 0.046 | 0.04 | 0.87 | 0.0463 | 0.0405 | 0.87 | 2.3 | No | No |
| BMS-P | 0.0002 | 0.003 | 15 | 0.01 | 0.059 | 5.9 | 3.7 | Yes | Yes |
| BMS-G | 0.023 | 0.002 | 0.08 | 0.105 | 0.108 | 1.03 | 1.2 | No | Yes |
| BMS-J | 0.095 | 0.892 | 9.39 | 0.036 | 0.0426 | 1.18 | 1.2 | No | yes |
Qualitative assessment of correlation to human FE based on human FE ratio categorization of positive FE (ratio >1.2); no FE (ratio 0.8–1.2); negative FE (ratio <0.8) as described in Table III
Table III.
In Vitro, In Vivo, and Human Correlations for FE risk
| In Vitro dissolution FE ratio | Dog PK FE ratio | Human PK FE ratio |
|
|---|---|---|---|
| Positive FE risk | >1.5 (9) | >1.5 (8) | >1.2 (11) |
| No FE risk | 0.8–1.5 (9) | 0.8–1.5 (5) | 0.8–1.2 (8) |
| Negative FE risk | <0.8 (2) | <0.8 (2) | <0.8 (3) |
Parenthesis denotes the number of compounds that fall in each category
Dog-Human FE Correlation
The FE ratio in the dog was compared with the clinical FE ratio for 15 compounds with known sensitivity to food. In Fig. 4a, b, dog FE ratio correlated linearly with the human FE ratio. With the omission of one outlier, BMS-G, the correlation coefficient was 0.74, the RMSE was 0.65, and %NRMSE was 16.25% (Fig. 4b). The linear correlation is described by the equation: y = 0.62x + 0.44. When BMS-G marked in red was included in the IVIVR (Fig. 4a), the equation changed to y = 0.38x + 0.78, R2 = 0.41. The Cmax-based correlations excluding BMS-G trended in a similar manner with the R2 = 0.71, RMSE 0.75, and the %normalized error 22.57% (data not shown). As mentioned before, AUC was the PK parameter of greatest relevance.
Fig. 4.
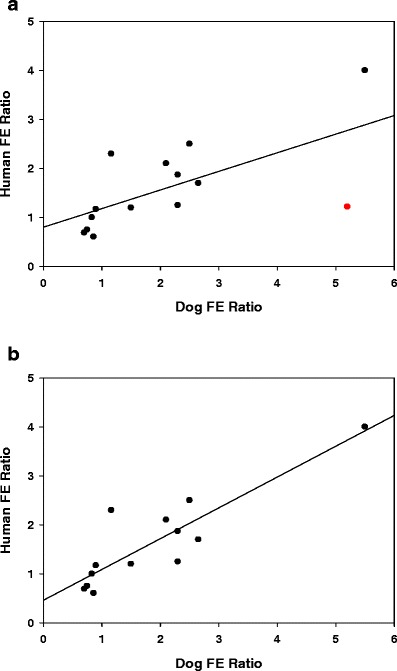
Dog-clinical FE relationship for predicting human FE for 15 compounds (Fig. 4a) and excluding the outlier, BMS-G (Fig. 4b)
Log P—Clinical FE Correlation
The correlation of a drug’s lipophilicity to its propensity to show a FE was explored. As seen in Fig. 5, the trend towards a relationship is aligned with the BCS class (marked by unique colors/symbols). Generally, the low log P drugs (log P<2) show a greater chance for negative FE or no FE. This group typically comprises, BCS class 3 compounds (red circles). BCS-2 compounds (filled squares) with a log P ≥ 2 appear to be scattered over a wide range of clinical FE ratio from 1.2–3.7. The dose, type of drug (free form or salt), type of dosage form, formulation excipients, and mechanism of FE all play a major role in determining the extent (2,3). BCS-4 drugs (open triangles) show no discernible trend, although only 2 compounds in this data set fall in this category.
Fig. 5.
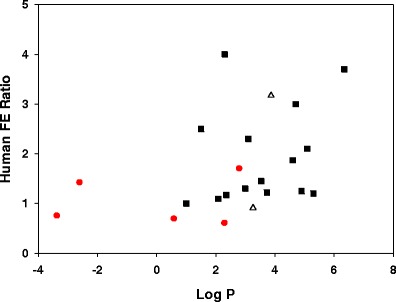
Plot of Log P with the human FE ratio for 20 compounds. Data points are colored to indicate the BCS class: BCS-2 (filled squares), BCS-3 (red circles), BCS-4 (open triangles)
DISCUSSION
With the broad use of in vitro dissolution models and in vivo PK models to predict performance in the human GI tract (4,5) and their general acceptance with regulatory agencies, it has become increasingly important to understand the relationship of data from these models to the clinical experience. Factoring the ethics of animal experimentation in the context of the 3Rs (Replacement, Reduction, and Refinement) to minimize animal use, in vitro dissolution is often prioritized as a screening tool to assess biopharmaceutical performance risks such as food effect. In this retrospective study, we attempt to consolidate learning’s from biorelevant in vitro dissolution and in vivo dog FE PK studies and their correlation to the human FE for 22 drugs, emphasizing the mechanistic approach to study the underlying reason for the FE risk.
