

Abstract
Molecular imaging allows the non-invasive assessment of membrane transporter expression and function in living subjects. Such technologies have the potential to become diagnostic and prognostic tools, allowing detection, localization, and prediction of response of tumors and their metastases to therapy. Beyond tumors, imaging can also help understand the role of transporters in adverse drug effects and drug clearance. Here, we review molecular imaging technologies that monitor transporter-mediated processes. We emphasize emerging probe substrates and potential clinical applications of imaging the function of membrane transporters in cancer.
KEY WORDS: membrane transporters, molecular imaging, multidrug resistance, near infrared, optical imaging
INTRODUCTION
Alterations in the expression of membrane transporters are hallmarks of neoplastic cells. Changes in transporter expression and their functional activity may result in altered uptake of nutrients by tumor cells and can also affect the distribution of chemotherapeutic drugs into their targets within tumors (1–4). Hence, assessing transporter-related processes can potentially assist in tumor localization, monitoring, and predicting response to therapy and in drug development. In this setting, whole body molecular imaging offers a very attractive, non-invasive tool for tracking altered transporter function and for directly evaluating drug distribution within tumors over time. Moreover, whole body imaging can provide information on the role of transporters in drug distribution, elimination, and toxicity. The most prominent example of how transporter activity is being utilized for improving cancer diagnosis and for monitoring treatment is the use of positron emission tomography (PET) and [18F]-fluorodeoxyglucose ([18F]-FDG). The major proteins associated with altered [18F]-FDG uptake in tumors and metastases are glucose transporters, primarily GLUT-1, and hexokinases (5,6). However, glucose transporters have not been directly associated with drug disposition and, therefore, will not be further discussed here.
The goal of this paper is to review the available imaging techniques and their respective transporter probes. Particularly, we focus on molecular imaging of intact living subjects and on probes that can serve as biomarkers for drug therapy or for tumor localization. Detailed discussions of microscopic imaging technologies and specific imaging modalities are available elsewhere (7,8).
THE TUMOR BARRIERS
To exert an effect on tumor cells, drugs need to achieve sufficient concentrations throughout the tumor and its metastases. Systemic distribution and elimination are important factors that can limit the amount of drug reaching the tumor, especially for tumors (or their metastases) located at sanctuaries such as the brain. Within tumor tissue, the barriers affecting drug and probe distribution are highly dynamic and poorly understood (9). Tumors are usually characterized by microenvironment composed of a heterogeneous mixture of cells and a chaotic structure of blood vessels, resulting from disorganized angiogenesis. This is accompanied by hemorrhages, high interstitial pressure, and hypoxia. Tumors may also be poorly penetrable by drugs because of a massive stromal component and physical distance from blood vessels (9). Tumor cells themselves express a variety of uptake and efflux transporters which can affect the cellular distribution of drugs and probes and contribute to the development of drug resistance (1–4). The majority of these transporters are also expressed in normal tissues involved in systemic drug disposition, such as the intestine, the liver, the kidneys, and the capillary endothelial cells that form the blood brain barrier (10,11).
The major transporters involved in drug disposition were highlighted in a series of white papers by the International Transporter Consortium (e.g., 12–14). Among these are the adenosine triphosphate-binding cassette (ABC) transporters P-glycoprotein (P-gp; encoded by the gene ABCB1), members of the multidrug resistance-associated proteins (MRP) family (ABCC), and the breast cancer resistance protein (BCRP; ABCG2) (11), as well as members of the solute carrier (SLC) superfamily: the organic cation transporters (OCTs) 1 (SLC22A1) and 2 (SLC22A2); the organic anion transporters (OATs) 1 (SLC22A6) and 3 (SLC22A8); the organic anion transporting polypeptides (OATPs) 1B1 (SLCO1B1), 1B3 (SLCO1B3), and 1A2 (SLCO1A2); and the multidrug and toxin extrusion proteins (MATEs, SLC47A). The equilibrative nucleoside transporters (ENTs) 1 (SLC29A1) and 2 (SLC29A2) were also described.
P-gp is the most extensively studied ABC transporter. P-gp overexpression has been associated with chemotherapy failure in many hematological and solid tumors, including lymphomas and leukemias and kidney, colon, and liver cancers (2). P-gp substrates include chemotherapeutic agents central to most chemotherapeutic regimens, such as vinca alkaloids, anthracyclines, taxanes, epipodophyllotoxins, camptothecins, and tyrosine kinase inhibitors (11). Generally, P-gp substrates are highly lipophilic, which may lead to retention in tissues with high fat content and subsequent high background in some imaging studies.
MRPs are widely expressed in various types of cancers including myeloma, breast cancer, lung cancer, sarcoma, and in chronic lymphocytic and prolymphocytic leukemia. MRP overexpression has been correlated with drug resistance in prostate, lung, and breast cancer (2). MRPs confer resistance to methotrexate, thiopurine analogs, and camptothecins (11).
BCRP has been associated with drug resistance in breast cancer and leukemia (2). BCRP substrates include mitoxantrone and camptothecins. There is overlapping substrate specificity of BCRP with both P-gp and the MRPs (11).
Similarly to ABC transporters, members of the SLC superfamily are expressed ubiquitously in normal tissues such as the brain, kidneys, liver, placenta, and lungs and in various tumors. These transporters are particularly important for the cellular uptake of water-soluble molecules, such as folate antagonists (OATPs, OATs) (4,15), nucleoside analogs (ENTs) (16,17), and platinum-based drugs (OCTs) (18). Additional substrates include taxanes, camptothecins (OATPs) (4), tyrosine kinase inhibitors (OCTN2, encoded by SLC22A5, and OATPs) (4,18), and the antimetabolite melphalan (the system L transporter type 1; LAT1; SLC7A5) (19).
Altered expression of SLC transporters has been identified in a variety of tumors. For example, the expression of OATP1B1 and OATP1B3 tends to be reduced in poorly differentiated hepatocellular carcinomas (HCCs) (4), whereas highly differentiated HCCs may be characterized by elevated OATP expression (20). OATP1A2 expression is enhanced in malignant breast tissue (4). Two amino acid transporters, LAT1 and the system alanine–serine–cysteine transporter type 2 (ASCT2; SLC1A5), are upregulated in many human cancers and their expression is associated with a poorer prognosis (18,21). In the treatment of pancreatic cancer, both ENT1 (16) and the concentrative nucleoside transporter 1 (CNT1; SLC28A1) (3) have been highly implicated in gemcitabine chemoresistance and patient outcome. In addition, transporters of the SLC superfamily, e.g., OATPs and LAT1, are key players in cancer pathology, as they may provide hormones and other nutrients to hormone-dependent tumors to maintain the proliferation of cancer cells (4,18).
Due to the complexity of biological systems, in vivo studies remain a key step in identifying which transporters play roles in drug disposition (22). Nonetheless, conventional methods that involve tissue sampling are not suitable for investigating the function of transporters in non-clearance organs due to ethical issues (10). Non-invasive molecular imaging methodologies are expected to render these issues irrelevant and, therefore, are an emerging field in transporter biology. Importantly, imaging allows using the statistical power of longitudinal studies, thereby reducing the numbers of patients or animals studied (8,23) and the numbers of tissue or body fluid samples per subject required for analysis (hence, "a picture is worth a thousand tubes"; Table I).
Table I.
Comparison of Imaging Methods vs. Traditional In Vivo/In Vitro Methods
| Characteristic | Imaging methods | Traditional methods | Comments | |
|---|---|---|---|---|
| Clinical applicability | Yes | Yes | Clinical applicability of bioluminescence imaging is unlikely | |
| General considerations | Invasiveness | No | Yes | Traditional methods require biopsy or sampling of body fluids from non-clearance organs or tumorsa |
| Repeat studies in same subject | Yes | No | Imaging studies allow minimizing the number of subjects and improve statistical power compared to conventional destructive biodistribution analyses performed by collecting tissue samples | |
| Health riskb | Potentially | No | Primarily if radioactivity is involved | |
| Whole body analysis possible | Yes | No | Restricted to small animals in optical imaging (limited depth of signal detectability) | |
| Direct cellular and subcellular distinction | Challenging | Yes | Modeling and simulations can be useful in imaging studies | |
| Permits studying cells in their natural environment | Yes | No | Least amount of perturbation of normal processes | |
| Requirement for specialized infrastructure | Yes | No | Infrastructure required for molecular imaging studies may include a cyclotron, scanners, multidisciplinary team | |
| Transporter expression | Expression level | Yes | Yes | |
| Transporter functional activity | Range of substrates studied | Narrow | Wide | Restricted to substrates detectable by the imaging modality or which can be labeled (e.g., by 11C for PET) |
| Assay sensitivity | Fento- to picomolar in PET | Picomolar | PET imaging allows the use of microdoses | |
| Drug metabolite distinction | No | Yes | Modeling may be useful in imaging studies | |
MOLECULAR IMAGING
Molecular imaging might be defined as the generation of images in which the intensity at each point is proportional to the local quantity of a specific molecular probe (23). Most imaging techniques can be performed non-invasively in the intact organism with adequate temporal and spatial resolution (Table II). Furthermore, multimodal imaging can correlate the parameter indicating transporter function with the anatomical findings or with other physiological parameters (23).
Table II.
