

Abstract
Programmed death-1 (PD-1) protein is a co-inhibitory receptor which negatively regulates immune cell activation and permits tumors to evade normal immune defense. Anti-PD-1 antibodies have been shown to restore immune cell activation and effector function—an exciting breakthrough in cancer immunotherapy. Recent reports have documented a soluble form of PD-1 (sPD-1) in the circulation of normal and disease state individuals. A clinical assay to quantify sPD-1 would contribute to the understanding of sPD-1-function and facilitate the development of anti-PD-1 drugs. Here, we report the development and validation of a sPD-1 protein assay. The assay validation followed the framework for full validation of a biotherapeutic pharmacokinetic assay. A purified recombinant human PD-1 protein was characterized extensively and was identified as the assay reference material which mimics the endogenous analyte in structure and function. The lower limit of quantitation (LLOQ) was determined to be 100 pg/mL, with a dynamic range spanning three logs to 10,000 pg/mL. The intra- and inter-assay imprecision were ≤15%, and the assay bias (percent deviation) was ≤10%. Potential matrix effects were investigated in sera from both normal healthy volunteers and selected cancer patients. Bulk-prepared frozen standards and pre-coated Streptavidin plates were used in the assay to ensure consistency in assay performance over time. This assay appears to specifically measure total sPD-1 protein since the human anti-PD-1 antibody, nivolumab, and the endogenous ligands of PD-1 protein, PDL-1 and PDL-2, do not interfere with the assay.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9762-4) contains supplementary material, which is available to authorized users.
KEY WORDS: biomarker, nivolumab, reagent characterization, reagent characterizationsoluble PD-1, reference standard
INTRODUCTION
Programmed death-1 (PD-1) protein is a negative regulator of immune responses and expressed on the surface of activated T and B lymphocytes as well as thymus resident cells (1). Binding of PD-1 with ligand proteins PDL-1 or PDL-2, which are expressed on tumor and stromal cells, transmits an inhibitory signal that inactivates immune cells (2). Recent progress in delineating mechanism of tumorigenesis supports the hypothesis that tumors can evade normal immune attack through the PD-1/PDL-1 interactions in PD-1 checkpoint pathway (2,3). Checkpoint blockade in the form of antibodies that block inhibitory receptors, such as PD-1, on immune effector cells has brought exciting new prospects for cancer immunotherapy (4). Consistent with this notion, anti-PD-1 antibodies, such as nivolumab (Opdivo®), has been shown to block the interactions between PD-1 and its ligands, thereby enhancing T cell responses in vitro (5) and eliciting anti-tumor activity in patients with solid tumors (6). Clinical assays to quantify soluble factors of the checkpoint pathway in normal and disease sera would improve our understanding of tumorigenesis and facilitate the development of new cancer therapy.
The human PD-1 gene encodes a 288 amino acid (aa) protein (~55 kDa) with a putative signal peptide, an extracellular region with one immunoglobulin like V-type domain, a transmembrane domain, and a cytoplasmic region (7,8). An alternative splice variant of PD-1 has been reported to produce an isoform that contains only the extracellular domain of the PD-1 protein and exists in sera of normal and rheumatoid arthritis (RA) individuals (9,10). There is some evidence that this soluble form of the PD-1 protein (sPD-1) may serve an autoantibody-like role in circulation that attenuates the negative regulatory effect of PD-1/PDL-1 on T cells (10). Interestingly, a recent report showed sPD-1 levels are elevated in early RA patients, and treatment that reduces disease activity concurrently reduces sPD-1 levels (11), raising the possibility of sPD-1 serving as a disease state biomarker of RA. Levels of sPD-1 protein in cancer patients have not been reported.
In published studies, the sPD-1 protein was measured with a research use only (RUO) commercial kit which has not been validated for clinical use. Here, we report the development and fit-for-purpose validation (12,13) of a sPD-1 ECL assay for support of nivolumab clinical studies. The analytical sensitivity of the assay was 100 pg/mL, with a dynamic range of 100–10,000 pg/mL. The intra- and inter-assay imprecisions were ≤15%. The assay bias (percent deviation) was ≤10%. The assay was capable of quantifying levels of sPD-1 in sera from normal healthy individuals and selected cancer patients in the presence of nivolumab or PD-1 ligands: PDL-1 and PDL-2. The assay was used for multiple clinical sample studies and was proven to be fit for purpose and robust.
MATERIALS AND METHODS
Materials
Human Serum Specimens
Sera from normal healthy individuals or patients with different cancer types (melanoma, renal cell carcinoma, squamous, and non-squamous lung cancer, Hodgkin’s lymphoma, multiple myeloma) were purchased from Bioreclamation (NY, USA).
Antibodies and Recombinant Proteins
Anti-PD-1 antibodies were purchased from commercial sources or produced internally. See Table S1 (Supplemental Materials) for detailed information. Recombinant human PDL-1 and PDL-2 proteins were purchased from R&D Systems (Minneapolis, MN, USA).
Anti-PD-1 antibody MIH4 (eBiosciences, San Diego, CA, USA) was labeled with Biotin, at 1:20 (antibody: biotin) molar ratio using a EZ-Link NHS-PEG4-Biotin kit (Thermo Fisher, Waltham, MA, USA). An anti-PD-1 antibody, AF1086 from R&D Systems, were labeled with ruthenium, at 1:12 (antibody:ruthenium) molar ratio, using the MSD SULFO-TAG NHS-Ester kit (Meso Scale Discovery, Rockville, MD, USA). Both were done according the kit’s instructions.
Construction of Expression Vectors for Soluble PD-1 Proteins
A cDNA clone encoding human PD-1 pre-protein residues 1-167 (NCBI mRNA RefSeq NM_005018) linked at the C-terminus with a Myc-TVMV-His fusion (-EQKLISEEDLGSSETVRFQGHHHHHH) was generated by PCR as a SalI-BamHI fragment and cloned into a pENTR2 vector. The protein derived from this construct [hPD1(25-167)-Myc-TVMV-His] was designated as hPD1(25-167)-His, reflecting the amino acid sequence of the extracellular domain of the human mature PD-1 protein.
In a similar fashion, a clone was constructed encoding the same region of human PD-1 fused at its C-terminus to a portion of human IgG1 Fc region (UniProt P01857) beginning from the hinge (with three Cys to Ser substitutions) and including the CH2 and CH3 domains. The protein derived from this construct was designated as hPD1(25-167)-3S-IG.
Both of these DNA sequences were introduced into a pTT22gate-based vector by Gateway® LR recombination.
HEK293 Expression of Soluble PD-1 Proteins
HEK293 cells (HEK293-6E) at 1 × 106 cells/mL were transfected with the hPD1(25-167)-His or hPD1(25-167)-3S-IG Gateway® destination vectors using a Durocher expression system with a 1:2 DNA:PEI (polyethylenimine) ratio. The transiently transfected cells were cultured in F17 expression medium, and the conditioned media were harvested by sedimentation 5 days post-transfection and filtered through 0.45-μm filters. For hPD1(25-167)-His, the cell density at harvest was ~4 × 106 cells/ml, and the cell viability was >95%. For hPD1 (25-167)-3S-IG, the cell density at harvest was ~3 × 106 cells/ml, and the cell viability was >90%.
