

The production of hydrogen (H2) from water is a robust method of storing renewable energy (solar, wind…) in chemical form. This energy can then be released on demand as electrical power through H2 oxidation in fuel-cells. Currently however, only noble metals meet technological specifications for fuel-cell applications. Inspiration for alternative catalysts based on Earth-abundant elements can be drawn from Nature, namely the hydrogenase enzymes, which only utilise nickel and/or iron, whilst rivalling platinum as catalysts for reversible H2 evolution/oxidation.[1]
In particular, detailed biochemical, structural and mechanistic information has been accumulated on NiFe hydrogenases (Figure 1). Their heterobimetallic active site contains a nickel ion ligated by four deprotonated (thiolate form) cystein residues, two of which bridge to an iron ion. One CO and two CN− ligands are also bound to the FeII center. Although several mimics of this active site were reported after the structure had been determined,[2] construction of models with structural relevance, that also replicate enzymatic activity, has long been a challenge for chemists (Figure 2). Indeed, up to 2009 none of these structural mimics were shown to be catalytically active, and only the use of organometallic ruthenium moieties as surrogates for the {FeII(CN)2(CO)} fragment allowed for the preparation of active catalysts first for H2 evolution[3] and later for its oxidation.[4] In both cases, the active intermediate contains a hydride ligand (H−) in a bridging mode, between the two metal centers,[3c, 3f, 4a] and is thus relevant to the Ni-C state of NiFe hydrogenases (Figure 1).[5]
Figure 1.
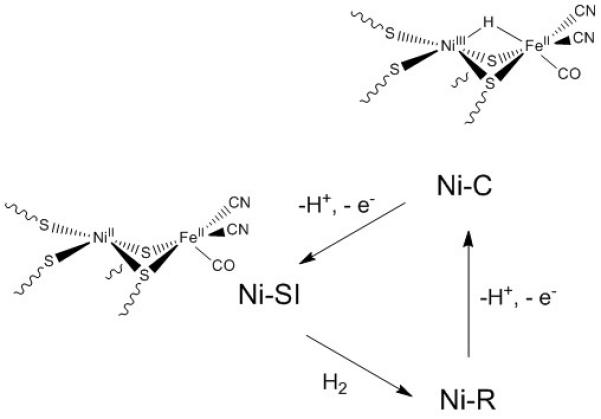
Mechanism for catalytic H2 oxidation mediated by [NiFe] hydrogenase.
Figure 2.
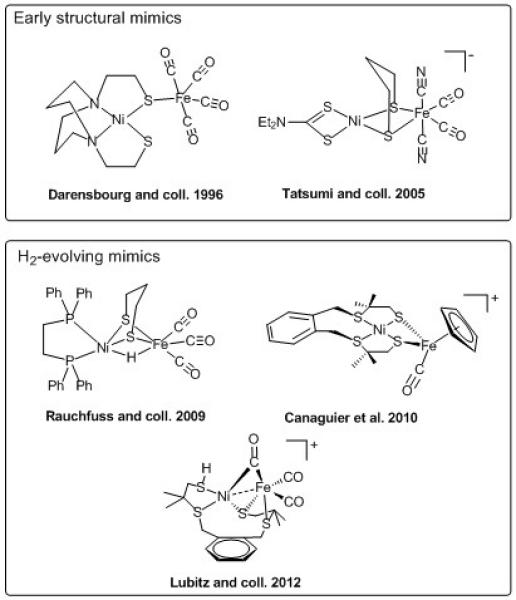
Selected structural models (top) and representation of the H2-evolving functional models (bottom) of the active site of [NiFe] hydrogenase (references lie in the text).
The next milestone was the design of nickel and iron-based functional models. Three such systems (Figure 2) were reported, albeit with catalytic activity restricted to H2 evolution,[6] once again with bridging hydride species as the active intermediate. Recent work from Ogo and co-workers has described a novel nickel-iron mimic (Figure 3) able to mediate both hydrogen evolution and oxidation, thus reproducing for the first time at a binuclear core, the bidirectional activity of the [NiFe]-hydrogenases.[7]
Figure 3.