Overview of Imaging Modalities Applied in Membrane Transporters Studies
| Technique | Modality | Spatial resolution | Temporal resolution | Depth of Penetration | Sensitivity | Used clinically |
|---|---|---|---|---|---|---|
| PET | High-energy gamma rays | 1–2 mm (preclinical) 5–7 mm (clinical) |
Seconds–minutes | Limitless | 10−11 to 10−12 M | Yes |
| SPECT | Low-energy gamma rays | 1–2 mm (preclinical) 8–10 mm (clinical) |
Minutes | Limitless | 10−10 to 10−11 M | Yes |
| MRI | Radiowaves | 25–100 μm (preclinical) ∼1 mm (clinical) |
Minutes–hours | Limitless | 10−3 to 10−5 M | Yes |
| Optical fluorescence imaging |
NIR | 2–3 mm | Seconds–minutes | <1 cm | 10−9 to 10−12 M | Emerging clinical utility |
| Optical bioluminescence imaging |
Bioluminescence | 3–5 mm | Seconds–minutes | 1–2 cm | 10−15 to 10−17 M | Low potential for clinical translation |
Imaging technologies can be grouped by the energy used to derive visual information (X-rays, positrons, photons, etc.), the type of information obtained (anatomic, physiological, and molecular/cellular), or the spatial resolution attained. Magnetic resonance imaging (MRI), PET, and single photon emission computed tomography (SPECT) are routinely used in clinical practice, whereas optical imaging technologies are currently employed mostly in preclinical studies or in the surgical suite (7) (Table II).
Desired characteristics of effective transporter probes include high affinity and selectivity for the transporter of interest, low nonspecific binding in tissues in which the transporter is functional, and chemical purity of the signal (absence of metabolites detectable by the respective imaging technique) (27). Genetic or pharmacological knockout of the transporter(s) of interest helps identify their contribution to signal intensity.
In imaging studies, intravascular, extracellular, and intracellular compartments, as well as stem cells within tumors, are indistinguishable. In addition, as in traditional pharmacokinetic studies, the probe signal from the tumor is related not only to the function of membrane transporters but also to its unbound concentration in plasma and to tumor retention. Correction for concentrations in plasma can be accomplished by direct blood sampling. In nuclear imaging, the signal intensity in blood pools such as the heart, the aorta, or other major arteries within the field of view is often used for such corrections.
Quantification of Imaging Data
The quantification of imaging data is presented here in brief. Further information is available in recent publications (24,27,28). Overall, data obtained from imaging studies can be analyzed by identification of regions of interest and use of non-compartmental or compartmental approaches. Alternatively, pixel-by-pixel analysis may be applied to the entire image (28). In many cases, concentrations measured in imaging studies are absolute (e.g., mCi/cc). However, in MRI and optical imaging studies, results often allow relative between-group comparisons only. In optical imaging, even between-tissue comparisons are semi-quantitative because light emission varies among organs and tissues, as described below.
Non-compartmental methods allow comparisons of parameters such as percent of injected dose (% ID), standard uptake values (SUV), and the area under the probe concentration-time curve (AUC). The %ID and SUV are used for normalizing the signal observed in the region of interest for the amount of tracer injected into the subject. The value of %ID can range from 0 to a value that exceeds 100% if a large percent of the dose accumulates into a site with a mass less than 1 g (or mL). The SUV, commonly used in nuclear medicine, takes into account, in addition to the injected dose, the body weight (SUV = local tissue radioactivity concentration/(administered radioactivity/body weight)), but represents a single "snapshot" of a dynamic process (28). In contrast, the AUC integrates the concentrations of probe activity in tissues over time. Furthermore, the tissue AUC values can be normalized by blood (or plasma) AUC to correct for alterations in systemic blood concentrations of the probe. In addition, AUC can be utilized in any type of imaging study in which a series of images is acquired over time, as previously described for PET (29), SPECT (30,31), and optical (32) imaging studies of P-gp activity. Non-compartmental analysis assumes linear kinetics, which is seldom a concern in PET studies, because PET ligands are usually administered in microdoses. However, this may be an issue in MRI or optical imaging.
The compartmental approach specifies a kinetic model and may provide insight into the mechanism of drug transfer across barriers and factors affecting data interpretation, such as probe metabolism (28). This approach is best applied to PET data because of the high temporal resolution (Table II), but may also be used for analysis of data obtained through other imaging modalities. As in traditional measurement methods, the models provide rate constants such as the one-compartment model’s K1 (net influx clearance of the probe from plasma or blood to tissue), k2 (an efflux rate constant), and the two compartment model’s K1 (distributional clearance into the tissue, CL12), k2 (the transport process from the tissue back into the blood), and k3 and k4 (the bidirectional exchange between the two tissue compartments). The microconstants can also be used to calculate macroparameters such as 'distribution volume' (DV) of the probe. The compartmental approach has been applied to data of [11C]-verapamil uptake in malignant tumors (33) and the fetus (34) as well as [11C]-verapamil and [11C]-N-desmethyl-loperamide ([11C]-dLop) uptake in the brain (35–38). Both [11C]-verapamil and [11C]-dLop are avid P-gp substrates; therefore, almost all of the effect attributed to P-gp is the decrease in K1 (block the entry of the radioligand to the intracellular compartment) (27,37). Importantly, because blood flow influences the initial transfer of many probes into tissues, transport (K1) may be normalized by flow.
The pixel-by-pixel analysis utilizes statistical methods, such as statistical parametric mapping (SPM). Each voxel can be analyzed using any standard statistical test. The resulting statistical parameters are assembled into an image—the SPM, which is used for making the statistical inferences. The advantage of the statistical mapping is that no assumption is made regarding the relevant regions and that it can be used to analyze the entire tissue observed in the image (e.g., brain) (28). Thus, it can detect changes outside a priori specified regions and is therefore useful for identification of regional changes in transporter activity, e.g., in epilepsy (39) and Alzheimer’s disease (40,41), and potentially also in tumors and their metastases.
NUCLEAR IMAGING
PET and SPECT monitor the distribution of systemically administered radiotracers within living subjects over time, with limitless depth of penetration (8) (Table II). The use of PET and SPECT allows a three-dimensional assessment of the distribution of the isotope (as opposed to the two-dimensional planar scintigraphy assessment). Although both PET and SPECT are commonly applied in the clinic, they involve the use of ionizing radiation, are technically demanding, and are costly. The advantages of PET over SPECT include higher sensitivity, better temporal and spatial resolution, and quantifiable recordings. However, in contrast to PET imaging, SPECT allows simultaneous imaging with two or more differently labeled radioisotopes. Furthermore, its cost is lower, and many of the probes are commercially available (23). Both imaging technologies are widely applied in the clinic (8).
PET radioisotopes commonly utilized for imaging transporter-related processes typically contain positron-emitting carbon-11 (11C) or fluorine-18 (18F), with radioactive half-lives of 20 and 110 min, respectively. Due to these short half-lives, PET radioligands are generated in a cyclotron and are rapidly incorporated into the molecule of interest immediately prior to their use (8). In SPECT, the commonly used isotope is technetium-99 m (99mTc; half-life, 6 h) (42). PET and SPECT radioligands are administered in subpharmacological trace quantities (nanogram to milligram range), which are unlikely to exert pharmacological effects. However, under non-linear conditions (for example, saturation of low-capacity transporters), the microdoses of radiolabeled drugs typically used in imaging studies may not be representative of their pharmacokinetics following administration of therapeutic doses.
PET
Nowadays, there are dozens of known PET radioligands used to image the expression and function of a variety of transporters (Table III). Probes can be radiolabelled transporter substrates, transporter inhibitors, or prodrugs converted into transporter substrates at the site of interest (42). PET imaging of drug transporter-mediated processes has been extensively reviewed in the past few years (e.g., 10,22,27,42) and will be discussed here briefly.
Table III.
Examples of Substrate Probes Used in Preclinical or Clinical Imaging Studies
| Transporter | Modality | |||||
|---|---|---|---|---|---|---|
| PET | SPECT | MRI | NIR | BLI | ||
| ABC superfamily | P-gp | [11C]-Daunorubicin (43) [11C]-N-Desmethyl-loperamide (48) [11C]-Elacridar (49,50) [11C]-Gefitinib (51) [11C]-Imatinib (10) [11C]-Paclitaxel (10) [11C]-Tariquidar (52) [11C]-Topotecan (53) [11C]-Verapamil (29) |
[99mTc]-Sestamibi (44,45) [99mTc]-Tetrofosmin (44) |
Rhodamine 800 (46) | Coelenterazine (47) | |
| BCRP | [11C]-Elacridar (49,50) [11C]-Gefitinib (51) [11C]-Imatinib (10) [11C]-Tariquidar (52) [11C]-Topotecan (53) |
d-Luciferin (54) Coelenterazine (55) |
||||
| MRPs | [11C]-Paclitaxel (10) | [99mTc]-Sestamibi (56) [99mTc]-Tetrofosmin (45) |
BOPTA (57) EOB-DTPA (59) |
ICG (58) | ||
| SLC superfamily | OATPs | [11C]-Rosuvastatin (60) [11C]-Telmisartan (10) [11C]-TIC (10) |
[99mTc]-MAG3 (61) [99mTc]-Mebrofenin (63) [99mTc]-PMT (66) |
BOPTA (57) EOB-DTPA (64) |
ICG (62) IR-783 (65) MHI-148 (65,67) |
|
| OATs | [11C]-Topotecan (53) | [99mTc]-MAG3 (61) | ||||
| OCTs | [123I]-MIBG (10) | |||||
| NTs | [18F]-FLT (68) | |||||
| LATs | [18F]-FACBC (21) | |||||
ABC adenosine triphosphate-binding cassette, BCRP breast cancer resistance protein, BLI bioluminescence imaging, BOPTA gadobenate dimeglumine, EOB-DTPA gadoxetate dimeglumine, [ 18 F]-FACBC [18F]-1-amino-3-fluorine 18-fluorocyclobutane-1-carboxylic acid, [ 18 F]-FLT [18F]-3′-deoxy-3′-fluorothymidine, ICG indocyanine green, LATs system L transporters, [ 99m Tc]-MAG3 [99mTc]-mercaptoacetylglycylglycylglycine, [ 123 I]-MIBG [123I]- metaiodobenzylguanidine, MRI magnetic resonance imaging, MRPs multidrug resistance-associated proteins, NIR near infrared, NTs nucleoside transporters, OATs organic anion transporters, OATPs organic anion transporting polypeptides, OCTs organic cation transporters, P-gp P-glycoprotein, PET positron emission tomography, [ 99m Tc]-PMT [99mTc]-N-pyridoxyl-5-methyltryptophan, SLC solute carrier, SPECT single photon emission computed tomography
The majority of probes synthesized to evaluate transporter activity target P-gp function. Among these, the most established are racemic [11C]-verapamil (29,36,69,70) and its (R)-enantiomer (39,71–73) and the radiolabeled loperamide metabolite, [11C]-dLop (48,74). In addition to these compounds, cytotoxic drugs, such as daunorubicin (43) and paclitaxel (75), have been radiolabelled and evaluated in various tumor models in rodents (Table III).