Purification of hPD1(25-167)-His Protein
The conditioned medium was concentrated and buffer exchanged by tangential flow filtration into phosphate buffered saline (PBS) with 200 mM additional NaCl and 20 mM imidazole (pH 8) using a Pellicon-2 TFF system and Ultracel-5 membrane (Millipore, Billerica, MA, USA). This sample was loaded onto 2 × 5 ml HisTrap HP columns (GE Healthcare, Princeton, NJ, USA) connected in series, washed with the same buffer, and eluted with a steep linear gradient to 300 mM imidazole in PBS with 200 mM additional NaCl (pH 8). The protein eluted with high imidazole was pooled, concentrated by centrifugal ultrafiltration to ~8 mg/ml, and loaded onto a 26 mm × 600 mm Superdex 200 preparative size exclusion chromatography (SEC) (GE Healthcare) equilibrated with PBS (pH 7.5). Based on absorption at 280 nm, there was a small amount of large molecular-sized material that eluted early, and this portion was discarded. The majority of protein eluted from the SEC column in a peak centered at 225 ml. The hPD1(25–167)-His protein fractions from this peak were pooled, divided into aliquots at 1.5 mg/ml, flash frozen in liquid nitrogen, and stored at −80°C.
Purification of hPD1(25-167)-3S-IG Protein
The conditioned medium was concentrated and buffer exchanged by tangential flow filtration into PBS (pH 7.4) using a Pellicon-2 TFF system and Ultracel-10 membrane (Millipore). This sample was loaded onto 2 × 5 ml HisTrap rProtein A columns (GE Healthcare) connected in series, and the column was washed with PBS (pH 7.4). The hPD1(25-167)-3S-IG protein was eluted with 80 mM sodium acetate (pH 3.0). The pH of the solution containing the eluted protein was immediately adjusted to ~pH 7.5–8.0 by the dropwise addition of ~1/8th volume of 1 M Tris-HCl (pH 8.0). The protein was concentrated to ~17 mg/ml by centrifugal ultrafiltration and loaded onto a 16 mm × 600 mm Superdex 200 preparative SEC equilibrated with PBS (pH 7.4). As observed by absorption at 280 nm, the protein eluted from the preparative SEC in a large peak was centered on 74 ml with essentially no large aggregate in earlier eluting fractions. The pool of peak fractions containing purified hPD1(25-167)-3S-IG protein at 1.9 mg/ml in PBS (pH 7.4) was divided into aliquots, flash frozen in liquid nitrogen, and stored at −80°C.
SDS-PAGE Analysis of Purified hPD1(25-167)-His Protein
Purified hPD1(25-167)-His protein was diluted with 4X LDS sample buffer (Invitrogen, Carlsbad, CA, USA) and 1 M dithiothreitol (DTT) to a final concentration of 1X LDS/100 mM DTT. The sample was heated to 65°C for 30 min, cooled on ice, and then loaded onto a NuPAGE 4–12% acrylamide Bis-Tris gel (Invitrogen) with Novex Sharp Pre-stained molecular weight standards (Invitrogen). The gel was electrophoresed at ~200 V for 35 min and then removed and stained with Simply Blue SafeStain (Invitrogen) according to the manufacturer’s protocol.
SEC-MALS Analysis of Purified hPD1(25-167)-His Protein
The molecular weight and carbohydrate content of the purified hPD1(25-167)-His protein were studied by analytical SEC coupled with ultraviolet absorption, refractive index, and multi-angle light scattering detection (MALS). Isocratic separations were performed on a Shodex Protein KW 803 column (Shodex, New York, NY, USA) connected to a UFLC system (Shimadzu, Kyoto, Japan) consisting of a degasser, isocratic pump, chilled sample holder with injector, UV/visible detector, and column oven, in a buffer containing 200 mM K2HPO4 and 150 mM NaCl (pH 6.8), with 0.02% sodium azide running at 0.5 ml/min. Data were obtained from three online detectors connected in series: the Shimadzu SPD-20 dual wavelength UV/visible spectrophotometer monitoring 280 nm, followed by a mini-Dawn TREOS three angle laser light scattering detector and an Optilab TrEX interferometric refractometer (Wyatt Technologies, Santa Barbara, CA, USA). Data were collected and analyzed using ASTRA 6 (Wyatt Technologies) and Lab Solutions (Shimadzu) software.
The carbohydrate content was determined by standard procedures in ASTRA 6 based upon the contributions of protein and carbohydrate to the absorption at 280 nm and the refractive index signal observed for the glycoprotein as it eluted from the SEC.
High-Resolution Mass Spectrometry (HRMS) Analysis of Purified hPD1(25-167)-His Protein
Purified hPD1(25-167)-His protein (8 μg) was incubated with 4 μL of protein deglycosylation mix (Promega, WI, USA), prepared according to the manufacturer’s specifications, at 37°C for 16 hours (h) with constant shaking.
Fifteen microliters of the resultant solution was mixed with 10.0 μL of 10 mM tri(2-carboxyethyl)-phosphine (TCEP). The mixture was then injected (5 μL) into an Acquity H-class Bio ultra-high pressure liquid chromatography system (UPLC, Waters Co.) fitted with a Kinetex C8 column (2.1 × 50 mm, 1.7 μm, Phenomenex Inc.). The mobile phases of 0.1% formic acid aqueous (A) and 0.1% formic acid in acetonitrile (B) were delivered under a gradient program: 20% B to 80% B over 7.0 min (curve factor 4), 80% B to 90% B over 0.5 min (curve factor 4), followed by re-equilibration. The flow rate was set to 0.3 mL/min, and the column was held at 80°C. The column eluent was introduced into a Bruker Daltonik MaXis 4G q-time-of-flight (TOF) mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany). The ionization source was set to positive polarity mode with a capillary voltage at 4.5 kV, nebulizer pressure at 1.6 bar, dry gas flow at 9.0 L/min, and temperature at 220°C. The mass analyzer was calibrated between 300 and 2900 m/z, and spectra were collected at 1.0 Hz. Profile mass spectra were summed from 3.4 to 3.7 min, smoothed, baseline subtracted, and deconvoluted using a maximum entropy equation in Compass DataAnalysis 4.2 (Bruker Daltonik).
Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) Analysis of Purified hPD1(25-167)-His Protein
A saturated solution of 2,5-dihydroxy acetophenone (2,5-DHA) (Sigma-Aldrich, MO, USA) was made by adding the matrix to 1.0 mL of an acetonitrile:water:trifluoroacetic acid (80:20:0.1) solution. The solution was sonicated and deemed saturated when solids still remained after 5 min of sonication. Purified hPD1(25-167)-His protein (1.0 μL) was spotted onto a stainless steel 384MTP target plate (Bruker Daltonik). The saturated 2,5-DHA solution (1.0 μL) was added to the sample spot on target plate and mixed by pipetting the solution up and down for three cycles. The resultant spot was allowed to dry before analyzing. Samples were then analyzed on a Bruker AutoFlex III MALDI-TOFMS (Bruker Daltonik) equipped with a 355 nm YAG SmartbeamTM laser. Spectra were collected in linear positive ion mode.