In Vitro Dissolution
The kinetic dissolution profile in FaSSIF and FeSSIF can inform the propensity of APIs to wet, dissolve and remain is solution through a typical absorption phase. This is more so for lipophilic, poor wetting, slow dissolving drugs. For example, the BMS-P dissolution profile (Fig. 1) is slow in FaSSIF reaching 6 μg/mL in 3 h, but faster in FeSSIF reaching 15 μg/mL. As with the API, the clinical tablet also showed <5% dissolution in 1 h in pH 1.2 SGF (data not shown). The slow dissolution likely contributed to 25.5 h median Tmax in the fasted state, while the improved dissolution rate and extent in fed state aided faster absorption with a median Tmax of 5 h. BMS-C also exhibits differential dissolution characteristics in FaSSIF and FeSSIF (Fig. 2), but reaches a reasonably high solubility plateau of about 70 μg/mL in both fluids within 2 h. Accordingly, the clinical Tmax difference in the fed state vs. fasted state was nominal (data not shown).
The importance of dissolution kinetics is evident for positive FE compounds with the tendency to supersaturate through bile and phospholipid mediated solubilization in FaSSIF and FeSSIF. As supersaturation is a transient state, the peak kinetic solubility can be higher than the corresponding equilibrium solubility (Table II). By definition, equilibrium solubility is a single point solubility measurement taken at 24–48 h when solid is in equilibrium with the fluid media. Consequently, any changes in the dissolution profile are missed prior to the equilibrium. Lower equilibrium solubility values are usually a product of a transitioned API physical state to a form with reduced solubility, which can be confirmed through X-ray diffraction, Raman spectroscopy, and NMR analysis. Supersaturation provides the driving force for passive absorption; hence, those compounds with a greater tendency to supersaturate are more likely to have kinetic solubility rather than equilibrium solubility represent the human situation. In Table II, the equilibrium FeSSIF/FaSSIF solubility ratio qualitatively indicates that 4 out of 9 compounds are predictive of the human FE ratio, scored by correlating to the human FE range categories listed in Table III: positive FE (ratio >1.2), no FE (ratio 0.8–1.2), and negative FE (ratio <0.8). Using the same criteria, the peak kinetic dissolution solubility identifies 8 out of 9 to be reflective of the human FE ratio which underscores the value of kinetic biorelevant dissolution in making correlations to the dynamic nature of drug absorption process in vivo.
In Vitro-In Vivo Relationship (IVIVR)
For a diverse set of 22 compounds comprising of proprietary development compounds and commercially available drugs (Table I), the in vitro dissolution model’s measure of FE risk (the FeSSIF:FaSSIF AUC ratio) correlated linearly with the human FE ratio (fed:fasted AUC ratio) with a correlation coefficient of 0.66 (Fig. 3b). The root mean square error (RMSE) and % normalized error of 0.53 and 20.7%, respectively, suggest that the in vitro FE ratio is a reasonable predictor of the human FE ratio. This correlation analysis treats triclabendazole, a drug with an unusually high in vitro FE ratio of 10, to be an outlier. The data set covers human FE ratio that ranges from 0.6 to 3.7. These comprise of 3 negative FE drugs (FE ratio <0.8), 6 no FE drugs (FE ratio 0.8–1.2), and 11 positive FE drugs (>1.2). Using the same FE ratio definitions, the in vitro dissolution qualitatively pointed out 2 with negative FE, 7 with no FE, and 11 with positive FE. The mean in vitro dissolution FE ratio for the no FE group was 1.4, while human FE data from the same group was 1.13. With this in mind, we consider the following guide to bin the FE risk into 3 categories based on the in vitro dissolution FE model (Table III): Negative FE (FE ratio <0.8), no FE (FE ratio 0.8–1.5), and positive FE (FE ratio >1.5), with the severity of the positive FE risk increasing as the value of the FE ratio increases.