Several radiolabelled P-gp and/or BCRP inhibitors, including [11C]-tariquidar (52), [11C]-laniquidar (76), and [11C]-elacridar (49,50), were developed as markers of P-gp/BCRP expression. However, in vivo experiments and further in vitro characterization suggested that these compounds are also transported substrates of P-gp, BCRP, or both (42).
Indeed, many radiolabelled agents are known to interact with more than one transporter. Examples are [11C]-gefetinib (51), a dual P-gp/BCRP substrate; [11C]-topotecan (53), a substrate of P-gp, BCRP, MRP4, MATE1, and MATE2-K (10); [11C]-glyburide, a substrate of OATPs, P-gp, and BCRP (77); and [11C]-rosuvastatin (transported by OATPs, NTCP, MRP2, and BCRP) (60). In such cases, the impact of an individual transporter may be masked by the contribution of other transporters to the probe’s whole body or cellular kinetics. Furthermore, the impact of inhibition of dual or multiple transporters on the probe kinetics is not necessarily the sum of the effects of the individual transporters inhibition or knockout. For example, topotecan CNS exposure was increased 1.5-fold in Bcrp- and Mdr1a/1b-knockout mice respectively, but 12-fold in mice lacking both Bcrp and P-gp (78).
Some of the abovementioned compounds were used for imaging transporter activity in human cancer, although the paucity of data and the differences in study design and reported parameters make it difficult to compare the findings among studies. Racemic-[11C]-verapamil was first used in five cancer patients. In this study, 0.9% of the injected dose accumulated in the tumor. In comparison, the % ID in the lungs and the heart was 43% and 1.3%, respectively. The peak plasma concentration was less than 0.01% ID/mL (79). Later on, the same radioligand was administered to ten soft tissue sarcoma patients, in addition to PET markers of cellular proliferation and hypoxic volume (33). [11C]-verapamil tumor uptake was assessed by a simple 1-tissue compartmental model using the first 10 min of the uptake data to determine the initial transport (K1; mL/g/min). The results were normalized to blood flow. The tumor uptake parameters of [11C]-verapamil varied between patients and with respect to the uptake parameters of other probes in individual patients, suggesting unique tumor biology within each patient. Flow-normalized verapamil transport values in sarcomas were considerably higher than those previously calculated for the brain, likely due to higher blood flow in the normal brain than in tumors. Similarly to the initial [11C]-verapamil study, average tumor distribution of [18F]-fluoropaclitaxel in breast cancer patients was reported as percent of the injected dose, 0.01% at 1 h after injection (80).
[11C]-Docetaxel has been evaluated in patients with advanced lung tumors. Tumor radioactivity uptake was moderate, related to tumor perfusion, and highly variable between and within tumors. In a subgroup of patients who subsequently received docetaxel therapy, relatively high [11C]-docetaxel uptake was associated with improved tumor response. Less than 1% of the infused therapeutic dose of docetaxel was finally taken up by tumor tissue (81). High hepatic accumulation of both [11C]-docetaxel and [18F]-fluoropaclitaxel makes these radioligands suitable candidates for studies on thoracic, but not abdominal tumors (80,81).
Radiolabelled amino acids and nucleoside analogs have been used for human cancer imaging for many years. The most established radiolabelled amino acid analogs are LAT1 substrates, whereas the thymidine analog [18F]-3′-deoxy-3′-fluorothymidine ([18F]-FLT) is a substrate of ENT1, ENT2, CNT1, and CNT3 (SLC28A3) (68). For these compounds, tumor uptake, as demonstrated by imaging, reflects both their transport and their incorporation into cellular metabolic pathways, similar to [18F]-FDG. In recent years, LAT1 probes have been used to follow the degree of LAT1 function in response to therapy (21).
SPECT and Gamma Scintigraphy
One of the first radiopharmaceuticals utilized in vivo as a P-gp substrate was the cationic tracer [99mTc]-sestamibi (hexakis-methoxyisobutyl isonitrile; MIBI) (56) (Table III). Consequently, several clinical studies demonstrated increased [99mTc]-sestamibi accumulation in tumors following P-gp inhibition and a correlation between [99mTc]-sestamibi efflux from tumors and P-gp expression (30,31,82,83). For example, in a phase I trial of the P-gp inhibitor PSC 833 in nine patients with metastatic renal carcinoma, the tumors in two of the patients could be seen only during treatment with PSC 833. In the others, the mean tumor/heart AUC ratio was significantly higher in the presence of PSC 833 as compared to baseline (31). In 13 of 17 patients with metastatic cancers, the tumor/heart [99mTc]-sestamibi AUC increased after the administration of the P-gp inhibitor tariquidar, with an increase of 36%–263% seen in eight of the patients (30). Breast cancer tumors expressing high amounts of P-gp displayed a 2.7-fold higher efflux rate for [99mTc]-sestamibi compared to tumors expressing little or no P-gp (82).
Alternative P-gp substrates for SPECT imaging are [99mTc]-tetrofosmin (44) and other metalloorganic complexes (42,84). It is noteworthy that [99mTc]-sestamibi and [99mTc]-tetrofosmin are also substrates of MRP1 (45) (Table III). In addition, they accumulate reversibly within mitochondria, which have electronegative membrane potentials (84,85). Some 99mtechnecium-based probes have been demonstrated to be SLC transporters substrates. The organic anion [99mTc]-mercaptoacetylglycylglycylglycine (MAG3), used in renal function studies, is a substrate of OAT1 (61) and the hepatobiliary scintigraphy tracers [99mTc]-N-pyridoxyl-5-methyltryptophan ([99mTc]-PMT) (66) and [99mTc]-mebrofenin (86) are OATP1B1 and OATP1B3 substrates. Following uptake into hepatocytes, [99mTc]-mebrofenin is excreted in the bile canaliculi by MRP2 and MRP3 (87).
Despite the involvement of SLC transporters in tracer kinetics, the abovementioned studies have confirmed that [99mTc]-sestamibi efflux rate can serve as a tool for predicting the degree of MDR and patient response to therapy. The utility of SPECT for assessment of the function of other transporters has not been established yet.
MRI
MRI is a non-invasive tomographic imaging modality that uses a powerful magnet and radiofrequency energy to visualize the body’s internal structures. MRI offers high spatial resolution with limitless depth of penetration and excellent soft tissue contrast (Table II). The technique’s sensitivity is lower in orders of magnitude than that of radionucleotide and optical imaging (8), and only certain nuclei, such as hydrogen or phosphorous, are detectable. Accordingly, imaging typically involves the detection of water protons because water is abundant in tissues. Nonetheless, variation in resonance frequencies according to the nuclei electronic environment (e.g., water vs. lactate molecules) also allows assessment of many metabolites and amino acids in tissue spectra. Furthermore, administration of an appropriate contrast agent such as paramagnetic gadolinium (Gd) complexes allows the imaging of various tumor processes. Due to the MRI’s low sensitivity, the target blood concentrations of the contrast agents are in the millimolar range and toxicity as well as transporter saturation issues are important considerations. In addition, signal intensities recorded on MR images are not linearly related to concentrations. The time for image acquisition depends in part on the resolution required and is typically several minutes.
Many Gd chelates, such as gadopentetate dimeglumine (Gd-DTPA; Magnevis), gadoterate meglumine (Gd-DOTA; Dotarem), and gadodiamide (Gd-DTPA-BMA; Omniscan), distribute nonspecifically throughout the plasma and the interstitial space (with the exception of the brain) and thus have been classified as "extracellular" (88). A typical use of these compounds is the detection of brain tumors based on increased blood–brain barrier permeability, which results in extravasation of the contrast agents and enhancement of tumor contrast (89). Thus, the interaction of MRI probes with membrane transporters has been mostly investigated with regard to “hepatobiliary” contrast agents such as gadoxetate dimeglumine (EOB-DTPA; Eovist) and gadobenate dimeglumine (BOPTA; MultiHance) used for visualizing the morphology of the liver and diagnosing focused lesions (87,90). These compounds are mostly negatively charged and have been demonstrated to act as substrates of organic anion transporters (87). BOPTA is a substrate of rOATP1A1, rOATP1A4, rOATP1B1, and rMRP2 (57), and its hepatic accumulation is inhibited in a dose-dependent manner by the OATP inhibitor rifampin (91) (Table III). EOB-DTPA is a substrate of OATP1B1, OATP1B3, the human sodium-taurocholate-co-transporting polypeptide (hNTCP) (64), and the ABC transporters MRP2 and MRP3 (59) (Table III). These compounds enter the hepatocytes through sinusoidal OATPs and are removed through biliary excretion or efflux back into sinusoids by MRP2 and MRP3, respectively (87). The OATP-mediated hepatic uptake of EOB-DTPA has been suggested to be the basis for its use for detection and characterization of focal hepatic lesions. The majority of the differences between lesions and their respective backgrounds are observed in the hepatobiliary phase of distribution (entry of contrast agent into hepatocytes; 20 min after EOB-DTPA injection). Most moderately to poorly differentiated hepatocellular carcinomas show hypointensity (are darker) compared to background liver, likely due to low tumor OATP1B3 expression. However, some hepatocellular carcinomas that are well differentiated are hyper-intense because the tumors overexpress functional OATPs, allowing for the contrast agent to enter into cells and become trapped within tumor cells or bile ductules (87). Indeed, several studies demonstrated that intensity is correlated with the tumoral OATP1B3 expression (63,92). In one of these studies, intensity was also related to the expression pattern of MRP2 within the nodules (63).
