

Abstract
Despite recent advances, the role of ROS in mediating hypertrophic and apoptotic responses in cardiac myocytes elicited by norepinephrine (NE) is rather poorly understood. We demonstrate through our experiments that H9c2 cardiac myoblasts treated with 2 µM NE (hypertrophic dose) generate DCFH-DA positive ROS only for 2 h; while those treated with 100 µM NE (apoptotic dose) sustains generation for 48 h, followed by apoptosis. Though the levels of DCFH fluorescence were comparable at early time points in the two treatment sets, its quenching by DPI, catalase and MnTmPyP suggested the existence of a different repertoire of ROS. Both doses of NE also induced moderate levels of H2O2 but with different kinetics. Sustained but intermittent generation of highly reactive species detectable by HPF was seen in both treatment sets but no peroxynitrite was generated in either conditions. Sustained generation of hydroxyl radicals with no appreciable differences were noticed in both treatment sets. Nevertheless, despite similar profile of ROS generation between the two conditions, extensive DNA damage as evident from the increase in 8-OH-dG content, formation of γ-H2AX and PARP cleavage was seen only in cells treated with the higher dose of NE. We therefore conclude that hypertrophic and apoptotic doses of NE generate distinct but comparable repertoire of ROS/RNS leading to two very distinct downstream responses.
Keywords: Cardiac myocyte, Hypertrophy, Apoptosis, Redox signaling, Reactive oxygen species, 8−OH-dG
Abbreviations: DCFH-DA, dichloro-dihydro-fluorescein diacetate; DHBA, dihydroxybenzoic acid; DHE, dihydroethidium; DPI, diphenyleneiodonium; ER, endoplasmic reticulum; HDAC, histone deacetylases; HPF, hydroxyphenyl fluorescein; NE, norepineprine; 8-OH-dG, 8-hydroxy-2′-deoxyguanosine; PF1, peroxyfluor1; ROS, reactive oxygen species; SOD, superoxide dismutase
Graphical abstract
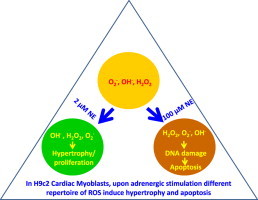
Highlights
-
●
H9c2 myoblasts upon treatment with 2 and 100 µM NE induces hypertrophy and apoptosis.
-
●
Both treatments show comparable levels of DCFH fluorescence with different kinetics.
-
●
Both treatments show comparable levels of HPF fluorescence in an oscillating manner.
-
●
More hydroxyl radical was generated in 100 µM NE treated set.
-
●
DNA damage and apoptosis occurs only in 100 µM NE treated sets.
Introduction
Oxidative stress has long been attributed to age related degenerative diseases [1]. Highly oxidized cellular milieu has been a common denominator for cardiovascular disorders like endothelial dysfunction, hypertrophy, myocyte loss, ischemia reperfusion injury and heart failure [2–4]. However, alleviating those conditions by antioxidants have largely been unsuccessful [5]. Recent years have seen paradigm shifts wherein apart from their deleterious effects, reactive oxygen species (ROS) have been accepted as the bona fide mediators of normal physiological signals [6]. The exact regulatory roles of the two prevalent reactive species, superoxide (O2•−) and hydrogen peroxide (H2O2) in cardiovascular biology have only emerged as a result of recent research [7,8].
Superoxide is generated in distinct cellular locations from NAPDH, xanthine and monoamine oxidases; uncoupled nitric oxide synthases and mitochondrial electron transport complexes. Upon enzymatic dismutation, it is converted to H2O2, a signaling molecule [9]. Notably, the consequences of the generation of reactive species not only depend upon their concentration and the duration of generation; but also on their locations in subcellular microdomains, surrounding antioxidant system, presence of heavy metals, and other physicochemical parameters like pH [10,11]. Based upon these determinants, reactive species may lead to the oxidation of cysteine thiols to sulfenic, sulfinic and sulfonic acids; formation of intra/intermolecular disulfide bonds; S-nitrosylation; S-glutathionylation; nitration of tryptophan and tyrosine; hydroxylation of proline and arginine etc. Such modifications affect the conformation, stability and function of respective proteins with diverse consequences [12,13]. Although a large number of studies suggest the importance of the source(s) and spatio-temporal distribution of ROS in determining the pathobiological outcomes, very little insight as to their mechanisms of actions is known even now [14].