The slope of the correlation curve is 0.74 (excluding the outlier), lower than one-to-one correlation where the slope approaches unity. Consequently, the IVIVR tends to skew the correlation towards in vitro dissolution stretching (over-estimating) the magnitude of the FE risk. There are 2 possible reasons for this: the in vitro dissolution was conducted with API powders, while clinical studies are conducted with formulated dosage forms; and secondly, the lack of sink conditions for dissolution and drug removal can exaggerate dissolution differences between the fasted and fed state. Triclabendazole stands out with an exceedingly high in vitro FE ratio of 10 that is not matched in the human (FE ratio 3.7). Its FE mechanism has been reported to be due to significant solubilization in fed state (26). Triclabendazole is a lipophilic (log P = 6.34), low aqueous solubility drug (0.2 μg/mL in phosphate buffer), with a >20-fold higher solubility in FeSSIF compared to FaSSIF. It preferentially form colloids in FeSSIF (27), possibly aggregating in the presence of bile salts/phospholipid to supersaturate much like other antifungal drugs (itraconazole, albendazole, mebendazole) also known to show strong FEs (26). In humans, triclabendazole undergoes extensive and rapid first-pass metabolism to its active sulphonated metabolites which can confound FE ratio of the parent drug (26).
From a mechanistic point of view, there are a couple of reasons why a linear correlation (Fig. 3b) is surprising: first, it suggests that a simple biorelevant kinetic dissolution method can be reasonably predictive of the human FE risk for a chemically diverse set of compounds. Secondly, the correlation suggests that dissolution/precipitation in the fasted and fed state (the assumed premise from in vitro dissolution methodology) is the predominant mechanism for the observed food effect for many of the APIs tested. In other words, most of the APIs that fall on or near the correlation line exhibit a FE that is primarily driven by dissolution/precipitation in the fed and fasted state. APIs that fall significantly above the correlation line have a higher human FE ratio, which suggests that physiological mechanisms such as prolonged window of absorption (due to slowed GI motility in the fed state), increased splanchnic blood flow, food-induced competition, or inhibition of transporters/enzymes play a role that is added to the role of solubilization.
APIs that fall significantly below the correlation line (Fig. 3b) over-estimate the role of dissolution which is not represented to the same degree in clinical PK data. In this scenario, dissolution method artifacts more so than events in the human GI tract are possible, for example, FaSSIF dissolution that is uncharacteristically poor (lower baseline) or bile-induced supersaturation in FeSSIF that is surprisingly high. The former can be alluded to factors like poor powder wetting, a feature of unfavorable surface properties, and/or particle size effects (aggregation of hydrophobic particles), both of which manifest as floating powder on the top of the dissolution media in the vial. Paying close attention to the in vitro dissolution profiles and visual observation on API wetting can flag such unusual behavior. BMS-O is an example of a poor wetted API that shows unexpectedly low dissolution in FaSSIF, hence, an in vitro FE ratio of 2.3 while the human FE ratio is 1.2. Similarly, nelfinavir dissolution was reported to be uncharacteristically low in FaSSIF which over-stated the FE risk, a disconnection from in vivo performance (28).
On the flip side, bile salt in FeSSIF can induce supersaturation that is unexpectedly high. In humans, the fed-state GI lumen may have a dynamic, viscous, partially digested meal that can disrupt the supersaturated solubility of drugs perhaps by triggering nucleation and/or particle growth which in turn reduces the extent of FE (29). Consistent with this scenario, Shono et al. (30) found the biorelevant dissolution over-predicted the FE risk for celecoxib by 7-fold, while the human FE ratio was only 1.3. Similarly, Bevernage et al. (31) reported significantly higher FeSSIF dissolution for several compound compared to dissolution in GI aspirates under different nutritional states. Taken together, even though the dissolution studies were performed using API while clinical studies used formulated dosage forms, in vitro dissolution correlates well with human FE ratio, therefore, can be an effective screen to flag the FE risk potential of new NCEs, as long as careful analysis of the dissolution profile is conducted to rule out methodology-related over/under-estimation of the risk level.
Dog-to-Human FE Prediction
Our experience with the dog FE model is in full agreement with previously published reports that the beagle dog model is an effective model to study the risk of FE in humans (14,15,32). We found that dogs dosed with the high-fat meal supplement (Boost® with Coffeemate®) predict the whole range of FEs, resulting in an IVIVR that correlates well with human FE ratio (correlation coefficient = 0.74, RMSE = 0.5, and % error = 12.5%). One distinct outlier (BMS-G) was apparent from this group of 15 compounds. Its inclusion dropped the correlation coefficient to 0.4; therefore, it was omitted from the regression analysis. The uniqueness of BMS-G is discussed in the mechanism section below.