OPTICAL IMAGING
Optical imaging of living subjects can be divided into fluorescence and bioluminescence imaging. Both imaging modalities are highly sensitive and, since they involve the detection of low-energy photos, are relatively safe. However, this also means that the depth of penetration is limited (Table II). Nevertheless, clinical translation is feasible where depth of penetration is not an issue, for example, in the endoscopy setting and when intraoperative devices are involved (8). In vivo optical imaging is easily accessible, with relatively low instrumentation costs and is easy to perform: animals are anesthetized, the respective substrate is injected, and the animal is placed in a dark chamber with a charged coupled device (CCD) camera. Simplicity and applicability make optical imaging techniques particularly suited for drug development and validation processes (23).
In bioluminescence imaging (BLI), light is generated by a chemiluminescent reaction between a luciferase enzyme (usually the firefly luciferase in ADMET assays) and its substrates. The photons emitted from this reaction extend into tissue-penetrating red and near-infrared (NIR) wavelengths (approximately 600 nm) (93).
Tomographic fluorescence systems (fluorescence molecular tomography, FMT) produce three-dimensional maps of fluorochromes and are quantitative. FMT is often combined with computed tomography or MRI to improve image visualization and provide anatomical information. More common systems, which obtain planar images, are useful for imaging events in surface tumors (xenografts), surgically exposed organs, or for intraoperative imaging. Using fluorochromes with emission in the NIR part of the electromagnetic spectrum (650–900 nm) helps minimize light attenuation and scattering within living organisms and decrease tissue autofluorescence, thereby increasing imaging depth and sensitivity, yet many NIR fluorochromes suffer from poor photostability, high plasma protein binding rate (which in fact may enhance emission intensity), and aggregation and quenching at high concentrations (Fig. 1). Quantification of fluorescent optical imaging data is confounded by “surface weighting” problems (meaning that anything closer to the surface will appear brighter) and variations in the extent of light attenuation in different organs and tissues (8). Many NIR fluorophores are also prone to non-specific binding to tissues due to their hydrophobicity, thus typically resulting in increased background fluorescence. In addition, the doses of fluorescent agents needed for imaging are considerably larger than for nuclear imaging. In preclinical studies, intervals in injection and positioning time between simultaneously imaged animals should be taken into account during image analysis (Fig. 2).
Fig. 1.
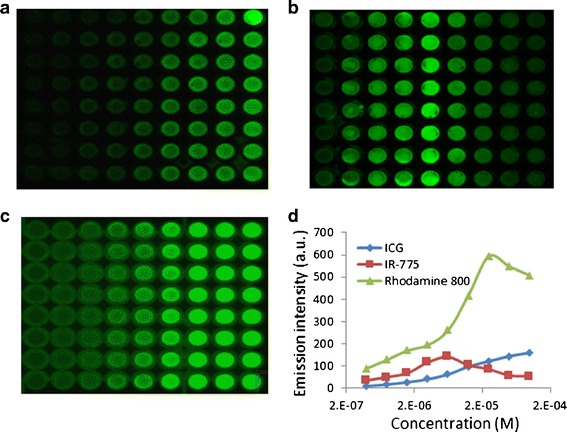
Non-linearity of the near-infrared emission intensity in vitro. a–c Representative images of plates incubated with increasing concentrations of indocyanine green (ICG) (a), IR-775 (b), or rhodamine 800 (c). d Quantification of emission intensity. MDCK II cells were seeded in 96-well plates at 8 × 104 cells/well. When achieved monolayers, cells were incubated with the indicated concentrations of the dyes for 1 h, then scanned by a molecular imager. At the studied concentrations, IR-775 and rhodamine 800 exhibited a quenching phenomenon which resulted in decreased signal intensity at higher concentrations. As the dye solution is more saturated, the distance between the fluorochrome molecules decreases and fluorescence resonance energy transfer (FRET) takes place (94). Considering that emission intensity is an indirect measurement of dye’s quantities, this artifact implies lesser concentrations in the cell, while, in fact, the concentrations are higher
Fig. 2.
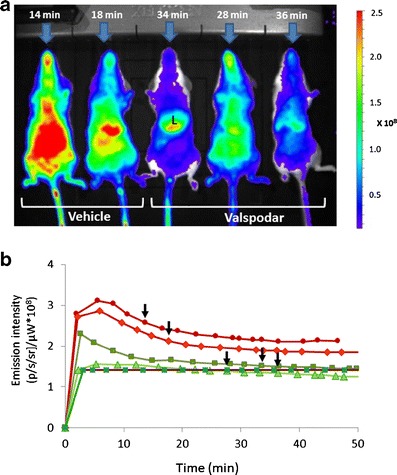
Whole body images of mice treated with indocyanine green in the presence and the absence of valspodar. a A representative image showing five female mice treated with indocyanine green (ICG; 8 mg/kg, i.v.). Forty-five minutes before ICG injection, mice were treated i.p. with valspodar (12.5 mg/kg, i.p.) or the vehicle. Shown is the time elapsed (min) between ICG injection and image acquisition for each mouse. Also indicated is the liver (L). b Concentration-time curves of liver emission intensity. Visual inspection of the image may lead to bias due to the different time intervals between injection and image acquisition for each mouse (indicated by arrows). The time intervals result from the need to inject the probe and induce anesthesia before placement in the dark chamber
Unlike fluorescence techniques, there is no inherent background with bioluminescence, which makes this technique highly sensitive. However, in contrast to fluorescence optical imaging, the clinical translation of this method is very limited because mammalian cells do not naturally express luciferases (8).
NIR Imaging
Organic cyanine dyes are the most widely used NIR probes. Among these, indocyanine green (ICG), the only FDA-approved cyanine dye, has been used for decades as a non-targeting contrast agent for optical imaging. ICG is approved for determining cardiac output, liver blood flow and hepatic function, and for ophthalmic angiography. In addition, it is used “off-label” to visualize structures that are filled with fluids such as lymph (95). Due to its wide clinical application, ICG is the best characterized cyanine dye in vivo. ICG binds to plasma proteins, distributes uniformly in the blood within 2 to 3 min after intravenous injection, and is exclusively cleared unchanged by the liver (96). Hepatic clearance is limited by blood flow (90). Uptake into hepatocytes is mediated by OATP1B3 and the human Na+-taurocholate co-transporting polypeptide (NTCP) (62), and its excretion into the bile is mostly through MDR3 (ABCB4) and MRP2 (ABCC2) (58) (Table III). Under pathophysiological circumstances, ICG transport capacity may be lowered because of reduced blood flow or OATPs’ downregulation (97,98). Accordingly, ICG clearance from blood, which can be simply measured by an optical sensor placed on the finger, reflects hepatic blood flow and hepatic uptake and efflux transporter activity (90). Another recently suggested application of ICG is visualizing the resection limits of hepatocellular carcinomas during surgery by a fluorescence camera. This is because, similar to EOB-DTPA, ICG tends to accumulate in well-differentiated hepatocellular carcinomas (20). Interestingly, ICG accumulation in HCC was positively correlated with the expression of NTCP and OATP1B3 (86).
Recent studies demonstrated a role for OATPs in tumor accumulation of additional cyanine dyes. For example, the OATP inhibitor bromosulfophthalein (BSP) suppressed the accumulation in tumor cells of IR-783 (65), MHI-148 (65,67), and IR-780 iodide (99), primarily through inhibition of OATP1B3 (67,99) (Table III). Further studies indicated that OATP1B3 expression and related MHI-148 accumulation were induced by hypoxia (67). In addition to diagnostic applications, IR-780’s accumulation within tumor cells was also associated with inhibition of cell growth in vitro and of tumor recurrence in the mouse syngeneic Lewis lung carcinoma xenograft model, suggesting that IR-780 could be used as a theranostic agent for the suppression of tumor recurrence (100). An interesting attempt to utilize near-infrared conjugates of 2-deoxyglucose as [18F]-FDG analogs for optical imaging revealed that conjugation of large bulky fluorophores to 2-deoxyglucose disrupts the facilitated transport and retention of these probes in cells. Thus, these probes cannot replace [18F]-FDG PET imaging as biomarkers of tumor cell viability and metabolism (101).
Optical imaging was also used for evaluation of P-gp or BCRP-mediated efflux at the blood–brain barrier. In mice, P-gp inhibition enhanced the cerebral emission intensity of rhodamine 800 (46), and, to a lesser extent, that of ICG (32). In a recent optical imaging study, synthetic human β-amyloid1–40 peptide labeled with Cy5.5 accumulated to a greater extent in the brains of Abcb1 and Abcg2 knockout mice compared to wild-type controls (102).
BLI
BLI modalities acquire macroscopic information in vivo through two basic strategies: (1) genetically encoded reporters (luciferase assays) for studying the regulation of a gene of interest and (2) assays of injected luciferin derivative for studying the functional activity of membrane transporters (103).
In genetic reporter assays, a luciferase complementary DNA (cDNA) is placed under the transcriptional control of the gene of interest’s regulatory elements in a plasmid vector used to transfect mouse embryonic cells. The first transgenic mouse reporter model was based on a firefly luciferase (fLUC) cDNA inserted into the murine mdr1a genetic locus by homologous recombination and allowed the imaging of the mdr1a gene expression (104). Using this model, it was demonstrated that abdominal luminescence intensity reflects basal and pregnenolone-16α-carbonitrile (PCN)-induced intestinal mdr1a and fLUC mRNA expression. Later on, the same group crossed mdr1a.fLUC mice with a genetically knockout pregnane X receptor (PXR; a master regulator of inducible MDR1 expression) strain (105). Luminescence intensity was enhanced upon treatment with PCN, a strong mouse PXR ligand, and two therapeutically relevant taxanes, but only PCN and docetaxel appeared to act primarily via PXR. Therefore, these mice models offer modular tools for preclinical studies on the extent and the kinetics of MDR1 induction by pharmacological agents or under various physiological and pathological conditions.