Calibration Standards and Quality Control Samples Preparations
The hPD1(25-167)-His protein stock in PBS-10% glycerol was first diluted with 1X Reagent Diluent (R&D Systems) and then with SeraSub (CST Technologies, NY, USA) to prepare sPD-1 standards at concentrations of 100, 200, 400, 1000, 2000, 4000, and 10,000 pg/mL. These standards were aliquoted and kept at −80°C until use.
Low-, mid-, and high-level quality control samples (LQC, MQC, and HQC) were prepared by spiking sPD-1 [hPD1(25-167)-His] reference standards into a normal human serum pool (prescreened for low sPD-1 levels) to prepare QCs of 500, 1500, and 5000 pg/mL, respectively. In addition, a low limit of quantification QC (LLOQ) sample was generated using a normal human serum pool with the targeted endogenous sPD-1 concentration of ~100 pg/mL. All QC aliquots were kept frozen at −80°C until use.
Surface Plasmon Resonance Studies
Binding of Human PDL-1 to hPD1(25-167)-His
Surface plasmon resonance (SPR) experiments to characterize the binding of hPDL-1 to hPD1(25-167)-His were performed using a ProteOn XPR36 instrument (BioRad, Hercules, CA, USA). The surface was activated by injecting nickel (II) ions at 30 μl/min for 120 s. All analyses were performed at 25°C in a PBS 0.05% Tween solution.
To determine binding affinity of hPDL-1 to soluble PD-1, the hPD1(25-167)-His protein was captured via its His-tag on a HTG surface. Different concentrations (6.25–100 μg/ml) of hPD1(25-167)-His were injected at 30 μl/min for 300 s to generate five different surface densities. The ligand capture response levels ranged from 480 to 1200 RU. The analyte hPDL-1 protein (0.125–2 μM) was injected over the surface at 30 μl/min for 180 s. The surface was regenerated with 300 mM EDTA (pH 8.5) following BioRad’s recommendations. The binding data were analyzed using the ProteOn Manager Software from BioRad.
Binding of Anti-PD-1 Antibody MIH4 to hPD1(25-167)-His Protein
SPR experiments were performed to characterize the binding of hPD1(25-167)-His to MIH4, a mouse monoclonal antibody against human PD-1. The experiments were performed at 25°C using a Biacore T200 instrument (GE Healthcare). All the consumables were obtained from GE Healthcare. Running buffer for all steps was HBS-EP (0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20). The hPD1(25-167)-His protein was coupled to a Series S Sensor Chip CM5 utilizing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) direct coupling. Target ligand densities were 300 to 400 RU for kinetic measurements.
For each flow cell, different concentrations (0.195–50 nM) of MIH4 were injected at 30 μl/min for 240 s to generate eight different surface plasmon sonograms for kinetic calculations. The flow cells were regenerated with Glycine (pH 2.0) between each injection of anti-PD-1. The response levels obtained for the analyte ranged from 0.2 to 49 RU. The data were analyzed with the Biacore T200 Evaluation software. Kinetic fit data with χ2 values less than 0.1 were accepted.
sPD-1 MSD Assay Procedure
Streptavidin plates (MSD, Rockville, MD, USA) were incubated with Blocker Casein in PBS at room temperature for 1 h and washed four times with a wash buffer (PBS with 0.05% Tween-20). These plates were then coated with biotinylated anti-PD1 MIH4 (4.0 μg/mL in PBS) at room temperature for 2 h on the day of study (fresh plate) or prepared prior to the study and treated with StabilCoat per manufacturer’s recommendations (Surmodics, Eden Prairie, MN, USA). These pre-coated plates were stored at 4°C in a sealed airtight pouch with desiccant.
During assay development and validation, each analytical run consisted of one set of standards, a blank, QCs (HQC, MQC, LQC, and LLOQ) and study samples. On the day of the assay, the standards, QCs, and study samples were diluted 1:4 with Diluent 2 (MSD). All samples were analyzed in duplicates unless specified otherwise. Fifty microliters of standard, blank, QC, or study sample was incubated in a MIH4-biotin-coated plate at room temperature for 2 h with shaking (~600 rpm). The plate was washed and incubated with 50 μL of a Sulfo-tagged anti-PD-1 detection antibody (3.0 μg/mL in Blocker Casein) at room temperature for 1 h with shaking. Following a wash step, 150 μL 2X Read Buffer Solution (MSD) was added into each well; the plate was then read on a MSD 6000 Imager.
Data Analysis
The conversion of ECL signal units to concentrations for the study samples and QCs was performed using SoftMax Pro (version 5.4.1, Molecular Devices, CA, USA). The ECL signal vs. concentration relationship was regressed according to a four-parameter logistic regression model with a weighting factor of 1/y2. Goodness of curve fit was determined on the following basis: 1) significance of the parameters included in the model (95% confidence limits that do not contain “0” for the estimate of maximum response, EC50 and slope); 2) p value for the general linear hypothesis test for goodness of model fit >0.001 (R2 for these curves were greater than 99% so the lack of fit test was conducted at a low significance level to prevent the test from being too sensitive to small departures); 3) residual plots from the regression model that show random scatter around “0”; 4) %RE (Relative Error) values within 20% over the range of the standards. The residual plots also provided information on the appropriate weight—the unweighted model and model with 1/y weights showed variances in the responses that increased with concentration; the weight of 1/y2 provided residual plots that showed greater randomness of the residuals around “0.” Based on these criteria, the four-parameter logistic regression model with a weight of 1/y2 was considered appropriate.
Variance components methods (14) in the context of analysis of variance (ANOVA) models were used for estimates of assay precision, expressed as coefficient of variation (CV) relative to the overall mean predicted concentration [%CV = 100(StdDev/mean)1/2]. Intra-assay (within replicates on the same plate) or inter-assay (between different plates) imprecision was defined by variance derived from back-calculated concentrations of QCs. Specifically, The estimates of inter- and intra-assay %CV were calculated as the “ANOVA Variance Components” for “Intrabatch” (sw2) and “Interbatch” (sb2) variance, respectively (12). Total variation in Table III is the sum of the two variance components (sb2 + sw2).
Table III.
Assay Accuracy and Precision of QC Samples in Human Sera
| LLOQ | LQC | MQC | HQC | |
|---|---|---|---|---|
| Nominal concentration (ng/mL) | n/a | 500 | 1500 | 5000 |
| Mean observed concentration (ng/mL) | 101.0 | 526.8 | 1583.2 | 5482.5 |
| %Deviation | n/a | 5.3 | 5.5 | 9.7 |
| Inter-assay imprecision (%CV) | 14.8 | 5.1 | 1.6 | 5.2 |
| Intra-assay imprecision (%CV) | 22.0 | 4.2 | 2.7 | 5.4 |
| Total variation (%CV) | 26.5 | 12.9 | 6.9 | 7.5 |
| Total error (%) | n/a | 18.3 | 12.5 | 17.1 |
| n | 51 | 24 | 24 | 24 |
| Number of runs | 7 | 7 | 7 | 7 |
See Materials and Methods section for calculations of %CV, total variance, and total error
n/a not applicable
%RE was calculated as 100[(observed value-nominal value)/nominal value]. Total error (TE) was calculated as the sum of the absolute value of %RE and the total variance. All analyses were performed using SAS version 9.2.