The responses of the cardiovascular system to diverse pathophysiological cues are highly complex, both in terms of clinical manifestations as well as the changes in molecular and biochemical parameters. It has now become evident that many well characterized modes of cellular responses including phosphorylation–dephosphorylation, subcellular trafficking, mRNA–protein turnover, redox-based modifications of cellular proteome etc also play an important role in the development of cardiac hypertrophy and heart failure [15]. In ex vivo myocytes as well as in whole heart stimulated with various pathophysiological agonists, the targets of ROS dependent modulation are as diverse as the signaling kinases (like Raf, MEK, ERK, PKA, PKG and CaMKII), sarcolemmal Na+/Ca++ exchangers, Na+ and L-type Ca++ channels, ryanodine receptor, myofilaments, transcription factors and epigenetic regulator HDAC [16–20].
Norepinephrine (NE) released from the sympathetic nervous system and its cognate receptors (α and β adrenergic, with multiple subtypes) play a critical role in cardiac performance and homeostasis [21,22]. Ex vivo cardiac myocytes upon limited adrenergic stimulation (≤10 µM NE) elicit hypertrophic response, while a higher dose (≥50 µM NE) induces apoptosis, providing a framework of a comparative analysis of two distinct responses culminating to a common pathology i.e., heart failure [23–25]. Number of laboratories have attributed ROS as the common denominator of both the responses [26–28].
We have been investigating the role of ROS in differential adrenergic signaling in cardiac myoblasts as paradigmatic in understanding the redox biology of heart failure. Earlier we have demonstrated that in H9c2 cardiac myoblasts, hypertrophic and apoptotic responses elicited by NE are initiated by a low intensity generation of ROS, followed by respective gene expression programs [24,29]. In the present study, we have extended those observations by investigating the nature and kinetics of ROS generation under the two treatments. We demonstrate that upon stimulation by two different concentrations of NE, multiple ROS are generated at comparable levels but with distinctive kinetics. However, only the cells treated with the higher dose of NE leads to DNA damage and cell death. Thus, both hypertrophic and apoptotic responses are consequences of distinctive redox signaling rather than of difference in threshold level of ROS generation. Our study thus reinforces the concept that dynamic and distinctive pattern of sustained ROS generation contribute to both hypertrophic and apoptotic responses, which are the two discrete arms of heart failure.
Materials and methods
Cell culture
Rat H9c2 cardiac myoblasts (obtained from National Centre for Cell Sciences, Pune, India; originally from ATCC, USA) were cultured as monolayer in Dulbecco's modified Eagle medium (DMEM; 500 mg/l glucose, 2 mmol/l glutamine) supplemented with 10% FBS (Sigma Aldrich, USA), 90 U/ml Penicillin, 90 µg/ml streptomycin and 5 µg/ml amphotericin B in humidified, 5% CO2 containing incubator at 37 °C.
Fluorescence imaging of ROS/RNS generation
H9c2 cells were grown on poly-l-lysine coated glass cover slips in 12 well plates to 70% confluence and kept in serum-free medium for 12–16 h followed by agonist treatment (2 and 100 µM of NE) in the presence and absence of various ROS/RNS inhibitors for the requisite period of time. Thirty minutes prior to imaging, cells were fed with phenol red free DMEM, loaded with various fluoroprobes including, DCFH-DA (5 µM: Sigma Aldrich), DHE (10 µM; Sigma Aldrich), HPF (10 µM: Cayman Chemicals), PF1 (5 µM; synthesized in the laboratory) in dark and kept in a CO2 incubator at 37 °C. Cells were then washed with PBS and examined under Nikon Eclipse Ti-E fluorescence microscope using excitation and emission settings as specified in literature for respective probes. The mean intensity of fluorescence was then measured by NIS-Elements software (Nikon).
Measurement of H2O2 generation
Generation of H2O2 was measured by horseradish peroxidase-linked Amplex red fluorescence assay. Following treatment with NE, the media was removed after due time and the cells were washed with 1× PBS. The cells were then incubated with 10 µM of Amplex red and 0.5 U/ml HRP (horseradish peroxidase) for 15 min at 37 °C. Fluorescence of the supernatant was recorded in Spectramax M2 plate reader (SpincoLab, USA) at 563 nm excitation and 587 nm emission wavelengths.
Estimation of hydroxyl radical generation
Generation of hydroxyl radical was estimated by measuring the conversion of salicylic acid into 2,5-DHBA [30]. H9c2 cells were treated with NE for appropriate duration and then loaded with salicylic acid (100 µM). After 2 h, cells were homogenized with ice-cold PBS (0.5 ml) and deproteinized by the addition of 10% perchloric acid containing 1 mM EDTA and 100 mM sodium pyrosulfite (1.5 ml). The deproteinized extracts were then centrifuged at 9000g for 10 min at 4 °C and 1 M HCl (0.4 ml) was added to the supernatant. The resulting solution was then extracted with 3 ml diethyl ether by vertexing for 1 min. The organic phase was separated by centrifuging at 3000g for 5 min, collected and evaporated to dryness under vacuum. The dried residue was dissolved in 0.03 M citrate/acetate buffer, pH 3.6 (the mobile-phase) and analyzed by HPLC using a zobrax C18 column (150×4.6 mm, 1220 Infinity LC, Agilent technologies, USA). The flow rate was kept at 1.0 ml/min. The 2,5-DHBA was detected at a wavelength of 315 nm and its content was expressed as nmol of DHBA/µmol of salicylic acid. The assay was validated by the estimation of hydroxyl radical generated by treating cells with 20 µM CuSO4 and 1 mM l-ascorbate at 37 °C for 4 h [31].