The transporters which determine the pharmacokinetics of luciferin and its derivatives are beginning to be identified. First, BCRP expression and function were found to have substantial influence on d-luciferin-dependent bioluminescent output in vivo in mice bearing BCRP-overexpressing and control tumor xenografts (54) (Fig. 3; Table III). The BLI-based assay was later shown to be an efficient method to identify new inhibitors of BCRP (106). The same group also demonstrated that d-luciferin cellular accumulation is facilitated by an uptake mechanism, likely OCTN1 (107). The work of Bakhsheshian et al. (108) showed that d-luciferin entry into the brain is restricted by BCRP and that this process correlates with BCRP expression and function. This study provided the groundwork for a novel method to examine transporter function at the blood–brain barrier. Most recently, studies in cell lines and in mouse xenografts demonstrated that coexpression of Oatp1 with luciferase increased luciferin luminescence intensity by several folds. The authors suggested that coexpression of Oatp1 could also be used to reduce the amount of luciferin required for BLI studies (109). In addition, the imidazolopyrazine derivative of luciferin, coelenterazine, was reported to be a substrate of P-gp (47) and BCRP (55) (Table III).
Fig. 3.
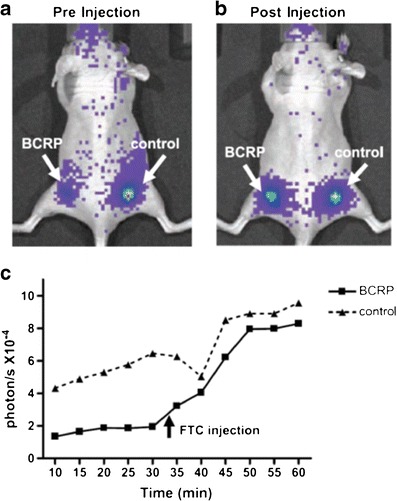
The effect of BCRP on d-luciferin bioluminescence in living mice. HEK-293 cells stably transfected with BCRP or empty vector (control) were implanted to the flanks (left and right, respectively) of immunocompromised mice. a A representative mouse showing bioluminescence imaging (BLI) acquired 30 min after i.p. administration of d-luciferin, immediately before administration of the BCRP inhibitor fumitremorgin C (FTC). b The same mouse as in a imaged 12 min after i.v. administration of FTC. c Time course of BLI signal from control and transgenic ABCG2/BCRP-expressing tumors. The BLI signal from the BCRP-transfected tumor was 70% less than the control tumor before FTC injection and increased 4-fold after FTC injection to levels comparable to the control tumor. Reprinted with permission from (106)
VARIATION IN DRUG TRANSPORTER FUNCTION
Variation in drug transporter expression, activity, or both is now increasingly recognized as a determinant of between- and within-subject variability in response to commonly prescribed drugs. Clearly, factors that affect transporter-based variability in drug response may also apply to variation in the signal intensity of substrate probes. Such factors include genetic polymorphisms in genes encoding transporter proteins (usually affecting the systemic disposition of the probe), acquired changes under disease states and interactions with medications, which may affect both the systemic disposition and the tumor uptake of the probe. Table IV describes examples of the contribution these factors make to variability in probes’ signal. Correction of tumor signal for the systemic blood concentration of the probe, as described above, can account for an altered signal due to changes in blood concentrations.
Table IV.
Examples of Probe-Drug Interactions Relevant to Cancer Therapy in Humans
| Classification | Example | Mechanism | Potential outcome | References |
|---|---|---|---|---|
| Modulation of transporter function in probe-eliminating organs | Rifamycin-ICG | OATP-inhibition | Reduced bile duct visibility | (110) |
| Altered tumor perfusion | Bevacizumab-[11C]-docetaxel | Reduced angiogenesis/blood flow | Reduced [11C]-docetaxel signal | (111) |
| Inhibition of tumoral membrane transporters | Lapatinib-gadoxetate disodium (EOB) | OATP1B1 inhibition | Compromised ability to diagnose liver tumors | (112) |
| Altered tumoral membrane transporter expression | Capecytabine-[18F]-FLT | ENT1 redistribution to the cell membrane | Increased [18F]-FLT uptake | (113) |
| Altered probe distribution or metabolism within cells | Tariquidar-[11C]-dLop | Probe-inhibitor competition on lysosomal accumulation | Reduced signal in tissues other than the brain; reduced probe accumulation suggestive of potential reduction of the cellular uptake of cytotoxic drugs | (74) |
[ 11 C]-dLop [11C]-N-desmethyl-loperamide, [ 18 F]-FLT '3′-deoxy-3′-[18F]-fluorothymidine, ICG indocyanine green, OATP organic anion transporting polypeptide
IN VIVO APPLICATIONS OF MOLECULAR IMAGING
Imaging has become a central tool in studying cancer biology and in clinical diagnosis, prognosis, and treatment. This section provides examples for potential uses of imaging of membrane transporter activity to improve cancer therapy (summarized in Table V).
Table V.
Molecular Imaging of Transporter-Related Processes: Suggested Uses to Improve Cancer Therapy
| Utility | Example | References |
|---|---|---|
| Evaluating/predicting response to chemotherapy | [99mTc]-Sestamibi for predicting MDR | (31) |
| Investigating chemotherapy pharmacokinetics | Studying docetaxel disposition using [11C]-docetaxel | (81) |
| Enhancing the visual differences between tumor and normal tissue during surgery | ICG for labeling hepatocellular carcinomas | (95) |
| Prediction of transporter expression or activity in humans based on preclinical data | [11C]-verapamil for comparing data obtained in preclinical studies to data in humans | (114) |
ICG indocyanine green, P-gp P-glycoprotein
Evaluating and Predicting Response to Therapy
Increased ABC transporters’ expression has been associated with impaired response to chemotherapy and lower patient survival (2). Non-invasive imaging of ABC transporter functional activity has the potential to aid in more individualized, targeted treatment and to assist in the development of new agents. For example, SPECT scans with [99mTc]-sestamibi predicted response to chemotherapy in a variety of tumors. Such studies can also improve the outcomes of clinical trials with inhibitors of MDR transporters (27,30,31). In addition, PET imaging with amino acid derivatives as PET tracers in patients can indicate the degree of inhibition of LAT1 function during therapy, thus guiding the dose regimens required to achieve significant inhibition during therapy (10,21).
Investigating the Pharmacokinetics of Chemotherapeutic Drugs
In addition to information on tumor distribution and retention, non-invasive measurements of tissue concentrations of drugs can help in understanding mechanisms of their clearance and their distribution into non-target organs. Tissue imaging studies can clarify the role of pharmacogenomics, disease states, and drug (or food) interactions in the pharmacokinetics of chemotherapeutic drugs and predict adverse drug reactions. For example, recent studies demonstrated that OCT2 inhibition protects against severe cisplatin-induced nephrotoxicity without sacrificing cisplatin’s antitumor effects (115). The effectiveness of OCT inhibition in terms of renal drug distribution can potentially be evaluated through imaging studies.
Intraoperative Imaging of Tumors or Their Metastases
Insufficient visual differences between tumor and normal tissue when using white light reflectance alone have been associated with increased local, potentially preventable tumor recurrence and poor prognoses. On the other hand, preservation of important structures such as blood vessels, ureters, and bile ducts during tumor resection is almost equally important in achieving optimal patient outcome. Thus, enhancing the visual differences between tissues could advance the visual capability of the operating surgeon and provide an opportunity to improve surgical outcomes (95). This can be accomplished by the design of probes that accumulate in tumors (e.g., OATP substrates) or ones that are preferably excreted in bile or urine to identity these structures during surgery. The limited depth penetration of fluorescence imaging, which is a limitation of whole body scanning, is less of a drawback during surgery (95). For example, 5-aminolevulinic acid (5-ALA) has been increasingly used as a fluorescent dye for fluorescence-guided tumor resections (116). 5-ALA is a precursor of heme biosynthesis that leads after oral administration to selective accumulation of fluorescing protoporphyrin IX (PpIX) in malignant tumor tissue. In 1998, Stummer et al. (117) reported the clinical application of 5-ALA during resection of ten suspected malignant gliomas. Glioma tissue was intraoperatively characterized by strong PpIX fluorescence, whereas normal tissue revealed no visible PpIX. A more recent study demonstrated that the accumulation of 5-ALA-mediated PpIX in glioma cells is enhanced by increased expression of the ABC mitochondrial porphyrin importer ABCB6, and that ABCB6 expression was greatly elevated in human gliomas compared with normal brain tissue (118).
Prediction of Transporter Expression or Activity in Humans
Molecular imaging is an ideal translational tool to assess differences in transporter expression and function between species during drug development. It allows for direct assessment of the clinical relevance of drug interactions identified in vitro or in animal models. Accumulating data from studies correlating transporter expression and activity as well as studies evaluating the roles of transporters in various species indicate that scaling transporter expression from cell lines and rodents to human tissue is a reasonable approach. For example, based on data obtained from studies in rats, Hsiao et al. utilized in vitro data to predict an increase of 129% in [11C]-verapamil distribution into the human brain, a value similar to that observed (79%) using PET (114).
CONCLUSION
Measurements of probes kinetics within tumors usually reflect an interplay between influx and efflux membrane transport systems as well as local blood flow and cellular binding and metabolism. Appropriate study design and data analysis can help differentiate the various contributors to the probe’s signal from each other. The ongoing development of novel probes and molecular imaging methodologies is expected to enhance our understanding of transporter biology and contribute to personalized cancer medicine.
Acknowledgments
The authors acknowledge the support of the Israeli Cancer Research Foundation (ICRF) and the Israel Science Foundation (ISF) Grant 506/13.
Conflict of Interest
None.