RESULTS
Characterization of the sPD-1 Reference Standards
To generate human sPD-1 reference material, we expressed two different recombinant human sPD-1 fusion proteins, hPD1(25-167)-His and hPD1(25-167)-3S-IG, that contain the extracellular domains of PD-1 (aa 25-167) and a hexa-His or a human IgG1 Fc domain at the C-terminus, respectively. The hPD1(25-167)-3S-IG protein is similar to the commercially available PD-1 Fc chimera protein which has been used in published studies (10,11). Both recombinant sPD-1 proteins were transiently transfected into HEK293 cells and purified. Analysis of the purified proteins by SEC-MALS indicated that both proteins were glycosylated, consistent with the presence of four predicted N-linked glycosylation sites in the human PD-1 extracellular domain and their expression in HEK293 cells. The SEC-MALS analysis indicated that hPD1(25-167)-His and hPD1 (25-167)-3S-IG in PBS solution were glycosylated monomeric and glycosylated dimeric proteins, with estimated mass of 20.0 kDa and 93.0 kDa, respectively (Table I). Since structural and biophysical studies have shown that PD-1 is monomeric both in solution as well as on cell surface (15), the monomeric hPD1(25-167)-His protein was selected as the reference standard for the sPD-1 assay to closely mimic the endogenous protein. SDS-PAGE analysis showed that the purified hPD1(25-167)-His protein is of a single form with >95% purity and an apparent molecular weight of ~45 kDa (Fig. 1a).
Table I.
Characterization of hPD1(25-167)-His and hPD1(25-167)-3S-IG
| hPD1(25-167)-His | hPD1 (25-167)-3S-IG | |
|---|---|---|
| Predicted monomer M.W. | 19.0 kDa | 42.6 kDa |
| Predicted dimer M.W. | 38.0 kDa | 85.3 kDa |
| Glyco-conjugate M.W. by SEC-MALS | 33.1 kDa | 114.0 kDa |
| Protein M.W. by SEC-MALS | 20.0 kDa | 93. 0 kDa |
| %Mass as carbohydrate by SEC-MALS | 39.6% | 18.4% |
| Mass fraction recovered by SEC-MALS | 90.3% | 99.9% |
| Glyco-conjugate M.W. by MALDI-TOF | 30.9 kDa | |
| Protein M.W. by HRMS | 19.0 kDa | |
| %Mass as carbohydrate by MALDI-TOF and HRMS | 38.5% |
The molecular weight (M.W.) and carbohydrate content estimate of the purified hPD1[25-167]-His and hPD-1-3S-IG proteins were determined by SEC-MALS or MALDI-TOF as described in the Materials and Methods section
Fig. 1.
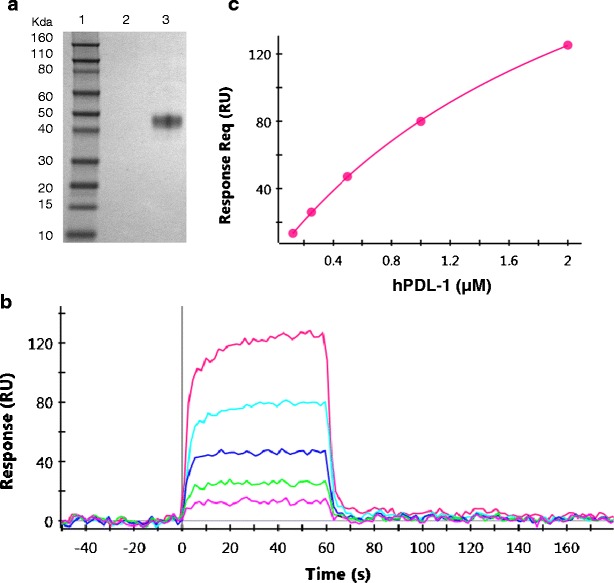
a–c Characterization of the assay reference standard-recombinant hPD1(25-167)-His protein. a SDS-PAGE analysis of the purified hPD1(25-167)-His protein expressed in HEK293 cells. Lane 1: molecular weight marker, lane 2: blank, lane 3: 1 μg of the purified hPD1(25-167)-His protein. The gel was stained with Simply Blue SafeStain. b Concentration series of hPDL-1 (0.125–2 μM) binding to captured hPD1(25-167)-His protein. c Equilibrium analysis of hPDL-1 binding to hPD1(25-167)-His yields a K D of 2.8 μM (average value over multiple surfaces)
High-resolution mass spectrometry (HRMS) was used to obtain more accurate mass of the hPD1(25-167)-His protein. The protein was subjected to deglycosylation using PNGase F (Promega) and injected onto an LC-MS system equipped with an ESI source and q-TOF mass analyzer. The experimentally observed mass of 19,009.8620 Da represents a mass accuracy of <3 ppm as compared to the calculated average mass [M + H] ion for the amino acid sequence. The average mass value of the glycosylated hPD1(25-167)-His protein was determined to be 30.9 kDa using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS), therefore, the carbohydrate content was calculated to be 38.5% by mass.
The functional binding affinity of hPD1(25-167)-His protein to human PDL-1 was assessed using SPR. In this study, hPD1(25-167)-His protein was captured on a HTG chip surface by its His-tag. Soluble human PDL-1 was injected into the mobile phase as an analyte. The observed KD of 2.8 μM was similar to the previously reported affinity of PDL-1 to PD-1 protein (Fig. 1b, c) (16).
Selection of Antibody Pair and Concentrations
We evaluated several anti-human PD-1 antibodies produced either internally or from commercial sources. These antibodies were assessed for binding to both monomeric and dimeric forms in the presence of nivolumab (see Table S1 in Supplemental Materials). Two antibodies, MIH4 and AF1086, which can bind both monomeric and dimeric forms of PD-1 in the presence of Nivolumab were selected as capture and detection antibodies, respectively. The binding affinity of hPD1(25-167)-His to MIH4 was 2.8 nM as determined by SPR analysis (Table II). The effect of PDL-1 on the binding of hPD1(25-167)-His to MIH4 was also evaluated using SPR. When the hPD1(25-167)-His protein was immobilized on the chip, its binding affinity to MIH4 antibody is 2.9 nM in the presence of 2 M excess PDL-1 protein, suggesting non-interference of PDL-1 for hPD1(25-167)-His and MIH4 binding.
Table II.