Synthesis of boronate based peroxyfluor1 (PF1) for detecting peroxynitrite
PF1 was synthesized by palladium-catalyzed transmetalation of fluoran1 in the presence of bis(pinacolato)diboron and potassium acetate [32]. Efficacy and specificity of the probe was confirmed by using SIN-1 (3-Morpholino-sydnonimine, Calbiochem), a specific inducer of peroxynitrite and H2O2, a general oxidant (Fig. S1).
Flow cytometric determination of oxidative DNA damage and apoptosis
Apoptosis and DNA damage were analyzed using the Apoptosis, DNA Damage, and Cell Proliferation Kit (Cat. No. 562253, BD Biosciences, USA). The cells were harvested by trypsin treatment, fixed and permeabilized using BD Cytofix/CytopermTM Fixation/Permeabilization solution. Finally, washing was done using BD Perm/WashTM Buffer. Intracellular staining was done with Alexa Fluor® 647 Mouse Anti-H2AX (pS139), PE Anti-Cleaved PARP (Asp214) antibodies. For the flow cytometric analysis, cells were resuspended in 0.5 ml of staining buffer (3% fetal bovine serum in PBS). Data acquisition and analysis were performed on a flow cytometer (BD FACSCalibur™ Flow Cytometer System) with dual laser. Data were presented by using the software provided with the flow cytometer by the manufacturer.
Estimation of 8-hydroxy-2′-deoxyguanosine
Following the NE treatment for appropriate duration, the genomic DNA was extracted in 100 mM NaCl, 10 mM TrisCl (pH 8.0), 25 mM EDTA, 0.5% SDS and 0.2 mg/ml Proteinase K. The content of oxidative DNA adduct 8-OH-dG was then estimated by competitive in vitro ELISA using anti-8-OH-dG monoclonal antibody [33].
Assay of catalase activity
Following treatment with 2 and 100 µM NE, cells were washed with ice-cold PBS and lysed in 50 mM potassium phosphate buffer (pH 7.8) using Branson ultrasonic cleaner (USA). The supernatant was then used as the source of catalase and the activity was measured by the method originally described by Beers and Sizer [34]. The assay reaction contained 50 mM potassium phosphate buffer (pH 7.8), 30 mM H2O2 and ~300 µg of protein in a total volume of 1.0 ml which measured the removal of hydrogen peroxide at 25 °C in a spectrophotometer (Cary 100 BIO UV Spectrophotometer, Sunnyvale, CA) at 240 nm wavelength. Activity was calculated using the formula described by Weydert and Cullen [35] and it was expressed as mU/mg protein. One unit is the amount of catalase necessary to decompose 1.0 µM of H2O2 per minute at pH 7.8 at 25 °C.
Assay of superoxide dismutase activity
Superoxide dismutase activity was estimated by measuring the inhibition of autoxidation of pyrogallol by the enzyme [36]. NE treated cells were harvested at different time points and lysed in 20 mM Tris (pH 8.2) using Branson ultrasonic cleaner (US). Total SOD activity in the cell extract was measured in 50 mM Tris, pH 8.2; 100 mM EDTA; 8 mM pyrogallol (prepared in 1 mM HCl) and ~300 µg protein. Autoxidation of pyrogallol was monitored for 5 min at room temperature using a UV–visible spectrophotometer (Ultrospec 21000pro, GE Healthcare Life Sciences, UK) at 420 nm. To calculate SOD activity, percentage of inhibition was determined using the following formula: % inhibition=[(control rate−sample rate)/(control rate)] ⁎100.
Statistical analysis
Statistical analyses were performed using GraphPad Prism, PC version 5 (GraphPad software) or Sigma Plot, PC version 12.0. Data are expressed as mean±SEM. Each experiment was performed at least in triplicate unless otherwise specified. Statistical analyses were also performed using one-way ANOVA, t test, followed by Tukey's post hoc test. Values of P<0.05 were considered statistically significant.
Results
Repertoires of ROS generated by the hypertrophic and apoptotic doses of NE are quantitatively comparable but qualitatively different.