References
- 1.El-Gebali S, Bentz S, Hediger MA, Anderle P. Solute carriers (SLCs) in cancer. Mol Aspects Med. 2013;34:719–34. doi: 10.1016/j.mam.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 3.Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320:138–49. doi: 10.1016/j.canlet.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obaidat A, Roth M, Hagenbuch B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol. 2012;52:135–51. doi: 10.1146/annurev-pharmtox-010510-100556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jadvar H, Alavi A, Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med. 2009;50:1820–7. doi: 10.2967/jnumed.108.054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu S, Alavi A. Unparalleled contribution of 18F-FDG PET to medicine over 3 decades. J Nucl Med. 2008;49:17N–21. doi: 10.2967/jnumed.108.050401. [DOI] [PubMed] [Google Scholar]
- 7.Condeelis J, Weissleder R. In vivo imaging in cancer. Cold Spring Harb Perspect Biol. 2010;2:a003848. doi: 10.1101/cshperspect.a003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James ML, Gambhir SS. A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev. 2012;92:897–965. doi: 10.1152/physrev.00049.2010. [DOI] [PubMed] [Google Scholar]
- 9.Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol. 2013;3:154. doi: 10.3389/fonc.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kusuhara H. Imaging in the study of membrane transporters. Clin Pharmacol Ther. 2013;94:33–6. doi: 10.1038/clpt.2013.85. [DOI] [PubMed] [Google Scholar]
- 11.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 12.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al. International Transporter Consortium. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94:52–63. doi: 10.1038/clpt.2013.74. [DOI] [PubMed] [Google Scholar]
- 14.Huang SM, Zhang L, Giacomini KM. The International Transporter Consortium: a collaborative group of scientists from academia, industry, and the FDA. Clin Pharmacol Ther. 2010;87:32–6. doi: 10.1038/clpt.2009.236. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Sweet DH. Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J. 2013;15:53–69. doi: 10.1208/s12248-012-9413-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackey JR, Yao SY, Smith KM, Karpinski E, Baldwin SA, Cass CE, et al. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst. 1999;91:1876–81. doi: 10.1093/jnci/91.21.1876. [DOI] [PubMed] [Google Scholar]
- 17.Bhutia YD, Hung SW, Patel B, Lovin D, Govindarajan R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011;71:1825–35. doi: 10.1158/0008-5472.CAN-10-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakanishi T, Tamai I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J Pharm Sci. 2011;100:3731–50. doi: 10.1002/jps.22576. [DOI] [PubMed] [Google Scholar]
- 19.Del Amo E, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur J Pharm Sci. 2008;35:161–74. doi: 10.1016/j.ejps.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 20.Gotoh K, Yamada T, Ishikawa O, Takahashi H, Eguchi H, Yano M, et al. A novel image-guided surgery of hepatocellular carcinoma by indocyanine green fluorescence imaging navigation. J Surg Oncol. 2009;100:75–9. doi: 10.1002/jso.21272. [DOI] [PubMed] [Google Scholar]
- 21.Huang C, McConathy J. Radiolabeled amino acids for oncologic imaging. J Nucl Med. 2013;54:1007–10. doi: 10.2967/jnumed.112.113100. [DOI] [PubMed] [Google Scholar]
- 22.Wulkersdorfer B, Wanek T, Bauer M, Zeitlinger M, Müller M, Langer O. Using positron emission tomography to study transporter-mediated drug-drug interactions in tissues. Clin Pharmacol Ther. 2014;96:206–13. doi: 10.1038/clpt.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 24.Chu X, Korzekwa K, Elsby R, Fenner K, Galetin A, Lai Y, et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther. 2013;94:126–41. doi: 10.1038/clpt.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lappin G, Garner RC. Big physics, small doses: the use of AMS and PET in human microdosing of development drugs. Nat Rev Drug Discov. 2003;2:233–40. doi: 10.1038/nrd1037. [DOI] [PubMed] [Google Scholar]
- 26.Hargreaves RJ. The role of molecular imaging in drug discovery and development. Clin Pharmacol Ther. 2008;83:349–53. doi: 10.1038/sj.clpt.6100467. [DOI] [PubMed] [Google Scholar]
- 27.Kannan P, John C, Zoghbi SS, Halldin C, Gottesman MM, Innis RB, et al. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86:368–77. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unadkat JD, Ke A, Eyal S. The impact of the BBB on the pharmacokinetics of drugs: investigations by PET. In: Elsinga PH, van Waarde A, Paans AMJ, Dierckx RA, editors. Trends on the role of PET in drug development. 1. Singapore: World Scientific Publishing Co.; 2012. [Google Scholar]
- 29.Sasongko L, Link JM, Muzi M, Mankoff DA, Yang X, Collier AC, et al. Imaging P-glycoprotein transport activity at the human blood–brain barrier with positron emission tomography. Clin Pharmacol Ther. 2005;77:503–14. doi: 10.1016/j.clpt.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal M, Abraham J, Balis FM, Edgerly M, Stein WD, Bates S, et al. Increased 99mTc-sestamibi accumulation in normal liver and drug-resistant tumors after the administration of the glycoprotein inhibitor, XR9576. Clin Cancer Res. 2003;9:650–6. [PubMed] [Google Scholar]
- 31.Chen CC, Meadows B, Regis J, Kalafsky G, Fojo T, Carrasquillo JA, et al. Detection of in vivo P-glycoprotein inhibition by PSC 833 using Tc-99 m sestamibi. Clin Cancer Res. 1997;3:545–52. [PubMed] [Google Scholar]
- 32.Portnoy E, Gurina M, Magdassi S, Eyal S. Evaluation of the near infrared compound indocyanine green as a probe substrate of P-glycoprotein. Mol Pharm. 2012;9:3595–601. doi: 10.1021/mp300472y. [DOI] [PubMed] [Google Scholar]
- 33.Eary JF, Link JM, Muzi M, Conrad EU, Mankoff DA, White JK, et al. Multiagent PET for risk characterization in sarcoma. J Nucl Med. 2011;52:541–6. doi: 10.2967/jnumed.110.083717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ke AB, Eyal S, Chung FS, Link JM, Mankoff DA, Muzi M, et al. Modeling cyclosporine A inhibition of the distribution of a P-glycoprotein PET ligand, 11C-verapamil, into the maternal brain and fetal liver of the pregnant nonhuman primate: impact of tissue blood flow and site of inhibition. J Nucl Med. 2013;54:437–46. doi: 10.2967/jnumed.112.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eyal S, Ke B, Muzi M, Link JM, Mankoff DA, Collier AC, et al. Regional P-glycoprotein activity and inhibition at the human blood–brain barrier as imaged by positron emission tomography. Clin Pharmacol Ther. 2010;87:579–85. doi: 10.1038/clpt.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikoma Y, Takano A, Ito H, Kusuhara H, Sugiyama Y, Arakawa R, et al. Quantitative analysis of 11C-verapamil transfer at the human blood–brain barrier for evaluation of P-glycoprotein function. J Nucl Med. 2006;47:1531–7. [PubMed] [Google Scholar]
- 37.Muzi M, Mankoff DA, Link JM, Shoner S, Collier AC, Sasongko L, et al. Imaging of cyclosporine inhibition of P-glycoprotein activity using 11C-verapamil in the brain: studies of healthy humans. J Nucl Med. 2009;50:1267–75. doi: 10.2967/jnumed.108.059162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2004;1:121–5. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldmann M, Asselin MC, Liu J, Wang S, McMahon A, Anton-Rodriguez J, et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: a case–control study. Lancet Neurol. 2013;12:777–85. doi: 10.1016/S1474-4422(13)70109-1. [DOI] [PubMed] [Google Scholar]
- 40.Deo AK, Borson S, Link JM, Domino K, Eary JF, Ke B, et al. Activity of P-glycoprotein, a β-amyloid transporter at the blood–brain barrier, is compromised in patients with mild Alzheimer disease. J Nucl Med. 2014;55:1106–11. doi: 10.2967/jnumed.113.130161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Assema DM, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–9. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- 42.Mairinger S, Erker T, Muller M, Langer O. PET and SPECT radiotracers to assess function and expression of ABC transporters in vivo. Curr Drug Metab. 2011;12:774–92. doi: 10.2174/138920011798356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hendrikse N, de Vries E, Eriks-Fluks L, van der Graaf W, Hospers G, Willemsen A, et al. A new in vivo method to study P-glycoprotein transport in tumors and the blood–brain barrier. Cancer Res. 1999;59:2411–6. [PubMed] [Google Scholar]
- 44.Chen WS, Luker KE, Dahlheimer JL, Pica CM, Luker GD, Piwnica-Worms D. Effects of MDR1 and MDR3 P-glycoproteins, MRP1, and BCRP/MXR/ABCP on the transport of (99 m)Tc-tetrofosmin. Biochem Pharmacol. 2000;60:413–26. doi: 10.1016/S0006-2952(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 45.Gomes CM, Abrunhosa AJ, Pauwels EK, Botelho MF. P-glycoprotein versus MRP1 on transport kinetics of cationic lipophilic substrates: a comparative study using [99mTc]sestamibi and [99mTc]tetrofosmin. Cancer Biother Radiopharm. 2009;24:215–27. doi: 10.1089/cbr.2008.0539. [DOI] [PubMed] [Google Scholar]