Binding Affinity of Anti-PD-1 Antibody MIH4 for Recombinant Human sPD-1 Proteins
| Ligand (on chip) | 2 M excess PDL-1 | k a (1/Ms) | k d (1/s) | K D, nM | Chi2 |
|---|---|---|---|---|---|
| hPD1(25-167)-His | No | 1.3E + 5 | 3.8E-4 | 2.8 | 0.16 |
| hPD1(25-167)-His | Yes | 1.2E + 05 | 3.4E-04 | 2.9 | 0.08 |
| hPD1(25-167)-3S-IG | No | 4.8E + 5 | 2.4E-4 | 0.5 | 0.14 |
Association constant (k a), dissociation constant (k d), and binding constant (K D) for monomeric (hPD1(25-167)-His) and dimeric (hPD1(25-167)-3S-IG) sPD-1 proteins. Chi2: Goodness of fit
In order to achieve desired assay sensitivity and dynamic range in the presence of nivolumab, we conducted a design of experiments (DoE) study using the central composite design to determine the optimal concentrations of the capture and detection antibodies. A wide range of capture and detection antibody concentrations were evaluated in this study (data not shown). With the aid of multi-variance analysis utilizing JMP 8.0 (SAS, NC, USA), the capture and detection antibody concentration was selected at 4 and 3 μg/mL, respectively.
Standard Curve and Assay Range
Method development and validation were conducted according to the framework for full validation of a biotherapeutic pharmacokinetic assay and published white papers (12,13,17). Seven validation runs were performed over four different days to determine the analytical range of the assay. Each run consisted of one set of standards and blank and at least two replicates of the QC and LLOQ samples. The validated analytical range of the assay was 100–10,000 pg/mL, defined by the lowest and highest non-zero points of the standard curve with acceptable precision and accuracy (Figure S1 in Supplemental Materials). The LLOQ was selected close to the concentration of endogenous sPD-1 levels in normal human sera (~100 pg/mL). This assay was designed and proven to measure sPD-1 in the presence of nivolumab (see last section of Results), the levels of total sPD-1 post drug treatment may increase significantly since the clearance of analyte–antibody complex were expected to be slower based on our experience with other circulating proteins (18). Therefore, the ULOQ was selected at 10,000 pg/mL so that the calibration curve covers a wide range of concentrations. The standard curve imprecision was ≤15% in total variance, and accuracy was ≤15% in %RE for all back-calculated standard concentrations (Table S2).
Assay Precision and Sensitivity
High-, mid-, and low-QC samples were used to evaluate assay precision and accuracy during the validation runs. The results of these studies are summarized in Table III. The intra- and inter-assay imprecision was ≤15%, which was defined by intra- and inter-assay variance derived from back-calculated concentrations of QCs. The assay bias or percent deviation was ≤10%. The accuracy of the assay, defined by percent of total error of LQC, MQC, and HQC, was <20% which satisfied the targeted criteria based on our internal SOP.
A LLOQ sample was also included in the same set of validation runs and was used to define the assay sensitivity as 100 pg/mL. The intra- and inter-assay imprecision of the LLOQ was ≤25% with a total variance of 26.5%.
Detection of Endogenous sPD-1 Protein and Study of Matrix Effect in Normal and Cancer Sera
We surveyed commercial serum samples from 15 normal individuals and 58 cancer patients from 6 cancer types. Greater than 98% of all tested samples had measurable levels of sPD-1. In this small set of samples, serum sPD-1 levels in melanoma and renal cell carcinoma patients appeared to be higher than those in normal individuals (Fig. 2a). The mean ± SD values of sPD-1 levels in normal, melanoma, and renal cell carcinoma patients are 188.0 ± 16.1 (N = 15), 319.2 ± 45.6 (N = 15), and 278.7 ± 45.7 (N = 11) pg/mL, respectively (p < 0.05, normal vs. melanoma or renal cell carcinoma).
Fig. 2.
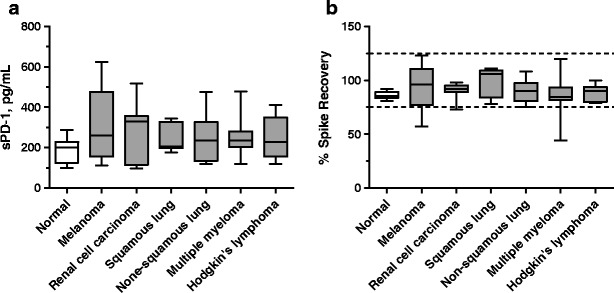
a–b Detection of endogenous sPD-1 protein and spike recovery in sera from normal and cancer individuals. a Levels of sPD-1 in serum samples from normal and cancer individuals: normal, n = 15; melanoma, n = 15; renal cell carcinoma, n = 11; squamous lung cancer, n = 7; non-squamous lung cancer, n = 8; multiple myeloma, n = 10; Hodgkin’s lymphoma, n = 7. b Spike and recovery study: 500 pg/mL hPD1(25-167)-His protein was spiked into serum samples from normal and cancer individuals: normal, n = 10; melanoma n = 8; renal cell carcinoma, n = 11; squamous lung cancer, n = 7; non-squamous lung cancer, n = 8; multiple myeloma, n = 10; Hodgkin’s lymphoma, n = 7. % Spike recovery = 100*(sample value post-spike − sample value pre-spike)/spiked value. Results are shown with box and whiskers graphs. The box included data within 5 to 95 percentiles with the median line in the middle and extended values in the whiskers. The dotted lines depict the nominal spiked value ± 25%
Potential effect of different sample matrices were studied by spiking 500 pg/mL hPD1(25-167)-His protein into serum samples from normal and cancer individuals. No significant bias was observed in % spike recovery in these samples, although % spike recovery in cancer sera appeared to be more variable, especially in samples from multiple myeloma patients (Fig. 2b).
Dilution Linearity in Normal and Cancer Sera
To assess sample dilution linearity, three melanoma serum samples with high levels of endogenous sPD-1 were diluted 1:1.5 to 1:8 in assay buffer. The dilution curve of these melanoma samples were parallel to that of the reference standard in SeraSub solution and diluted in the same concentration range (Fig. 3a). Dilution linearity was also observed in two serum samples with high levels of endogenous sPD-1 from hematologic malignancy patients which were diluted 1:4 to 1:128 (data not shown). Together, these results also demonstrated that concentration–response relationship of the endogenous sPD-1 protein in sample matrix was similar to that of reference standards in assay buffer [proof of “parallelism” (17)].
Fig. 3.
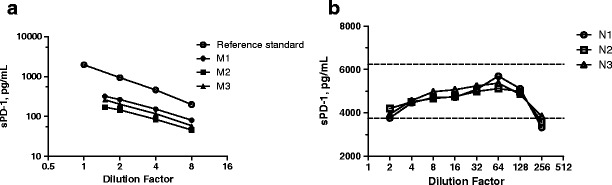
a–b Dilution linearity studies in melanoma and spiked normal sera. a Three melanoma serum samples (M1, M2, M3) were each diluted 1.5–8-fold with assay buffer; recombinant hPD1(25-167)-His protein standard in a SeraSub solution was diluted in same concentration range. b Three normal human serum samples (N1, N2, N3) were spiked with 5000 pg/mL of human hPD1(25-167)-His and were each diluted 2–256-fold with assay buffer. The back-calculated sPD-1 concentrations (observed concentrations times the dilution factor) were plotted against dilution factors. The dotted lines depict 75–125% range of the nominal spiked value
Dilution linearity was also demonstrated with three normal human serum samples each spiked with 5000 pg/mL hPD1(25-167)-His protein and then serial diluted in assay buffer starting from 1:2. The back-calculated concentrations of samples diluted 1:4 to 1:128 were within 25% deviation from the nominal (Fig. 3b), suggesting no significant matrix effect in this dilution range (13,17). Based on these results, the assay’s minimal required dilution factor (MRD) was determined as 4.