We had previously demonstrated that immediately (≤30 min) after treatment with 2 and 100 µM NE (hypertrophic and apoptotic doses respectively), H9c2 cardiac myoblasts induce low intensity ROS (DCFH sensitive) at a comparable level followed by cognate responses after 24 and 48 h respectively [24]. To connect the status of ROS vis-à-vis the two distinct outcomes, we monitored the kinetics of ROS generation for the entire duration.
Cells were kept in serum free medium overnight and treated with 2 and 100 µM NE. At different time intervals, cell permeable redox sensitive dye DCFH-DA (2′-7′-dichlorodihydrofluorescein diacetate) was added, followed by imaging after 30 min. As shown in Fig. 1A, in agreement with our previous observations; immediately after NE treatment, moderate increase in ROS levels (vis-à-vis untreated control) were seen in both 2 and 100 µM NE treated cells until 2 h. Thereafter, DCF fluorescence tapered off under both treatments but again picked up at 24 h onwards only in 100 µM NE treated cells. Such resurgence of ROS generation suggests a second phase of redox signaling followed by apoptosis. Faintly visible signals were also seen in controls cells at later time points, presumably due to ROS generation under sustained deprivation of serum. these data thus suggest that since the level of ROS generated under both condition are comparable, cells treated with 100 µM NE might not undergo apoptosis because of oxidative stress.
Fig. 1.
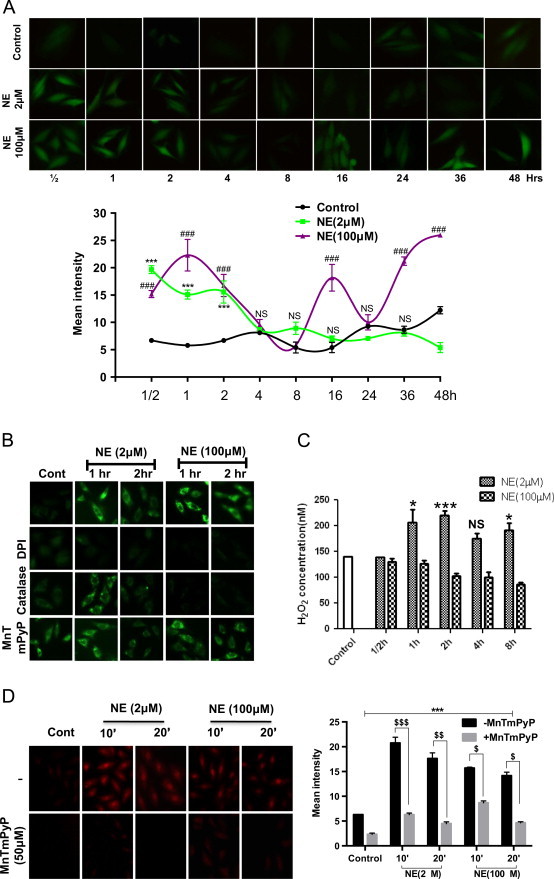
Different repertoire of DCFH-DA sensitive ROS is generated by two doses of NE. (A) H9c2 cells were treated with 2 and 100 µM NE and generation of ROS was monitored by adding 5 µM DCFH-DA 20 min prior to imaging at different time points as marked on each panel. Relative fluorescence intensity for each image is shown in the lower panel. (B) Following treatment with 2 and 100 µM NE, inhibitors of ROS generation viz., DPI (5 µM; inhibitor of flavoenzymes), PEG-catalase (100 µg/ml, quencher of H2O2), and MnTMPyP (50 µM; an SOD mimetic) were added either after 20 min (1 h readout) or 80 min (2 h readout). DCFHDA (5 µM) was added 20 min prior to imaging. (C) Following treatments with 2 and 100 µM NE for various durations, culture supernatants were removed and generation of hydrogen peroxide was estimated by Amplex red assay. (D) H9c2 cells were incubated with superoxide specific probe DHE (10 µM) with or without the SOD mimetic MnTmPyP (50 µM) and 20 min after, cells were also treated with 2 and 100 µM NE, followed by fluorescence imaging at indicated time points (left panel). Relative fluorescence intensity for each image is shown in the right panel. Data are expressed as the mean±SEM of three independent experiments performed in duplicate. *P≤0.05: control vs 2 µM NE; **P≤0.01: control vs 2 µM NE; ***P≤0.001: control vs 2 µM NE; #P≤0.05: control vs 100 µM NE; ##P≤0.01: control vs 100 µM NE and ###P≤0.001: control vs 100 µM NE; $P≤0.05: control vs 100 µM NE; $$P≤0.01: control vs 100 µM NE and $$$P≤0.001: control vs 100 µM NE; NS: not significant P value.