Drugs that fall on or near the correlation curve suggest that there may be common mechanisms in both human and dog species responsible for the FE. Data points that stray from the correlation line indicate deviations in the primary FE mechanistic drivers. These are likely to stem from differences in physiology that are exemplified for certain drugs. For example, in dogs, drugs that are susceptible to a narrow window of absorption tend to show enhanced absorption in the fed state as longer gastric emptying compared to humans prolongs the window of absorption (21). BMS-G and BMS-P are examples of drugs that show such distinctive behavior in dogs with FE ratios of 5.2 and 5.5. The corresponding human FE ratios are 1.2 and 3.7, respectively. Consequently, BMS-G benefits from delayed gastric emptying in the dog fed state that enhances its overall bioavailability and FE ratio. Another possible scenario for mechanistic differences stems from fed-state gastric pH being 3–4 units lower than in humans (21). Ionizable weak base drug, such as BMS-D that show strong pH-dependent dissolution at low fed-state gastric pH drives up the exposure and FE ratio to 2.65 compared to human FE ratio of 1.7.
In this study, we found that the dog FE ratio serves as a useful semi-quantitative gauge of the human FE risk. We consider the following guide to empirically bin the FE risk into three categories: positive FE, no FE, and negative FE (Table III). An FE ratio <0.8 represents negative FE, 0.8–1.5 represents the range for no FE, and values >1.5 represent positive FE. The range for each category is similar to that from the in vitro dissolution-IVIVR (Table III). The slope of the dog-to-human FE IVIVR is 0.62, a value close to the in vitro dissolution-human FE ratio IVIVR slope of 0.74 which suggests both models have more discriminatory power having slopes <1, a preferred attribute for an experimental model system.
Despite the physiological and biochemical differences between dogs and human GI tract, the dog model is well accepted as a reasonable surrogate model to predict human performance (5,14,15,22,33). Correlation analyses have shown it to be practical to study both APIs and formulations (33,34). Sutton (33) reported a good linear correlation between the relative bioavailability of 11 compounds administered to dogs and human colon in the fasted state. Comparing 9 drugs in the dog FE model, Lentz (14,34) noted that the model predicts PK changes that are qualitatively reflective of the change observed in humans, although the magnitude of the effect can be hard to replicate/predict. Similarly, Zane et al. (15) found that for 4 BCS-II drugs, the dog FE model is predictive of humans, except between the FE ratio range 0.5–2. The inability to discriminate the extent of human FE in this range was attributed to inherent PK variability with dog studies powered with small sample size (n = 4–6), difference in the diet, and/or differences in GI physiology. Our observations are similar to these studies, wherein, the differences contribute to increased dog PK variability. The expanded “no FE” range of 0.8–1.5 compared to the corresponding range in humans (0.8–1.2) is required to accommodate this PK variability (Table III).
Recognizing the differences between the dog model and human can be useful to help explain unique behavior and why the dog model can trend towards over-estimating the positive FE ratio and diminish the extent of a negative FE. An observation consistent with that noted by Lentz et al. (14) and Zane et al. (15). The beagle dog has a gastrointestinal physiology that is similar to the human for the most part (22,32). In the fasted state, two major differences being high resting gastric pH and the shorter GI transit time relative to the human GI tract. In the fed state, the key differences are 3–4 unit lower gastric pH, slower gastric emptying, greater mechanical forces, and shorter intestinal transit (5,21). In literature, there are conflicting reports of canine GI physiology (resting pH, fed-state pH, transit time) which can be dependent on the dog colony (21,32). Additionally, the method of studying the fed state, i.e., type of meal (dog chow, homogenized American breakfast, or high-fat meal supplement), and the meal amount which affects drug diffusivity in the GI milieu are other variables that can come into play (5,21,34). In our hands, the high-fat meal supplement provides commercially available components in a consistent, reproducible, and easy to dose meal type that simulates the caloric breakdown of a human high-fat meal. Administered as a liquid via oral gavage immediately after drug dosing, it provides a realistic volume (50 mL) of fed-state media for the drug to go through dissolution/solubilization process in the GI lumen while allowing a direct comparison with 50 mL water in the fasted state. Using pentagastrin pre-treatment helps control dog gastric pH in a range that is more representative of the human gastric condition, minimizing the PK variability for pH-sensitive drugs. While not the focus of this article, it is possible to further assess meal-based variations, such as light meal vs. standard meal, or low vs. high-fat meal by altering the meal volume or the amount of fat component, respectively.