QC Sample and Endogenous Analyte Stability
To support clinical studies, we evaluated short-term stability of QCs including the LLOQ sample (a pooled human serum sample with endogenous analyte) at different storage temperatures and through multiple freeze/thaw (FT) cycles, and results from each tested conditions were compared to QC ranges established during assay validation. As shown in Tables IV and V, benchtop stability was demonstrated at room temperature and 2–8°C for up to 24 h, at −20°C for up to 2 weeks, and at −80°C for up to 4 months. Freeze/thaw cycle stability was tested by comparing data from QC samples subjected to multiple F/T cycles to those from a freshly prepared control set and showed that both QC and test samples can withstand at least 5 freeze/thaw cycles. Together, these results informed sample handling during clinical studies.
Table IV.
Storage Condition on QC Sample Stability. Back-Calculated Concentration Values (pg/mL) of QC Samples Kept at Room Temperature (RT), −4, −20, or −80°C
| Validation Range, pg/mL | 24 h@RT | 24 h@4oC | 2 weeks@-20oC | 4 months@-80oC | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| High value | Low value | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | |
| LLOQ | 49 | 153 | NT | NT | 86 | 9.4 | NT | |||
| LQC | 412 | 600 | 447 | 2 | 482 | 3.4 | 504 | 3.3 | 474 | 1.3 |
| MQC | 1142 | 1784 | 1483 | 1.4 | 1478 | 1.1 | 1513 | 3.9 | 1464 | 0.7 |
| HQC | 4148 | 6150 | 5224 | 1.3 | 5198 | 2.9 | 5154 | 7.1 | 5180 | 1.8 |
The qc range from assay validation studies is included in each table. Results shown as mean concentration value and %CV (n = 2)
NT not tested
Table V.
Freeze/Thaw (F/T) Effects on QC Sample Stability. Back-Calculated Concentration Values of QC Samples Underwent Different Number of F/T cycles
| Validation range, pg/mL | 0 FT | 1X FT | 2X FT | 3X FT | 4X FT | 5X FT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low value | High value | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | pg/mL | %CV | |
| LLOQ | 49 | 153 | 84 | 16.1 | NT | 60 | 11.7 | 95 | 5.6 | 98 | 12.2 | 92 | 21.7 | |
| LQC | 412 | 600 | 505 | 0.7 | 469 | 1.1 | 503 | 5.7 | 539 | 1 | 538 | 1 | 546 | 2.8 |
| MQC | 1142 | 1784 | 1532 | 1 | 1358 | 1.4 | 1584 | 0.6 | 1507 | 1.8 | 1567 | 0.1 | 1606 | 0.5 |
| HQC | 4148 | 6150 | 5067 | 0.1 | 4954 | 1.2 | 5328 | 9.3 | 5435 | 7.3 | 5380 | 2.9 | 5310 | 6.9 |
The QC range from assay validation studies is included in each table. Results shown as mean concentration value and %CV (n = 2)
NT not tested
Method Optimization and Robustness
Large clinical studies often involve multiple sites and span several years. To ensure consistency of assay performance overtime and to reduce assay duration, we investigated whether assay plates can be coated with capture antibodies prior to testing. The analytical performance of the pre-coated Streptavidin plates were evaluated by comparing standard and QC sample performance using pre-coated plates with those using freshly coated Streptavidin plates. Results from these studies demonstrated that assay standard curve and QC performance of pre- and freshly coated plates were comparable as long as the pre-coated plates were treated with StabilCoat (Fig. 4a, b). The stability of these pre-coated plates with StabilCoat was evaluated by real-time assessment and an accelerated stability assessment approach (19,20). During the accelerated stability assessment, the pre-coated plates with StabilCoat were kept in an airtight pouch with desiccants for 1 or 2 weeks at 37°C and then the QC sample performance was evaluated. Results from this study demonstrated that the pre-coated plates with StabilCoat are stable for up to 2 weeks at 37°C. Based on the Arrhenius equation (19,20), the estimated stability of these plates stored at 2–8°C is approximately 1 year (Fig. 4c). In these studies, percent deviation (%Dev) of each QC was calculated against its mean value from validation studies using freshly coated plates.
Fig. 4.
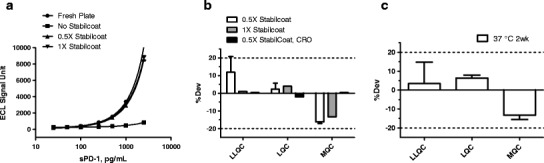
a–c Assay performance and stability of pre-coated and StabilCoat-treated Streptavidin plates. Standard curves (a) and QC performance (b) from analytical runs using fresh plates and pre-coated Streptavidin plates. The fresh plates were coated with the capture antibody on the day of the assay. The pre-coated plates were coated with capture antibody, treated with 0.5X or 1X StabilCoat and kept at 4°C or 37°C for 48 h (no differences between the 4°C and 37°C groups and the data were grouped together, n = 4). Assay QC performance using 0.5X StabilCoat-treated plates at a CRO were also presented (n = 18). (c) Accelerated stability study of pre-coated Streptavidin plates. QC performance in analytical runs using fresh or pre-coated plates (treated with 0.5X StabilCoat) stored at 37°C for 2 weeks. Results are shown as mean and SD (n = 4)
The use of bulk-prepared frozen reference standard samples can further improve the consistency of assay performance and reduce assay duration. The comparison between standard curves generated with bulk-prepared frozen standards and freshly made standards demonstrated that sPD-1 reference standards can be kept in aliquots at −80°C for up to 2 months without any significant changes in assay performance (Fig. 5a). In addition, Fig. 5a also showed consistent assay performance among analytical runs conducted by two different analysts on different days. Together, these results demonstrated the robustness of the assay.
Fig. 5.
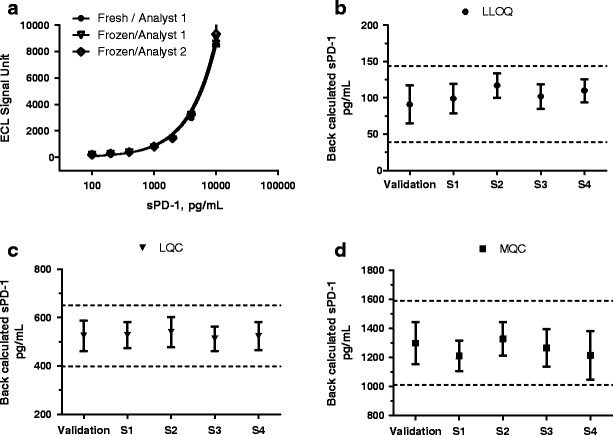
a–d Assay performance using fresh or frozen standards during validation and clinical studies. a Standard curves generated by two different analysts using freshly prepared standards (n = 2) or pre-prepared standards (n = 4) which were kept frozen at −80°C until use. Results are shown as mean ± standard error of mean. b–d In-study QC sample performance (using frozen standards). Back-calculated LLOQ, LQC, and MQC data were collected from assay validation and four clinical sample analyses (S1–S4) over a 3-month period. Results are shown as mean and SD, (n = 38–76). The dotted lines in the graph depict mean ± 2SD values for each QC
Further demonstration of assay robustness came from sample analyses of 56 analytical runs from 4 clinical studies. QC sample data from all 56 runs were within the acceptance criteria established during assay validation. These analytical runs were performed by three analysts and using two reference standard lots and three labeled coating and detection antibody lots (Fig. 5b–d). These clinical studies also showed that assay sensitivity, standard curve range, and QC levels were adequate for samples from multiple cancer types both pre- and post-nivolumab treatment (specific results not shown).