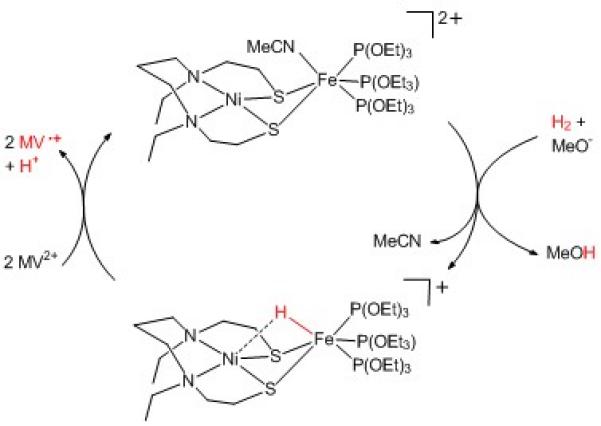
Structure and reactivity of the novel Ni-Fe mimic from Ogo’s group.
The novelty of this mimic, likely responsible for its functionality, is the use of three triethylphosphite (P(OEt)3) ligands to modulate the electronic properties of the iron center, so as to promote coordination of H2 as a first step towards its activation. Heterolytic splitting of H2 is promoted in the presence of methanolate, a strong base that captures a proton while a hydride ligand remains coordinated to the iron center. From this hydride species, oxidation via methylviologen (MV2+) and release of a proton regenerates the starting compound. The net reaction is the two-electron oxidation of molecular hydrogen, which is the process mediated by both hydrogenase and the anode of a H2 fuel-cell. This new nickel-iron compound achieves only a single turn-over with a 12% yield, although a better yield (45%) could be measured when using a stronger oxidant such as the ferrocenium ion. The system also operates far from the thermodynamic equilibrium, as evidenced by the requirement of a strong base to activate H2, while the natural process operates in water at neutral pH. Similarly, a strong acid is required to produce H2 from the hydride species, and so whilst this system shows promise as a catalyst for hydrogen evolution, the necessity of large overpotentials, as noted for other Ni-Fe mimics,[6a, 6c, 6d] remains an issue. Nonetheless, this novel nickel-iron compound mediates both hydrogen oxidation and evolution, behaviour so far restricted to a single series of mononuclear nickel catalysts.[8]
In line with this result is the recent report from Bullock and co-workers, that a mononuclear iron diphosphine complex can act as an electrocatalyst for H2 oxidation (Figure 4).[9] Here again, the electronic properties of the iron center have been tuned, thanks to a functionalised cyclopentadienyl (C6F5Cp−) ligand. Although similar systems reported by Bullock were shown to bind H2, they were only able to mediate H/D exchange from a mixture of H2 and D2.[10] Modification of the Cp− ligand with an electron withdrawing group, renders bound H2 suitably acidic to facilitate H2 oxidation. As with previous nickel complexes designed by D. L. DuBois,[8] an amine function has been incorporated in the diphosphine ligand, to act as a proton transfer relay and allow for fast deprotonation of the coordinated H2 molecule, with formation of a terminal hydride complex. This system catalyses electro-assisted H2 oxidation (1 atm. and 22°C) from C6H5F solutions of N-methylpyrrolidine with a turnover frequency of 0.66-2.0 s−1 and overpotential requirement estimated to 160-200 mV. Coordinating species must however be excluded from the media for catalysis to occur which precludes any direct utilisation in water.
Figure 4.