Case Studies on FE Mechanisms
BMS-G is a typical lipophilic, BCS-II drug with very low aqueous solubility (<1 μg/mL), which exhibits solubility-limited absorption in humans (35). The contact angle (θ), a measure of powder wetting property, is 130°, highlighting the poor wetting nature of the powder. However, bile salt aided dissolution in FaSSIF and FeSSIF was fairly rapid, reaching a plateau concentration ~100 μg/mL within 5–10 min. While the dissolution rate in FaSSIF was half that in FeSSIF, the overall in vitro FE risk was only 1.1 which was in agreement with clinical FE ratio of 1.2. The dog model predicted a FE ratio of 5.2 that was surprisingly high for the same capsule formulation as used in clinical studies. Therefore, lack of consistent wetting to initiate dissolution in vivo can be ruled out. We hypothesize that the high FE in the dog may be related to the fed-state dog physiology. In dogs, the potential for longer gastric residence in the fed state compared to humans (21,22) may prolong the window of absorption leading to improved absorption in the upper GI tract.
For this dataset, the propensity to elicit a FE based on the BCS class is consistent with previously published FE studies (10,18,25,34). BCS-I drugs typically dissolve fully and are well absorbed, thus not likely to show a FE. BCS-2 drugs tend to show a broad range of positive FEs by virtue of their solubility/dissolution-limited absorption. Permeability limited BCS-3 drugs trend towards a negative FE due to bile micellar/food complexation or transporter interaction. While BCS-4 drugs are erratic and the most challenging to predict due to confounding transporter and/or metabolism interactions (25). Shown in Fig. 5, the BCS classes trend in a similar fashion with respect to the FE ratio category. The BCS-2 group (filled squares) is the most intriguing as they typically have log P ≥ 2.0 and exhibit a wide range of FEs in human from 1.08 to 3.7. For instance, danazol, triclabendazole, BMS-B, and BMS-P all show a strong positive human FE ratio >3, where bile-mediated solubilization or improved dissolution/solubility in the fed state drives the overall FE ratio. The in vitro dissolution FE ratios were high for each of these drugs. On the other hand, there are BCS-2 drugs such as BMS-A, BMS-O, BMS-K, BMS-Q, and celecoxib that show a modest or no FE. Clearly, the dose of the drug is a critical factor. If the dose is solubilized in both food states, the FE ratio is likely to stay close to 1. When high dose drugs are not fully dissolved, solubilization in the meal can push the FE ratio towards strong positive FE. An example of this scenario is BMS-C (log P = 5.09), at a dose of 200 mg, the clinical FE ratio was 2.1, but as the dose increases, the human FE ratio increased up to 12 at 600 mg (36). BMS-J stands out as a low dose, lipophilic drug (log P 5.3), but with no appreciable clinical FE. It’s dissolution in both FaSSIF and FeSSIF was moderately high; therefore, in vitro dissolution correctly predicted no significant FE. Some lipophilic drugs do not benefit from bile micellar solubilization, perhaps due to sequestration in bile micelles or binding to bile salts. This scenario where bile can potentially limit the availability of free drug for absorption at the intestinal membrane surface was shown by Kawai et al. (37) when analyzing 40 approved drugs.
BCS-3 compounds (red circles) tend to be hydrophilic compounds with log P < 2 with no FE or a negative FE (Fig. 5). BMS-F and BMS-H are examples of such compounds where it is generally thought that the physical entrapment of the drug in the viscous, post-prandial GI fluid milieu leads to unavailability of free drug for absorption through the paracellular junctions (29). Kawai et al. (37) reported that hydrophilic drugs that dissolve rapidly are more likely to be limited by their permeation rates due to physical interaction with food components in the fed state. In this study, both BMS-F and BMS-H show rapid dissolution, although, the reason for reduced dissolution in FeSSIF was not investigated further. Heimbach et al. (10) reported a BCS-3 drug that showed a negative FE with a drop near 70% for both AUC and Cmax. While the drug was considered an OATP2B1 influx transporter substrate, they suggested that food components competed for transporter-based absorption reducing the systemic fed-state PK.
The correlation of human FE to log P (Fig. 5) is similar to the correlation reported by Singh (8) when statistically analyzing the physicochemical properties of 100 oral administered compounds. In that study, compounds with Log P > 2 were spread across a wide range of human FE AUC ratio from 0.5 to about 5 (8). Compounds with a Log P ≥ 3 were more likely to benefit from increased bioavailability with food. It is possible that this wide range for high log P compounds is linked to susceptibility of cytochrome P450 metabolism as suggested by Benet et al. (18). Examining 927 drugs, they found that the probability of extensive metabolism being about 80% when Log P > 2 and only about 25% when Log P < 1 (18). On similar lines, Gu et al. (7) studying >90 compounds found that compounds with Log D < 1 are statistically prone to show a negative FE and those with Log D > 1 show no FE. For each of these studies and our study, the trends in the data set do not apply to all drugs as exceptions exist. Probing the mechanism behind the unusual behavior can be deduced from in vitro dissolution, animal PK, or dissolution-absorption modeling.