Effects of PDL-1, PDL-2, and Nivolumab on the sPD-1 ECL Assay
To assess whether the endogenous ligands of PD-1 protein interfere with the assay, we measured sPD-1 protein in a melanoma serum sample in the absence or presence of varying concentrations of human PDL-1 and PDL-2 proteins. Soluble PDL-1 in human sera has been reported in the low nanogram per milliliter range (21); levels of soluble PDL-2 is currently not known. As shown in Fig. 6a, PDL-1 or PDL-2, up to 1 μg/mL (20 nM), did not have significant effect on sPD-1 measurement (Fig. 6a). This study was repeated with three additional melanoma serum samples, and similar results were observed. In a separate study, we evaluated the effect of nivolumab (1–100 μg/mL) on the assay. As shown in Fig. 6b, nivolumab at 100 μg/mL (685 nM) had minimal interference on the assay performance (~15–20% reduction in back-calculated QC values) (Fig. 6b). Together, these results demonstrated that the sPD-1 assay is a “total assay” which is capable of measuring circulating human sPD-1 regardless of the presence of nivolumab or PDL-1/2.
Fig. 6.
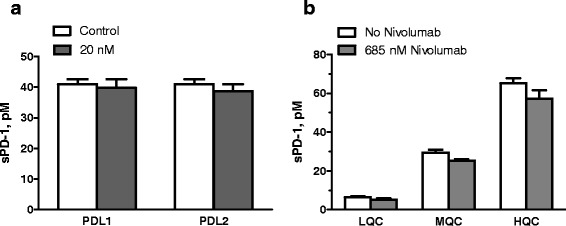
a–b Effect of PDL-1, PDL-2, and nivolumab on QC sample performance. a sPD-1 in a melanoma serum sample measured in the absence or presence of 20 nM (1 μg/mL) of human PDL-1 or PDL-2 proteins. b QC performance from analytical runs in the absence or presence of 685 nM (100 μg/mL) nivolumab. Results are shown as mean and SD of each group (n = 2–4)
DISCUSSION
We report here the development and “fit-for-purpose” validation of a sPD-1 protein assay on the MSD ECL platform. The assay used two commercial anti-PD-1 antibodies as capture and detection reagents and a recombinant human PD-1 monomeric protein as reference standard. The assay sensitivity (LLOQ) was 100 pg/mL and the dynamic range was 100–10,000 pg/mL. The intra- and inter-assay imprecision, defined by variance derived from back-calculated concentrations of LQC, MQC, and HQC was <15%. The accuracy of the assay, defined by total error of LQC, MQC and HQC, was <20%. The intra- and inter-assay imprecision of assay LLOQ of 100 pg/mL is ≤25%. Evaluations of a large number of serum samples from our clinical studies and a commercial source showed that the assay sensitivity and dynamic range were sufficient for sPD-1 measurement in normal and cancer subjects.
A LLOQ sample, made of a normal human serum pool with a low level of sPD1, is included in all validation and sample analysis runs since the baseline sPD-1 concentration in normal healthy individuals and some of the cancer patients, as determined using this assay, are close to the assay LLOQ. Both LQC and MQC samples were prepared by spiking recombinant sPD-1 protein into pooled human sera which mimic the actual sample matrix and serve as good monitors of assay variability. Consistently, high assay performance despite the use of frozen standards, pre-coated assay plates, as well as the use of different analysts, different plate washers, or plate readers is a testimony of the robustness of the assay. This assay appears to measure total sPD-1 in human serum since nivolumab and the endogenous PD-1 ligands, PDL-1 and PDL-2, do not interfere with the assay. Overall, the method validation follows the framework for full validation of a biotherapeutic pharmacokinetic assay.
Reagent Characterization
Careful selection and characterization of key assay reagents is critical to ensure immunoassay performance (22). During assay development, we evaluated two candidate reference standard proteins: hPD1(25-167)-His which contains the extracellular portion (aa 25-167) of the full length PD-1 receptor and hPD1 (25-167)-3S-IG, a PD-1 Fc chimeric protein which is similar to the commercial reagent used in published studies (10,11). Our goal was to gain information regarding protein identity, purity, and glycosylation content, and to have a reference standard which closely mimics the endogenous analyte in structure and function. Our SEC-MALS analysis showed that hPD1(25-167)-His was a glycosylated monomeric protein in solution phase. Further, its binding affinity to sPDL-1 was similar to published results (16). The same study also showed that hPD1 (25-167)-3S-IG formed a homodimer in solution. Since it has been documented that PD-1 is monomeric in solution and on the cell surface (15), the hPD1(25-167)-His was selected as reference standard for the assay.
Our reagent characterization effort also included antibody binding affinity assessments when it was possible. The binding affinity of the coating antibody MIH4 to sPD-1 was determined to be 2.8 nM by SPR. The binding affinity of AF1086 to sPD-1 was not examined since it was a polyclonal antibody. In addition, each lot of the biotinylated coating antibody and ruthenium-labeled detection antibody was examined for its purity and labeling reagent molar incorporation ratio using SEC and MALDI-TOF technology (data not shown). Together, these detailed characterization efforts enabled a better understanding of assay performance and ensured consistent, reproducible assay results across different reagent lots over time.
Matrix Effect
As for many high sensitivity biomarker assays, finding serum matrix with no detectable sPD-1 protein was challenging. Therefore, recombinant human sPD-1 proteins were reconstituted in a serum substitute SeraSub, a buffered synthetic polymer solution which is similar to serum and plasma with respect to specific gravity, viscosity, and osmolality. We have determined whether there is similar proportionality in immunoreactivity between the recombinant sPD-1 protein standard in SeraSub and the endogenous sPD-1 protein in serum matrix (“parallelism”). This was accomplished by a dilution linearity study using samples with high levels of endogenous sPD-1 proteins. Results from this study showed that the dilution curve of endogenous sPD-1 in sample matrix was parallel to that of the reference standard in SeraSub solution in the same concentration range. These findings support that recombinant sPD-1 standards in buffer offer a valid proportional scale for the relative quantitative measurement of the endogenous sPD-1 protein in serum.
Soluble factors in sample matrix may cause assay interference due to nonspecific binding (matrix effect). This was investigated by spike and recovery studies on 15 lots of normal human serum and ~10 serum lots each of 6 cancer types. No significant matrix effect was observed in normal or disease sera.