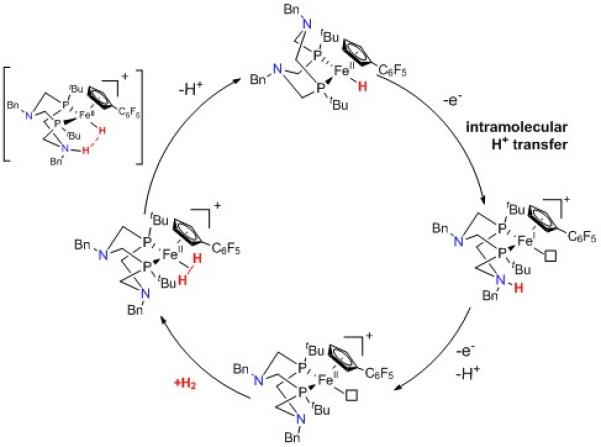
Catalytic mechanism for H2 oxidation mediated by the mononuclear iron diphosphine from Bullock and co-workers.
These two systems have several common properties: (i) they contain electron-rich, low spin d6 centers, coordinated by soft bidentate ligands, a nickel bisthiolate moiety in one case and a diphosphine in the other; (ii) they interact with H2 - in the case of Ogo’s system however, it has not been definitively demonstrated that Fe is the primary binding site, whilst H2 binding to NiII centers is now documented;[11] (iii) they stabilize an Fe-bound terminal hydride ligand; (iv) they can split molecular H2 into electrons and protons.
Despite the structural similarity of Ogo’s system with the active site of [NiFe] hydrogenase, direct comparison of catalytic mechanisms should be avoided. Other characterised[4a, 6a, 6b] or computed[3c, 3f, 6c, 12] dinuclear hydride derivatives, based on a {NiII(μ-SR)2M} core with M a low-spin d6 metal center, feature a bridging structure in contrast to the terminally bound hydride in question. One such Ni-Ru bridging hydride from Ogo[4a] is active for H2 oxidation, making it difficult to form a clear association between a terminal binding mode and H2 activation. Even if these two terminally Fe-bound hydrides are associated with H2 activation, it is not in accordance with the observation of a bridging hydride in the Ni-C state of the enzyme,[5] that being said, the production of a bridging hydride derivative that does replicate the NiIII/FeII electronic structure of the Ni-C state, remains elusive. The NiII/FeII center of Ogo’s hydride derivative may correspond to the Ni-R (Figure 1) state of the enzyme, however no definitive data exists as to the nature of the additional ligand (H2, H−…) nor to its binding mode to the {NiII(μ-SR)2FeII} core.
Very few systems based on first-row transition systems are known to catalyse hydrogen oxidation under technologically relevant conditions.[8, 13] These recent results from Ogo[7] and Bullock,[9] clearly indicate that chemists are making continued progress towards better understanding how Nature exploits abundant metals to achieve complex reactions, whilst channelling this knowledge into the design of original catalytic systems, which are now close to readiness for technological applications.
Footnotes
This work was supported by the French National Research Agency (ANR, NiFe–Cat ANR–10–BLAN–711 and Labex program ARCANE, 11–LABX–003) and the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n.306398
References
- [1].Cracknell JA, Vincent KA, Armstrong FA. Chem. Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- [2] a).Lai CH, Reibenspies JH, Darensbourg MY. Angew. Chem. Int. Ed. 1996;35:2390–2393. [Google Scholar]; b) Canaguier S, Artero V, Fontecave M. Dalton Trans. 2008:315–325. doi: 10.1039/b713567j. [DOI] [PubMed] [Google Scholar]; c) Tard C, Pickett CJ. Chem. Rev. 2009;109:2245–2274. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]; d Li ZL, Ohki Y, Tatsumi K. J. Am. Chem. Soc. 2005;127:8950–8951. doi: 10.1021/ja051590+. [DOI] [PubMed] [Google Scholar]