The chemistry of intracellular oxidation of DCFH is complex. It has been suggested that being a hydrophilic molecule, it might not penetrate all cellular compartments with equal efficiency [37]. Also, apart from getting directly oxidized by superoxide or hydrogen peroxide, it is oxidized by Fenton type and certain other unspecific enzymatic reactions [38]. Therefore, although both 2 and 100 µM doses of NE induced comparable levels of DCFH sensitive ROS, their nature, amplitude and sources is likely to be different, thereby explaining two distinct downstream signaling. To examine such possibility, cells were treated with NE and after 20 min (1 h readout) or 80 min (2 h readout), three different ROS quenchers i.e., Catalase, MnTmPyP (a SOD mimetic) and DPI (inhibitor of flavoenzymes including NADPH oxidase) were added. DCFHDA was then added after 20 min and imaging was done after 20 more minutes. As shown in Fig. 1B, treatment with DPI completely inhibited the fluorescence for both treatment sets at either time points. Catalase had no effects on fluorescence in 2 µM NE treated cells at 1 h while partially (~50%) inhibiting it at 2 h. In 100 µM NE treated cells, catalase completely suppressed ROS generation at both time points. MnTmPyP substantially increased fluorescence levels in untreated cells, did not affect it in 2 µM NE treated cells at 1 h but partially inhibited it at 2 h. MnTmPyP slightly inhibited it in 100 µM NE treated cells at both time points. Although such differential inhibition of DCFH fluorescence does not reveal the exact nature and quantity of ROS generated, it strongly suggests that their repertoire (H2O2, superoxide and other subspecies) are quite different and dynamic (temporally) across the two treatment sets.
Depending upon the stimuli superoxide and hydrogen peroxide can be generated both intra- and extracellularly which then enter into the cell inducing signals [39,40]. We tested whether either doses of NE generates extracellular hydrogen peroxide as a determinant of cognate responses. As shown in Fig. 1C, 1 h after treatment with 2 µM NE, extracellular hydrogen peroxide level increased by ~50% and remained high for 8 h, the last time point tested. In contrast, 100 µM NE treated cells showed a gradual decrease in H2O2 secretion that reached ~50% to that of baseline in 8 h. The steady state level of hydrogen peroxide is under homeostatic control involving SOD, catalase, and glutathione/thioredoxin peroxidases. Such increase in H2O2 level in 2 µM NE treated set suggests an alteration in their dynamics and further investigation will be needed for its elucidation. Nevertheless, the concentrations of H2O2 are in nM scale and it is likely to have signaling functions.
We also compared the extent of superoxide generation under the two sets of NE treatments. Dihydroethidium (DHE) is a widely used cell permeable fluoroprobe for superoxide. Upon oxidation in cytosol, it is converted into 2-hydroxyethidium (2-OH-E+), which intercalates with the genomic DNA and the nucleus becomes fluorescent red. When NE treated cells were probed with DHE, both the treatment sets showed nuclear fluorescence as expected. However, in the 2 µM NE treated cells, the fluorescence was increased ~1.2–1.4 fold as compared to the 100 µM NE treated set (Fig. 1D). Noticeably, in both sets, fluorescence was higher at the 10 min time point, which tapered off at 20 min. Although the oxidation of DHE by superoxide is well characterized in vitro, in the intracellular milieu, transition metal ions and other interfering oxidants lead to the generation of related sub-species with overlapping fluorescence spectrum [41]. We thus confirmed its identity by quenching with MnTmPyP, a SOD mimetic. Noticeably, it was substantially (~70–80%) quenched at all time points except at 30 min (~40%) suggesting the existence of some other subspecies. Nevertheless, comparable levels of nuclear fluorescence (albeit with different intensities) are proof that under two treatment sets, difference in superoxide generation is subtle and nuanced rather than absolute. Due to potential redundancy of conclusions, we did not pursue this assay for a longer duration as was done with the DCFHDA probe.
NE induces the generation of hydroxyl radicals but no peroxynitrite.