The Use of Bulk-Prepared Assay Reagents
Biomarker sample analysis supporting late-phase clinical trials often involves processing a large number of samples collected over many years and at different analytical labs. Consistency in assay performance over time and across different labs is important for delivering high-quality clinical data and is challenging. With this in mind, we used bulk-prepared pre-coated Streptavidin plates and frozen standards and QC samples in this assay. This allowed us to provide the same batches of standards, QCs, and assay plates to a contract research organization (CRO) to ensure consistent assay performance over time and across different laboratories. In addition, these approaches also increased assay efficiency by reducing assay duration from 8–9 h to 6–7 h.
We found that StabilCoat treating is critical for maintaining integrity of the pre-coated plates. Accelerated stability studies showed that the pre-coated plates after StabilCoat treatment can be kept at 37°C for up to 2 weeks. Based on previous studies, these plates are likely to maintain assay performance for ~1 year if kept at 4°C (20). The long-term stability of these plates has been conducted in real time, and thus far, we have confirmed they are stable for at least 4 months at 4°C.
Comparison with Existing Method and Clinical Application
sPD-1 protein levels have been reported to be elevated in rheumatoid arthritis and are associated with disease activities (10,11). During the submission of the present study, a new study was published that prospectively examined the relationship between plasma sPD-1 levels and hepatitis B virus load as well as the development of hepatocellular carcinoma (23). In these published studies, baseline sPD-1 levels in healthy control human sera were reported to be in the low nanogram per milliliter range, which is higher than what we observed (~200 pg/mL). This difference may be, at least in part, due to the differences in assay methodologies. In the published studies, levels of sPD-1 were measured with a research use only (RUO) commercial kit using a purified recombinant hPD-1 Fc chimera protein as reference standard (10,11). Based on our study on hPD1(25-167)-3S-IG, the hPD1 Fc chimera protein is likely to be a homodimer. In contrast, the hPD1(25-167)-His protein used as reference standard in our assay is a monomeric protein and a better mimic of the endogenous sPD-1 protein in human circulation (15). The commercial RUO assay and our assay also used different capture antibodies. Although the two assays shared the same detection antibody, they were labeled differently (biotin vs. ruthenium). In addition, the two assays also used different buffers. The RUO kit may have matrix interference issues if it were used in clinical studies, since we were unable to establish dilution linearity in several human serum samples using this kit (data not shown).
To our knowledge, this study provides the first report of sPD-1 levels in human cancer. The clinical utility of sPD-1 as a biomarker in cancer therapy is yet to be evaluated.
CONCLUSION
PD-1 protein is an important co-inhibitory receptor expressed on the surface of activated T and B lymphocytes. An immunoassay for the measurement of a soluble form of PD-1 protein has been developed and validated for supporting clinical studies. This total sPD-1 assay measures soluble PD-1 in normal and diseases human sera in the presence of the human anti-PD-1 antibody, nivolumab, and the endogenous ligands of PD-1 protein, PDL-1 and PDL-2.
Electronic supplementary material
(DOC 347 kb)
(DOC 38 kb)
(DOCX 13 kb)
Acknowledgments
The authors wish to thank Dr. J. Grosso for helpful discussions during assay development; Mian Gao, Lin Cheng, Kristina Moore, and Bozena Abramczyk for recombinant PD-1 cloning and expression; S Crawford for his assistance with the DoE study, F Nabbie and J Postelnek for their technical assistance during assay optimization and sample analyses. We also thank Drs. Melody Woods and Linda Robbie (Quintiles Inc.) for providing assay performance results included in Fig. 4b.
Financial & competing interests disclosure
All authors in this manuscript are employed by Bristol-Myers Squibb Co.
References
- 1.Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol. 1996;8(5):773–80. doi: 10.1093/intimm/8.5.773. [DOI] [PubMed] [Google Scholar]
- 2.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209(2):201–9. doi: 10.1084/jem.20112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, Thudium KB, Han M, Wang X-T, Huang H, Feingersh D, et al. In vitro characterization of the Anti-PD-1 Antibody Nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014. [DOI] [PubMed]
- 6.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubat T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–72. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 8.Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1) Genomics. 1994;23(3):704–6. doi: 10.1006/geno.1994.1562. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen C, Ohm-Laursen L, Barington T, Husby S, Lillevang ST. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005;235(2):109–16. doi: 10.1016/j.cellimm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Wan B, Nie H, Liu A, Feng G, He D, Xu R, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. 2006;177(12):8844–50. doi: 10.4049/jimmunol.177.12.8844. [DOI] [PubMed] [Google Scholar]
- 11.Greisen S, Rasmussen T, Stengaard-Pedersen K, Hetland M, Hørslev-Petersen K, Hvid M, et al. Increased soluble programmed death-1 (sPD-1) is associated with disease activity and radiographic progression in early rheumatoid arthritis. Scand J Rheumatol. 2014;43(2):101–8. doi: 10.3109/03009742.2013.823517. [DOI] [PubMed] [Google Scholar]
- 12.DeSilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res. 2003;20(11):1885–900. doi: 10.1023/B:PHAM.0000003390.51761.3d. [DOI] [PubMed] [Google Scholar]
- 13.Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23(2):312–28. doi: 10.1007/s11095-005-9045-3. [DOI] [PubMed] [Google Scholar]
- 14.Milliken GA, Johnson DE. Analysis of messy data, volume I: designed experiments. CA: Wadsworth, Inc.; 1984. pp. 211–45. [Google Scholar]
- 15.Zhang X, Schwartz J-CD, Guo X, Bhatia S, Cao E, Chen L, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20(3):337–47. doi: 10.1016/S1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
- 16.Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan S, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem. 2013. [DOI] [PMC free article] [PubMed]
- 17.Lee JW. Method validation and application of protein biomarkers: basic similarities and differences from biotherapeutics. Bioanalysis. 2009;1(8):1461–74. doi: 10.4155/bio.09.130. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, McCabe T, Condra JH, Ni YG, Peterson LB, Wang W, et al. An anti-PCSK9 antibody reduces LDL-cholesterol on top of a statin and suppresses hepatocyte SREBP-regulated genes. Int J Biol Sci. 2012;8(3):310–27. doi: 10.7150/ijbs.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson G, Scott M. Determination of product shelf life and activation energy for five drugs of abuse. Clin Chem. 1991;37(3):398–402. [PubMed] [Google Scholar]
- 20.Weiss WF, 4th, Young TM, Roberts CJ. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci. 2009;98(4):1246–77. doi: 10.1002/jps.21521. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, et al. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine. 2011;56(2):231–8. doi: 10.1016/j.cyto.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Haulenbeek J, Piccoli SP. Conjugated critical reagent characterization for ligand-binding assays: using MALDI-TOF-MS as an orthogonal tool to assess assay performance. Bioanalysis. 2014;6(7):983–92. doi: 10.4155/bio.14.65. [DOI] [PubMed] [Google Scholar]
- 23.Cheng HY, Kang PJ, Chuang YH, Wang YH, Jan MC, Wu CF, et al. Circulating programmed death-1 as a marker for sustained high hepatitis B viral load and risk of hepatocellular carcinoma. PLoS One. 2014;9(11):e95870. doi: 10.1371/journal.pone.0095870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 347 kb)
(DOC 38 kb)
(DOCX 13 kb)