- [3] a).Oudart Y, Artero V, Pécaut J, Fontecave M. Inorg. Chem. 2006;45:4334–4336. doi: 10.1021/ic060510f. [DOI] [PubMed] [Google Scholar]; b) Oudart Y, Artero V, Pécaut J, Lebrun C, Fontecave M. Eur. J. Inorg. Chem. 2007:2613–2626. [Google Scholar]; c) Canaguier S, Vaccaro L, Artero V, Ostermann R, Pécaut J, Field MJ, Fontecave M. Chem. Eur. J. 2009;15:9350–9365. doi: 10.1002/chem.200900854. [DOI] [PubMed] [Google Scholar]; d) Canaguier S, Fontecave M, Artero V. Eur. J. Inorg. Chem. 2011:1094–1099. [Google Scholar]; e) Canaguier S, Fourmond V, Perotto CU, Fize J, Pécaut J, Fontecave M, Field MJ, Artero V. Chem. Commun. 2013 doi: 10.1039/c3cc40987b. DOI:10.1039/C3CC40987B. [DOI] [PubMed] [Google Scholar]; f) Vaccaro L, Artero V, Canaguier S, Fontecave M, Field MJ. Dalton Trans. 2010;39:3043–3049. doi: 10.1039/b912690b. [DOI] [PubMed] [Google Scholar]
- [4] a).Ogo S, Kabe R, Uehara K, Kure B, Nishimura T, Menon SC, Harada R, Fukuzumi S, Higuchi Y, Ohhara T, Tamada T, Kuroki R. Science. 2007;316:585–587. doi: 10.1126/science.1138751. [DOI] [PubMed] [Google Scholar]; b) Matsumoto T, Kure B, Ogo S. Chem. Lett. 2008;37:970–971. [Google Scholar]
- [5].Brecht M, van Gastel M, Buhrke T, Friedrich B, Lubitz W. J. Am. Chem. Soc. 2003;125:13075–13083. doi: 10.1021/ja036624x. [DOI] [PubMed] [Google Scholar]
- [6] a).Barton BE, Whaley CM, Rauchfuss TB, Gray DL. J. Am. Chem. Soc. 2009;131:6942–6943. doi: 10.1021/ja902570u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barton BE, Rauchfuss TB. J. Am. Chem. Soc. 2010;132:14877–14885. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Canaguier S, Field M, Oudart Y, Pecaut J, Fontecave M, Artero V. Chem. Commun. 2010;46:5876–5878. doi: 10.1039/c001675f. [DOI] [PubMed] [Google Scholar]; d) Weber K, Krämer T, Shafaat HS, Weyhermüller T, Bill E, van Gastel M, Neese F, Lubitz W. J. Am. Chem. Soc. 2012;134:20745–20755. doi: 10.1021/ja309563p. [DOI] [PubMed] [Google Scholar]
- [7].Ogo S, Ichikawa K, Kishima T, Matsumoto T, Nakai H, Kusaka K, Ohhara T. Science. 2013;339:682–684. doi: 10.1126/science.1231345. [DOI] [PubMed] [Google Scholar]
- [8] a).Wilson AD, Newell RH, McNevin MJ, Muckerman JT, DuBois MR, DuBois DL. J. Am. Chem. Soc. 2006;128:358–366. doi: 10.1021/ja056442y. [DOI] [PubMed] [Google Scholar]; b) Smith SE, Yang JY, DuBois DL, Bullock RM. Angew. Chem. Int. Ed. 2012;51:3152–3155. doi: 10.1002/anie.201108461. [DOI] [PubMed] [Google Scholar]; c) Le Goff A, Artero V, Jousselme B, Tran PD, Guillet N, Metaye R, Fihri A, Palacin S, Fontecave M. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- [9].Liu T, DuBois DL, Bullock RM. Nat Chem. 2013;5:228–233. doi: 10.1038/nchem.1571. [DOI] [PubMed] [Google Scholar]
- [10].Liu TB, Chen ST, O’Hagan MJ, DuBois MR, Bullock RM, DuBois DL. J. Am. Chem. Soc. 2012;134:6257–6272. doi: 10.1021/ja211193j. [DOI] [PubMed] [Google Scholar]
- [11].Tsay C, Peters JC. Chem. Sci. 2012;3:1313–1318. [Google Scholar]
- [12].Fourmond V, Canaguier S, Golly B, Field MJ, Fontecave M, Artero V. Energy Environ. Sci. 2011;4:2417–2427. [Google Scholar]
- [13].Camara JM, Rauchfuss TB. Nature Chemistry. 2012;4:26–30. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]