In silico modeling is a well established tool to predict human FE and has been extensively published (9–11,36). An example to illustrate dissolution-absorption modeling to investigate the primary mechanism for a strong positive FE for BMS-C has been reported previously (36). Using GastroPlus™ software (Simulations Plus, Lancaster, CA), a retrospective analysis was conducted to probe possible reason for the strong positive FE based on the clinical oral plasma time course data, as well as the experimental in vitro and dog FE model data. Figure 6 shows the model simulated plasma profiles (the line) in relation to the clinical data points (symbols) when 600 mg was dosed in the fasted state in the morning and 12 h later re-dosed after a high-fat meal. The high-fat meal dramatically increased the AUC and Cmax by 12-fold. The equilibrium FaSSIF solubility was reasonably high at 0.11 mg/mL, 0.07 mg/mL in FeSSIF (Table II), and the overall dissolution FE ratio predicted a modest FE ratio of 2.05 at 200 mg dose. To experimentally simulate the high-fat environment solubilization for the high dose 10% dietary lipid (Microlipid) was added to FeSSIF which resulted in a 10-fold increase in dissolution/solubility. This enhanced solubility helped establish a better model fit for the high-fat meal clinical FE at 600 mg dose. Overall, BMS-C’s FE mechanism was driven by solubilization in high-fat meal, aided further by the delayed fed-state gastric emptying that prolongs the window of absorption (36). Using this knowledge, FE mitigation strategies such as the inclusion of surfactants and lipid formulation additives were found to show improved bioavailability in the fasted state (38).
Fig. 6.
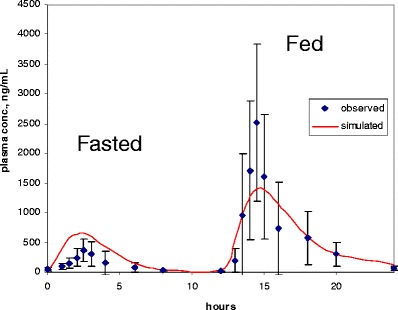
Plot of GastroPlus simulated profile and observed clinical data of BMS-C in the fasted and fed state
In summary, in vitro dissolution presents a preliminary, albeit critical, risk severity indicating clue to the mechanism of FE. Evidence of deviations from dissolution-controlled FE can be teased out with dog FE studies. Finally, in silico modeling built on the in vitro dissolution and in vivo PK data can be further used to diagnose the potential role of physiological mechanisms such as, delayed gastric emptying, altered gastric pH, the role of transporters/enzymes, or reduced availability of free drug in the meal.
Limitations of the In Vitro Models and Challenges
The most important assumption with the in vitro microdissolution studies is that dissolution is rate-limiting step and that altered dissolution in FaSSIF or FeSSIF is the reason for the observed in vitro FE. Therefore, it is likely to work best to predict the FE risk for solubility/dissolution-limited BCS-II compounds. In most cases, BCS-III drugs show high solubility in both FaSSIF and FeSSIF, trending towards no FE, except in specific cases where complexation to bile acids is observed. Predicting FE for BCS-IV drugs through in vitro dissolution is particularly challenging being confounded by transporter interaction or extensive metabolism (18,19). The microdissolution method is a small volume, non-sink assay; therefore, dissolved drug is not removed as is the fraction of drug absorbed in the GI lumen. This influences supersaturation and precipitation kinetics. Additionally, this test does not replicate the dosage form disintegration and excipient effects on dissolution rate, supersaturation maintenance, and precipitation kinetics, although, it is possible to rank-order dosage form formulation strategies as granulations, dispersions, and crushed tablets. The microdissolution is designed to provide an API-sparing and rapid method to assess the FE-risk, while capturing the kinetics of API supersaturation and precipitation in the fasted and fed state. When large FE ratio is observed, it is prudent to probe the underlying root cause. Unreasonably low dissolution in FaSSIF due to poor wetting should be ruled out, or on the flip side, unreasonably high bile micellar solubilization in FeSSIF should be carefully questioned. The former can be diagnosed by analytical techniques such as contact angle measurement for drug wettability. The latter can be tested for sequestration in bile, a situation for high log P drugs that show good bile micelle solubilization, but poor absorption at the epithelial surface due to unavailability of free drug (6,37).
CONCLUSION
The in vitro dissolution model and the dog FE model’s reported here correlate well with clinical FE ratio for a wide range of drugs, conferring confidence in these experimental tools to predict the human performance. We recommend the in vitro dissolution as an initial screening tool to get a preliminary read on risk severity, followed by a dog FE model to confirm the liability for high risk NCEs. Data from these models can be used as inputs in in silico dissolution-absorption modeling to simulate the FE risk in humans and test for scenarios such as sensitivity to dose, API properties (salts or free forms, particle size), and sensitivity to improved solubilization in the fasted and fed state. Conceivably, the combined use of in vitro, in vivo, and in silico tools facilitates the pharmaceutical scientist decision process for the FE risk identification and mitigation. This knowledge is useful to guide clinical FE trial planning and design and ultimately influence the prescribing information for a NCE, instructing patients and healthcare practitioners on dose administration with or without regard to food.