- 46.On NH, Chen F, Hinton M, Miller DW. Assessment of P-glycoprotein activity in the blood–brain barrier (BBB) using near infrared fluorescence (NIRF) imaging techniques. Pharm Res. 2011;28:2505–15. doi: 10.1007/s11095-011-0478-6. [DOI] [PubMed] [Google Scholar]
- 47.Pichler A, Prior JL, Piwnica-Worms D. Imaging reversal of multidrug resistance in living mice with bioluminescence: MDR1 P-glycoprotein transports coelenterazine. Proc Natl Acad Sci U S A. 2004;101:1702–7. doi: 10.1073/pnas.0304326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreisl WC, Liow JS, Kimura N, Seneca N, Zoghbi SS, Morse CL, et al. P-glycoprotein function at the blood–brain barrier in humans can be quantified with the substrate radiotracer 11C-N-desmethyl-loperamide. J Nucl Med. 2010;51:559–66. doi: 10.2967/jnumed.109.070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bauer M, Karch R, Zeitlinger M, Stanek J, Philippe C, Wadsak W, et al. Interaction of 11C-tariquidar and 11C-elacridar with P-glycoprotein and breast cancer resistance protein at the human blood–brain barrier. J Nucl Med. 2013;54:1181–7. doi: 10.2967/jnumed.112.118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawamura K, Yamasaki T, Konno F, Yui J, Hatori A, Yanamoto K, et al. Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [(11)C]GF120918 by PET in mice. Mol Imaging Biol. 2011;13:152–60. doi: 10.1007/s11307-010-0313-1. [DOI] [PubMed] [Google Scholar]
- 51.Kawamura K, Yamasaki T, Yui J, Hatori A, Konno F, Kumata K, et al. In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [(11)C]gefitinib. Nucl Med Biol. 2009;36:239–46. doi: 10.1016/j.nucmedbio.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 52.Kawamura K, Konno F, Yui J, Yamasaki T, Hatori A, Yanamoto K, et al. Synthesis and evaluation of [11C]XR9576 to assess the function of drug efflux transporters using PET. Ann Nucl Med. 2010;24:403–12. doi: 10.1007/s12149-010-0373-y. [DOI] [PubMed] [Google Scholar]
- 53.Yamasaki T, Fujinaga M, Kawamura K, Hatori A, Yui J, Nengaki N, et al. Evaluation of the P-glycoprotein- and breast cancer resistance protein-mediated brain penetration of 11C-labeled topotecan using small-animal positron emission tomography. Nucl Med Biol. 2011;38:707–14. doi: 10.1016/j.nucmedbio.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Bressler JP, Neal J, Lal B, Bhang HE, Laterra J, et al. ABCG2/BCRP expression modulates D-Luciferin based bioluminescence imaging. Cancer Res. 2007;67:9389–97. doi: 10.1158/0008-5472.CAN-07-0944. [DOI] [PubMed] [Google Scholar]
- 55.Huang R, Vider J, Serganova I, Blasberg RG. ATP-binding cassette transporters modulate both coelenterazine- and D-luciferin-based bioluminescence imaging. Mol Imaging. 2011;10:215–26. [PMC free article] [PubMed] [Google Scholar]
- 56.Piwnica-Worms D, Chiu ML, Budding M, Kronauge JF, Kramer RA, Croop JM. Functional imaging of multidrug-resistant P-glycoprotein with an organotechnetium complex. Cancer Res. 1993;53:977–84. [PubMed] [Google Scholar]
- 57.Planchamp C, Hadengue A, Stieger B, Bourquin J, Vonlaufen A, Frossard JL, et al. Function of both sinusoidal and canalicular transporters controls the concentration of organic anions within hepatocytes. Mol Pharmacol. 2007;71:1089–97. doi: 10.1124/mol.106.030759. [DOI] [PubMed] [Google Scholar]
- 58.Huang L, Vore M. Multidrug resistance p-glycoprotein 2 is essential for the biliary excretion of indocyanine green. Drug Metab Dispos. 2001;29:634–7. [PubMed] [Google Scholar]
- 59.Jia J, Puls D, Oswald S, Jedlitschky G, Kühn JP, Weitschies W, et al. Characterization of the intestinal and hepatic uptake/efflux transport of the magnetic resonance imaging contrast agent gadolinium-ethoxylbenzyl-diethylenetriamine-pentaacetic acid. Investig Radiol. 2014;49:78–86. doi: 10.1097/RLI.0b013e3182a70043. [DOI] [PubMed] [Google Scholar]
- 60.He J, Yu Y, Prasad B, Link J, Miyaoka RS, Chen X, et al. PET imaging of Oatp-mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol Pharm. 2014;11:2745–54. doi: 10.1021/mp500027c. [DOI] [PubMed] [Google Scholar]
- 61.Shikano N, Kanai Y, Kawai K, Ishikawa N, Endou H. Transport of 99mTc-MAG3 via rat renal organic anion transporter 1. J Nucl Med. 2004;45:80–5. [PubMed] [Google Scholar]
- 62.de Graaf W, Häusler S, Heger M, van Ginhoven TM, van Cappellen G, Bennink RJ, et al. Transporters involved in the hepatic uptake of (99 m)Tc-mebrofenin and indocyanine green. J Hepatol. 2011;54:738–45. doi: 10.1016/j.jhep.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 63.Tsuboyama T, Onishi H, Kim T, Akita H, Hori M, Tatsumi M, et al. Hepatocellular carcinoma: hepatocyte-selective enhancement at gadoxetic acid-enhanced MR imaging—correlation with expression of sinusoidal and canalicular transporters and bile accumulation. Radiology. 2010;255:824–33. doi: 10.1148/radiol.10091557. [DOI] [PubMed] [Google Scholar]
- 64.Leonhardt M, Keiser M, Oswald S, Kühn J, Jia J, Grube M, et al. Hepatic uptake of the magnetic resonance imaging contrast agent Gd-EOB-DTPA: role of human organic anion transporters. Drug Metab Dispos. 2010;38:1024–8. doi: 10.1124/dmd.110.032862. [DOI] [PubMed] [Google Scholar]
- 65.Yang X, Shi C, Tong R, Qian W, Zhau HE, Wang R, et al. Near IR heptamethine cyanine dye-mediated cancer imaging. Clin Cancer Res. 2010;16:2833–44. doi: 10.1158/1078-0432.CCR-10-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobayashi M, Nakanishi T, Nishi K, Higaki Y, Okudaira H, Ono M, et al. Transport mechanisms of hepatic uptake and bile excretion in clinical hepatobiliary scintigraphy with 99mTc-N-pyridoxyl-5-methyltryptophan. Nucl Med Biol. 2014;41:338–42. doi: 10.1016/j.nucmedbio.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 67.Wu JB, Shao C, Li X, Shi C, Li Q, Hu P, et al. Near-infrared fluorescence imaging of cancer mediated by tumor hypoxia and HIF1α/OATPs signaling axis. Biomaterials. 2014;35:8175–85. doi: 10.1016/j.biomaterials.2014.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paproski RJ, Ng AM, Yao SY, Graham K, Young JD, Cass CE. The role of human nucleoside transporters in uptake of 39-deoxy-39-fluorothymidine. Mol Pharmacol. 2008;74:1372–80. doi: 10.1124/mol.108.048900. [DOI] [PubMed] [Google Scholar]
- 69.Elsinga PH, Franssen EJ, Hendrikse NH, Fluks L, Weemaes AM, van der Graaf WT, et al. Carbon-11-labeled daunorubicin and verapamil for probing P-glycoprotein in tumors with PET. J Nucl Med. 1996;37:1571–5. [PubMed] [Google Scholar]
- 70.Eyal S, Chung FS, Muzi M, Link JM, Mankoff DA, Kaddoumi A, et al. Simultaneous PET imaging of P-plycoprotein inhibition in multiple tissues in the pregnant non-human primate. J Nucl Med. 2009;50:798–806. doi: 10.2967/jnumed.108.059360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bauer M, Karch R, Neumann F, Wagner CC, Kletter K, Müller M, et al. Assessment of regional differences in tariquidar-induced P-glycoprotein modulation at the human blood–brain barrier. J Cereb Blood Flow Metab. 2010;30:510–5. doi: 10.1038/jcbfm.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bauer M, Zeitlinger M, Karch R, Matzneller P, Stanek J, Jäger W, et al. Pgp-mediated interaction between (R)-[11C]verapamil and tariquidar at the human blood–brain barrier: a comparison with rat data. Clin Pharmacol Ther. 2012;91:227–33. doi: 10.1038/clpt.2011.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toornvliet R, van Berckel BN, Luurtsema G, Lubberink M, Geldof AA, Bosch TM, et al. Effect of age on functional P-glycoprotein in the blood–brain barrier measured by use of (R)-[(11)C]verapamil and positron emission tomography. Clin Pharmacol Ther. 2006;79:540–8. doi: 10.1016/j.clpt.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 74.Kannan P, Brimacombe KR, Kreisl WC, Liow JS, Zoghbi SS, Telu S, et al. Lysosomal trapping of a radiolabeled substrate of P-glycoprotein as a mechanism for signal amplification in PET. Proc Natl Acad Sci U S A. 2011;108:2593–8. doi: 10.1073/pnas.1014641108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gangloff A, Hsueh WA, Kesner AL, Kiesewetter DO, Pio BS, Pegram MD, et al. Estimation of paclitaxel biodistribution and uptake in human-derived xenografts in vivo with (18)F-fluoropaclitaxel. J Nucl Med. 2005;46:1866–71. [PubMed] [Google Scholar]
- 76.Luurtsema G, Schuit RC, Klok RP, Verbeek J, Leysen JE, Lammertsma AA, et al. Evaluation of [11C]laniquidar as a tracer of P-glycoprotein: radiosynthesis and biodistribution in rats. Nucl Med Biol. 2009;36:643–9. doi: 10.1016/j.nucmedbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 77.Tournier N, Saba W, Cisternino S, Peyonneau MA, Damont A, Goutal S, et al. Effects of selected OATP and/or ABC transporter inhibitors on the brain and whole-body distribution of glyburide. AAPS J. 2013;15:1082–90. doi: 10.1208/s12248-013-9514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–9. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 79.Hendrikse NH, de Vries EG, Franssen EJ, Vaalburg W, van der Graaf WT. In vivo measurement of [11C]verapamil kinetics in human tissues. Eur J Clin Pharmacol. 2001;56:827–9. doi: 10.1007/s002280000253. [DOI] [PubMed] [Google Scholar]