Apoptotic responses are often attributed to the generation of highly reactive species like peroxynitrite and hydroxyl radical [42,43]. We thus tested whether those highly reactive species are generated under NE treatment, especially at 100 µM (apoptotic dose). While DCFH is widely used fluoroprobe for milder oxidants like hydrogen peroxide, superoxide and their subspecies [37,38], hydroxyphenyl fluorescein (HPF) is a probe for the highly reactive species like peroxynitrite and hydroxyl radicals [44,45]. Cells were treated with 2 and 100 µM NE for different durations and HPF were added 30 min prior to imaging (4–48 h). As shown in Fig. 2A, HPF fluorescence was seen as early as 4 h of treatment with both doses of NE, but it was less intense in 100 µM NE treated cells. HPF fluorescence levels then sustained in an oscillating manner until 48 h which was the last tested time point. Noticeably, under both treatment conditions, the intensity of maximum fluorescence seen in the two treatment sets (i.e., at 24 h for 2 µM NE and at 16 h for 100 µM NE) was largely comparable, although their time kinetics differed. Under 2 µM NE treatment, the fluorescence intensities were high at 4, 24 and 48 h time intervals and it was low at the 8, 16 (lowest) and 36 h intervals. On the contrary, in 100 µM NE treated cells; highest fluorescence was seen at 8 and 16 h but it was low at 4, 24, 36 and 48 h. Such oscillation was consistently seen in repeated experiments done with two different stocks of cells and by two independent workers. It is thus anticipated that the HPF fluorescence is due to a dynamic generation of redox species involved in cell signaling rather than causing oxidative stress (also in agreement with the reappearance of DCFH fluorescence in 100 µM NE treated set).
Fig. 2.
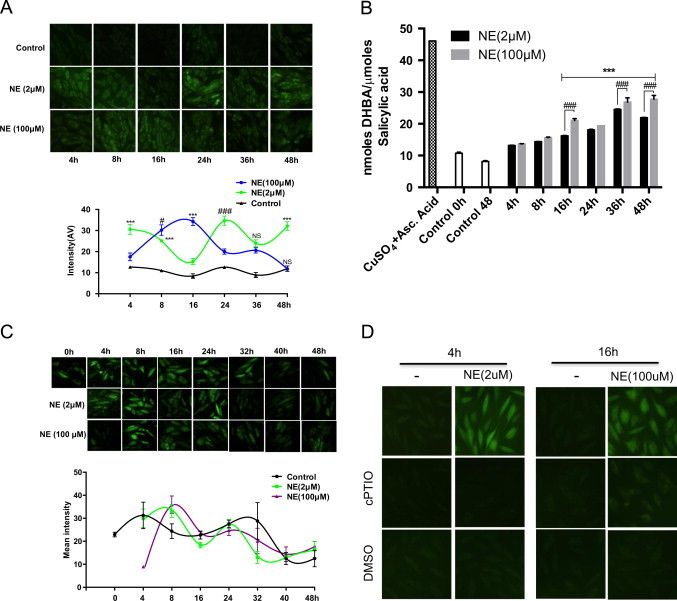
Different kinetics of HPF sensitive ROS generation by two different doses of NE. (A) H9c2 cells were treated with 2 and 100 µM of NE for the indicated time period. Highly reactive species sensitive fluoroprobe HPF (10 µM) was added half an hour prior to fluorescence imaging. Relative fluorescence intensity in each image is shown below. (B) H9c2 cells were treated with 2 and 100 µM NE for various time periods and assayed for the generation of hydroxyl radical. To validate the assay, cells treated with CuSO4/l-ascorbate were used as positive control. (C) H9c2 cells were treated with 2 and 100 µM NE for the indicated time and 5 min prior to imaging cells were also treated with the peroxynitrite specific probe PF1 (5 µM). Lower panel shows relative intensity of images shown in each panel. (D) H9c2 cells were treated with 2 and 100 µM of NE for the indicated time period. Cells were also treated with hydroxyl radical scavenger DMSO (75 mM) and nitric oxide scavenger cPTIO (50 µM) 2.5 h and 0.5 h prior to imaging while fluoroprobe HPF (10 µM) was added half an hour prior to imaging. Data are expressed as the mean±SEM of three independent experiments performed in duplicate. *P≤0.05: control vs 2 µM NE; **P≤0.01: control vs 2 µM NE; ***P≤0.001: control vs 2 µM NE; #P≤0.05: control vs 100 µM NE; ##P≤0.01: control vs 100 µM NE and ###P≤0.001: control vs 100 µM NE; NS: not significant P value.