REFERENCES
- 1.US Food and Drug Administration. Food-effect bioavailability and fed bioequivalence studies: guidance for industry. http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf. 2002.
- 2.Fleisher D, Li C, Zhou Y, Pao LH, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clin Pharmacokinet. 1999;36:233–254. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 3.Charman WN, Porter C, Methani S, Dressman JB. Physicochemical and physiological mechanisms for the effects of food on drug absorption: the role of pH and lipids. J Pharm Sci. 1997;86:269–282. doi: 10.1021/js960085v. [DOI] [PubMed] [Google Scholar]
- 4.Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–366. doi: 10.1016/j.ejps.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 5.Sjögren E, Abrahamsson B, Augustijns P, Becker D, Bolger MB, Brewster M, et al. In vivo methods for drug absorption—comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur J Pharm Sci. 2014;57:99–151. doi: 10.1016/j.ejps.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Sugano K, Kataoka M, Mathews C, Yamashita S. Prediction of food effect by bile micelles on oral drug absorption considering free fraction in the intestinal fluid. Eur J Pharm Sci. 2010;40:118–124. doi: 10.1016/j.ejps.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Gu CH, Li H, Levons J, Lentz K, Gandhi RB, Raghavan K, et al. Predicting effect of food on the extent of drug absorption based on physicochemical properties. Pharm Res. 2007;24:1118–1130. doi: 10.1007/s11095-007-9236-1. [DOI] [PubMed] [Google Scholar]
- 8.Singh BN. A quantitative approach to probe the dependence and correlation of food-effect with aqueous solubility, dose/solubility ratio and partition coefficient (Log P) for orally active drugs administered as immediate release formulations. Drug Dev Res. 2005;65:55–75. doi: 10.1002/ddr.20008. [DOI] [Google Scholar]
- 9.Parrott N, Lukava V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drug using physiologically based modeling: application to food effects. AAPS J. 2009;11:45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heimbach T, Binfeng X, Liu T, He H. Case studies for practical food effect assessment across BCS/BDDCS Class compounds using in silico, in vitro and preclinical in vivo data. AAPS J. 2012;15:143–158. doi: 10.1208/s12248-012-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:300–351. doi: 10.1016/j.ejps.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Dressman JB, Reppas C. In vitro-in vivo correlation for lipophilic, poorly soluble drugs. Eur J Pharm Sci. 2000;11:S73–S80. doi: 10.1016/S0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 13.Katoaka M, Itsubata S, Masaoka Y, Sakuma S, Yamashita S. In vitro dissolution/permeability system to predict the oral absorption of poorly water soluble drugs: effect of food and dose strength on it. Biol Pharm Bull. 2011;34:401–407. doi: 10.1248/bpb.34.401. [DOI] [PubMed] [Google Scholar]
- 14.Lentz K, Quitko M, Morgan DG, Grace JE, Gleason C, Marathe PH. Development and validation of a preclinical food effect model. J Pharm Sci. 2007;96:459–472. doi: 10.1002/jps.20767. [DOI] [PubMed] [Google Scholar]
- 15.Zane P, Guo Z, MacGerorge D, Vicat P, Ollier C. Use of the pentagastrin dog model to explore the food effects on formulations in early drug development. Eur J Pharm Sci. 2014;57:207–213. doi: 10.1016/j.ejps.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Balimane P, Han YH, Chong S. Current industrial practices in assessing permeability and P-glycoprotein interactions. AAPS J. 2006;8:E1–13. doi: 10.1208/aapsj080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lennernas H. Human jejunal effective permeability and its correlation with preclinical drug absorption models. J Pharm Pharmacol. 1997;49:627–638. doi: 10.1111/j.2042-7158.1997.tb06084.x. [DOI] [PubMed] [Google Scholar]
- 18.Benet LZ, Brocatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13:519–547. doi: 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ku S. Use of the biopharmaceutical classification system in early drug development. AAPS J. 2008;10:208–212. doi: 10.1208/s12248-008-9020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galia E, Nicolaides E, Hörter D, Löbenberg R, Reppas C, Dressman JB. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 1998;15:698–705. doi: 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- 21.Sagawa K, Fasheng LL, Liese R, Sutton S. Fed and fasted gastric pH and gastric residence time in conscious beagle dogs. J Pharm Sci. 2009;98:2494–2500. doi: 10.1002/jps.21602. [DOI] [PubMed] [Google Scholar]