- 80.Kurdziel KA, Kalen JD, Hirsch JI, Wilson JD, Bear HD, Logan J, et al. Human dosimetry and preliminary tumor distribution of 18F-fluoropaclitaxel in healthy volunteers and newly diagnosed breast cancer patients using PET/CT. J Nucl Med. 2011;52:1339–45. doi: 10.2967/jnumed.111.091587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van der Veldt AA, Lubberink M, Mathijssen RH, Loos WJ, Herder GJ, Greuter HN, et al. Toward prediction of efficacy of chemotherapy: a proof of concept study in lung cancer patients using [11C]docetaxel and positron emission tomography. Clin Cancer Res. 2013;19:4163–73. doi: 10.1158/1078-0432.CCR-12-3779. [DOI] [PubMed] [Google Scholar]
- 82.Del Vecchio S, Zannetti A, Aloj L, Salvatore M. MIBI as prognostic factor in breast cancer. Q J Nucl Med. 2003;47:46–50. [PubMed] [Google Scholar]
- 83.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 84.Swed A, Eyal S, Madar I, Zohar-Kontante H, Weiss L, Hoffman A. The role of P-glycoprotein in intestinal transport versus the BBB transport of tetraphenylphosphonium. Mol Pharm. 2009;6:1883–90. doi: 10.1021/mp900170y. [DOI] [PubMed] [Google Scholar]
- 85.Cattelotte J, Tournier N, Rizzo-Padoin N, Schinkel A, Scherrmann J, Cisternino S. Changes in dipole membrane potential at the mouse blood–brain barrier enhance the transport of Technetium Sestamibi more than inhibiting Abcb1, Abcc1, or Abcg2. J Neurochem. 2009;108:767–75. doi: 10.1111/j.1471-4159.2008.05832.x. [DOI] [PubMed] [Google Scholar]
- 86.Ghibellini G, Leslie EM, Pollack GM, Brouwer KL. Use of tc-99 m mebrofenin as a clinical probe to assess altered hepatobiliary transport: integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm Res. 2008;25:1851–60. doi: 10.1007/s11095-008-9597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pastor CM, Müllhaupt B, Stieger B. The role of organic anion transporters in diagnosing liver diseases by magnetic resonance imaging. Drug Metab Dispos. 2014;42:675–84. doi: 10.1124/dmd.113.055707. [DOI] [PubMed] [Google Scholar]
- 88.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev. 1999;99:2293–352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 89.Heye AK, Culling RD, Valdés Hernández Mdel C, Thrippleton MJ, Wardlaw JM. Assessment of blood–brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review. Neuroimage Clin. 2014;6:262–74. doi: 10.1016/j.nicl.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoekstra LT, de Graaf W, Nibourg GA, Heger M, Bennink RJ, Stieger B, et al. Physiological and biochemical basis of clinical liver function tests: a review. Ann Surg. 2013;257:27–36. doi: 10.1097/SLA.0b013e31825d5d47. [DOI] [PubMed] [Google Scholar]
- 91.Daali Y, Millet P, Dayer P, Pastor CM. Evidence of drug-drug interactions through uptake and efflux transport systems in rat hepatocytes: implications for cellular concentrations of competing drugs. Drug Metab Dispos. 2013;41:1548–56. doi: 10.1124/dmd.113.051870. [DOI] [PubMed] [Google Scholar]
- 92.Ichikawa T, Sano K, Morisaka H. Diagnosis of pathologically early HCC with EOB-MRI: experiences and current consensus. Liver Cancer. 2014;3:97–107. doi: 10.1159/000343865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adams STJ, Miller SC. Beyond D-luciferin: expanding the scope of bioluminescence imaging in vivo. Curr Opin Chem Biol. 2014;21C:112–20. doi: 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bremer C, Ntziachristos V, Weissleder R. Optical-based molecular imaging: contrast agents and potential medical applications. Eur Radiol. 2003;13:231–43. doi: 10.1007/s00330-002-1610-0. [DOI] [PubMed] [Google Scholar]
- 95.Nguyen QT, Tsien RY. Fluorescence-guided surgery with live molecular navigation—a new cutting edge. Nat Rev Cancer. 2013;13:653–62. doi: 10.1038/nrc3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cherrick GR, Stein SW, Leevy CM, Davidson CS. Indocyanine green: observations on its physical properties, plasma decay, and hepatic extraction. J Clin Invest. 1960;39:592–600. doi: 10.1172/JCI104072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283–308. doi: 10.1016/j.bbamcr.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 98.Ishizawa T, Masuda K, Urano Y, Kawaguchi Y, Satou S, Kaneko J, et al. Mechanistic background and clinical applications of indocyanine green fluorescence imaging of hepatocellular carcinoma. Ann Surg Oncol. 2014;21:440–8. doi: 10.1245/s10434-013-3360-4. [DOI] [PubMed] [Google Scholar]
- 99.Zhang E, Luo S, Tan X, Shi C. Mechanistic study of IR-780 dye as a potential tumor targeting and drug delivery agent. Biomaterials. 2014;35:771–8. doi: 10.1016/j.biomaterials.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Liu T, Zhang E, Luo S, Tan X, Shi C. Preferential accumulation of the near infrared heptamethine dye IR-780 in the mitochondria of drug-resistant lung cancer cells. Biomaterials. 2014;35:4116–24. doi: 10.1016/j.biomaterials.2014.01.061. [DOI] [PubMed] [Google Scholar]
- 101.Tseng JC, Wang Y, Banerjee P, Kung AL. Incongruity of imaging using fluorescent 2-DG conjugates compared to 18F-FDG in preclinical cancer models. Mol Imaging Biol. 2012;14:553–60. doi: 10.1007/s11307-012-0545-3. [DOI] [PubMed] [Google Scholar]
- 102.Zhang W, Xiong H, Callaghan D, Liu H, Jones A, Pei K, et al. Blood–brain barrier transport of amyloid beta peptides in efflux pump knock-out animals evaluated by in vivo optical imaging. Fluids Barriers CNS. 2013;10:13. doi: 10.1186/2045-8118-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cali JJ, Niles A, Valley MP, O’Brien MA, Riss TL, Shultz J. Bioluminescent assays for ADMET. Expert Opin Drug Metab Toxicol. 2008;4:103–20. doi: 10.1517/17425255.4.1.103. [DOI] [PubMed] [Google Scholar]
- 104.Gu L, Tsark WM, Brown DA, Blanchard S, Synold TW, Kane SE. A new model for studying tissue-specific mdr1a gene expression in vivo by live imaging. Proc Natl Acad Sci U S A. 2009;106:5394–9. doi: 10.1073/pnas.0807343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gu L, Chen J, Synold TW, Forman BM, Kane SE. Bioimaging real-time PXR-dependent mdr1a gene regulation in mdr1a.fLUC reporter mice. J Pharmacol Exp Ther. 2013;345:438–45. doi: 10.1124/jpet.113.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Y, Byun Y, Ren YR, Liu JO, Laterra J, Pomper MG. Identification of inhibitors of ABCG2 by a bioluminescence imaging-based high-throughput assay. Cancer Res. 2009;69:5867–75. doi: 10.1158/0008-5472.CAN-08-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y, Pullambhatla M, Laterra J, Pomper MG. Influence of bioluminescence imaging dynamics by D-luciferin uptake and efflux mechanisms. Mol Imaging. 2012;11:499–506. [PMC free article] [PubMed] [Google Scholar]
- 108.Bakhsheshian J, Wei BR, Chang KE, Shukla S, Ambudkar SV, Simpson RM, et al. Bioluminescent imaging of drug efflux at the blood–brain barrier mediated by the transporter ABCG2. Proc Natl Acad Sci U S A. 2013;110:20801–6. doi: 10.1073/pnas.1312159110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patrick PS, Lyons SK, Rodrigues TB, Brindle KM. Oatp1 enhances bioluminescence by acting as a plasma membrane transporter for D-luciferin. Mol Imaging Biol. 2014;16:626–34. doi: 10.1007/s11307-014-0741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Acocella G, Nicolis FB, Tenconi LT. The effect of an intravenous infusion of rifamycin SV on the excretion of bilirubin, bromsulphalein, and indocyanine green in man. Gastroenterology. 1965;49:521–5. [PubMed] [Google Scholar]
- 111.Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 112.Nakamura Y, Date S, Toyota N, Tani C, Honda Y, Komoto D, et al. Effect of lapatinib on hepatic parenchymal enhancement on gadoxetate disodium (EOB)-enhanced MRI scans. J Comput Assist Tomogr. 2011;35:351–2. doi: 10.1097/RCT.0b013e31821a02aa. [DOI] [PubMed] [Google Scholar]
- 113.Kenny LM, Contractor KB, Stebbing J, Al-Nahhas A, Palmieri C, Shousha S, et al. Altered tissue 3'-deoxy-3'-[18F]fluorothymidine pharmacokinetics in human breast cancer following capecitabine treatment detected by positron emission tomography. Clin Cancer Res. 2009;15:6649–57. doi: 10.1158/1078-0432.CCR-09-1213. [DOI] [PubMed] [Google Scholar]
- 114.Hsiao P, Bui T, Ho R, Unadkat J. In vitro to in vivo prediction of P-glycoprotein based drug interactions at the human and rodent blood–brain barrier. Drug Metab Dispos. 2008;36:481–4. doi: 10.1124/dmd.107.018176. [DOI] [PubMed] [Google Scholar]
- 115.Sprowl A, van Doorn L, Hu S, van Gerven L, de Bruijn P, Li L, et al. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance. Clin Pharmacol Ther. 2013;94:585–92. doi: 10.1038/clpt.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Widhalm G. Intra-operative visualization of brain tumors with 5-aminolevulinic acid-induced fluorescence. Clin Neuropathol. 2014;33:260–78. doi: 10.5414/NP300798. [DOI] [PubMed] [Google Scholar]
- 117.Stummer W, Stocker S, Wagner S, Stepp H, Fritsch C, Goetz C, et al. Intraoperative detection of malignant gliomas by 5-aminolevulinic acid-induced porphyrin fluorescence. Neurosurgery. 1998;42:518–25. doi: 10.1097/00006123-199803000-00017. [DOI] [PubMed] [Google Scholar]
- 118.Zhao SG, Chen XF, Wang LG, Yang G, Han DY, Teng L, et al. Increased expression of ABCB6 enhances protoporphyrin IX accumulation and photodynamic effect in human glioma. Ann Surg Oncol. 2013;20:4379–88. doi: 10.1245/s10434-011-2201-6. [DOI] [PubMed] [Google Scholar]