We then examined to what extent the HPF fluorescence can be attributed to peroxynitrite and/or hydroxyl radicals. Salicylic acid is a trapping agent for hydroxyl radical which converts it into dihydroxylated products 2,3-; 2,5-dihydroxybenzoic acid (DHB) and catechol; assayable by HPLC [46]. As shown in Fig. 2B, treatment with both doses of NE showed a slow but sustained increase in hydroxyl radical generation until 48 h when the net increase was ~3 fold. Nevertheless, though it was slightly higher in 100 µM NE treated set, especially at later time points; the difference did not appear to be substantial enough to be attributed to apoptosis induced by the higher dose of NE. We also tested whether the HPF fluorescence could be due to peroxynitrite generation. PF1 is a cell permeable boronate based probe specific for peroxynitrite. Since it is not commercially available, we synthesized it in the laboratory following the accepted published methodology [47] and confirmed its authenticity as shown in Supplemental Fig. S1. As shown in Fig. 2C, H9c2 cells without any NE treatment showed a baseline fluorescence with PF1 at all time points tested until the 32 h mark that receded from then on, until the 48 h mark. However, neither of the NE treated sets (2 and 100 µM) showed any increase in fluorescence. These two sets also showed a tapering off of fluorescence after 32 h like the untreated control. Therefore, HPF fluorescence induced by NE cannot be attributed to the generation of peroxynitrite. We also confirmed this observation by western blot analysis of cell lysate (2 and 100 µM NE) using nitrotyrosine antibody but could not detect any signal (data not shown). Since above assays did not unambiguously identify the reactive species responsible for the HPF fluorescence, we also tested the effects of specific ROS/RNS quenchers like DMSO, a quencher of hydroxyl radical and cPTIO, a scavenger of nitric oxide [48,49]. As shown in Fig. 2D, in cells treated with 2 µM NE for 4 h, HPF fluorescence was quenched by both DMSO and cPTIO. Similarly, in cells treated with 100 µM NE for 16 h, both DMSO and cPTIO fully quenched the HPF fluorescence. These two time points were chosen because the HPF fluorescence were maximum as well as exclusive for the respective doses of NE (Fig. 2A). Nevertheless, at other time points also, HPF fluorescence was quenched by cPTIO and DMSO (data not shown). Hence, the HPF fluorescence induced by NE is likely to be due to the generation of both hydroxyl radical and some unspecified reactive nitrogen species.
Oxidative DNA damage occurs only in cells undergoing apoptosis
DNA damage is a major reason for cells undergoing apoptosis under oxidative stress [50,51]. Flow cytometric analysis of H2AX phosphorylation at Ser-139 along with PARP cleavage is a reliable method for monitoring DNA damage induced apoptosis [52]. We performed flow cytometric analysis of H2AX phosphorylation and PARP cleavage, with cells treated with 2 and 100 µM NE. As shown in Fig. 3A, 30 h onwards, 100 µM treatment set had a higher proportion of cells positive for H2AX phosphorylation and PARP cleavage and at 40 h, it reached the maximum level. these data suggest that although both treatment sets showed comparable level of ROS/RNS generation (Figs. 1 and 2), DNA damage and apoptosis was prevalent in cells treated with only the higher dose of NE.
Fig. 3.
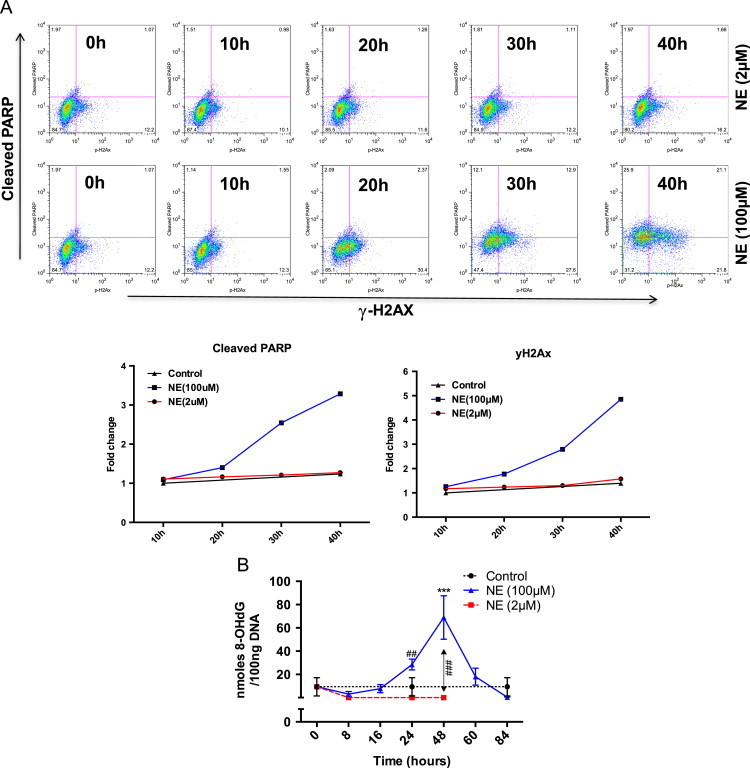
Oxidative DNA damage and apoptosis occurs only in cells treated with 100 µM NE. (A) H9c2 cells were treated with 2 and 100 µM of NE and cells were harvested and analyzed for oxidative DNA damage and apoptosis measuring cleaved PARP (Asp214) and phosphorylated H2AX at serine (S139) (upper panel). Fold changes in mean fluorescent intensity is shown in the lower panel. (B) H9c2 myoblasts were treated with 2 and 100 µM NE. Cells were harvested at various time points, genomic DNA was isolated and then assayed for 8-OH-dG by competitive ELISA using monoclonal antibody specific for 8-OH-dG (33). Data represented ±standard error mean of three independent experiments.