- 22.Dressman JB. Comparison of canine and human gastrointestinal physiologies. Pharm Res. 1986;3:123–131. doi: 10.1023/A:1016353705970. [DOI] [PubMed] [Google Scholar]
- 23.Braganza JM, Herman K, Hine P, Kay G. The effect of pentagastrin on peptic secretion in man. J Physiol. 1979;289:9–16. doi: 10.1113/jphysiol.1979.sp012721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh BN. Effects of food on clinical pharmacokinetics. Clin Pharmacokinet. 1999;37:213–255. doi: 10.2165/00003088-199937030-00003. [DOI] [PubMed] [Google Scholar]
- 25.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Phar Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 26.Lecaillon JB, Godbillon J, Campestrini J, Naquira C, Miranda L, Pacheco R, et al. Effect of food on the bioavailability of triclabendazole in patients with fascioliasis. Br J Clin Pharmacol. 1998;45:601–604. doi: 10.1046/j.1365-2125.1998.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doak AK, Wille H, Prusiner SB, Shoichet BK. Colloidal formation by drugs in simulated intestinal fluid. J Med Chem. 2010;53:4259–4265. doi: 10.1021/jm100254w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shono Y, Jantratid E, Dressman JB. Precipitation in the small intestine may play a more important role in the in vivo performance of poorly soluble weak base in the fasted state: case example nelfinavir. Eur J Pharm Sci. 2011;79:349–356. doi: 10.1016/j.ejpb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Cvijic S, Parojcic J, Langguth P. Viscosity-mediated negative food-effect on oral absorption of poorly-permeable drugs with an absorption window in the proximal intestine: In vitro experimental simulation and computational verification. Eu J Pharm Sci. 2014 doi: 10.1016/j.ejps.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Shono Y, Jantratid E, Janssen N, Kesisoglou F, Mao Y, Vertzoni M, et al. Prediction of food-effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur J Pharm Biopharm. 2009;73:107–114. doi: 10.1016/j.ejpb.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Bevernage J, Brouwers J, Clarysse S, Vertzoni M, Tack J, Annaert P, et al. Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J Pharm Sci. 2010;99:4525–4534. doi: 10.1002/jps.22154. [DOI] [PubMed] [Google Scholar]
- 32.Lui CY, Amidon GL, Berardi RR, Fleisher D, Yongberg C, Dressman JB. Comparison of gastrointestinal pH in dogs and humans—implications on the use of the beagle dog as a model for oral absorption in humans. J Pharm Sci. 1986;75:271–274. doi: 10.1002/jps.2600750313. [DOI] [PubMed] [Google Scholar]
- 33.Sutton SC. Companion animal physiology and dosage form performance. Adv Drug Del Rev. 2004;56:1383–1398. doi: 10.1016/j.addr.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Lentz KA. Current methods for predicting human food effect. AAPS J. 2008;10:282–288. doi: 10.1208/s12248-008-9025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao ZH, Hussain MA, Motheram R, Gray DB, Benedeck IH, Fiske WD, et al. Investigation of human pharmacoscintigraphic behavior of two tablets and a capsule formulation of a high dose, poorly water soluble/highly permeable drug. J Pharm Sci. 2007;96:2970–2977. doi: 10.1002/jps.20962. [DOI] [PubMed] [Google Scholar]
- 36.Mathias NR, Crison J. The use of modeling tools to drive efficient oral product design. AAPS J. 2012;14:591–600. doi: 10.1208/s12248-012-9372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawai Y, Fujii K, Tabata F, Ito J, Metsugi Y, Kameda A, et al. Profiling and trend analysis of food effects on the oral absorption considering micelle interaction and solubilization by bile micelles. Drug Metab Pharmacokinet. 2011;26:180–191. doi: 10.2133/dmpk.DMPK-10-RG-098. [DOI] [PubMed] [Google Scholar]
- 38.Pandey P, Hamey R, Bindra DS, Huang Z, Mathias NR, Eley T, et al. From bench to humans: formulation development of a poorly water soluble drug to mitigate food effect AAPS Pharm. Sci Tech. 2014;15:407–416. doi: 10.1208/s12249-013-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daneshmend TK, Warnock DW, Ene MD, Johnson EM, Potten MR, Richardson MD, et al. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob Agents Chemother. 1984;25:1–3. doi: 10.1128/AAC.25.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.US Food and Drug Administration. Ribavirin approval package label. 2004.
- 41.US Food and Drug Administration. Danazol approval package review. 1996.
- 42.US Food and Drug Administration. Celebrex approval package label. 1998.