Formation of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) is one of the consequences of hydroxyl radical generation and has often been used as a marker for cardiac oxidative stress and its amelioration by β-blockers [53–55]. Results shown above confirm DNA damage in 100 µM NE treated cells although the nature of ROS/RNS involved remains ambiguous. We thus measured the formation of 8-OH-dG if any, in cells treated with NE. As shown in Fig. 3B, while a significant increase in 8-OH-dG was observed in 100 µM NE treated cells, a decrease (vis-à-vis untreated control) was observed under treatment with 2 µM NE. It is possible that in untreated control, since cells are kept under serum free medium during the course of the experiment, low intensity generation of oxidants might cause mild DNA damage (followed by slow induction of apoptosis later). Being immortalized, H9c2 cells upon treatment with 2 µM NE undergo proliferation along with the induction of certain markers of hypertrophy [24]. Therefore, following treatment with 2 µM NE, it probably removes 8-OH-dG prior to replication [56]. Similar but lesser decrease in 8-OH-dG content was also seen in cells treated with 100 µM NE only until 16 h and thereafter, ~7 fold increase in 8-OH-dG content was seen until 48 h (followed by the onset of apoptosis). Subsequent decrease in 8-OH-dG content at 60 and 84 h time points represented cells (~20% of initial population) that survive apoptotic insults (a recurrent observation made over the years). These results, taken in conjunction with the HPF/DCFH imaging and H2AX phosphorylation/PARP cleavage strongly suggest that although comparable levels of multiple ROS/RNS are generated in 2 and 100 µM NE treated cells, DNA damage and apoptosis occurs only in those treated with the higher dose of NE.
Higher dose of NE decreases catalase but increases SOD activity
After the generation of ROS, various antioxidant enzymes like, CuZn/Mn superoxide dismutase (SOD), glutathione peroxidase and catalase attenuate them, preventing oxidative injury. Under certain experimental setups, a decrease in their activities has been associated with pathological consequences. In experiments on rats, sustained low-level administration of isoproterenol leads to a reduction in cardiac CuZn-SOD but no change in Mn-SOD, catalase and glutathione peroxidase [57]. Since the levels of various ROS in 2 and 100 µM NE treated cells did not show any appreciable differences in their profile, we investigated the activities of two widely used antioxidant enzymes, SOD and catalase, to see if their levels dropped significantly in cells undergoing apoptosis (100 µM NE treated cells). As shown in Fig. 4A, both doses of NE caused modulation of catalase activity. In cells treated with the lower dose, a slight decrease in activity was seen at the 8 h mark, followed by a small increase at 16 h. There was a spike (~85%) at 36 h, followed by a decrease at 48 h. In cells treated with 100 µM NE, a gradual decrease in catalase activity down to 50% was observed until 48 h. Changes in total SOD activity was also seen under both treatment conditions, but to a lesser extent (Fig. 4B). In 2 µM NE treated cells, it slightly increased at 8 h, reduced from 16 to 24 h and then got restored to the control level. In cells treated with the higher dose of NE, a slight increase in SOD activity was seen initially at 8 h, followed by a steady level until 24 h. Thereafter, a sharp increase (~100%) in activity was seen until 48 h. Such an up- and down-regulation of cardiac SOD has also been reported in rats injected with NE for 30 days [58]. Taken together, these results suggest a perturbation in catalase and SOD activities in NE treated cells, but this could not be attributed to any preferential loss of antioxidant levels in cells undergoing apoptosis.
Fig. 4.
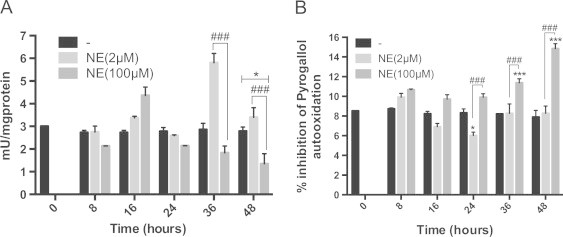
Higher dose of NE decreases catalase but increases SOD activity. (A) After NE treatment, cells were harvested and (A) catalase activity was assayed as described in “methods section”. Activity was expressed as mU/mg of protein. Data are expressed as the mean±SEM of three independent experiments performed in duplicate. *P≤0.05: control vs 2 µM NE; ###P≤0.001: 2 µM NE vs 100 µM NE. (B) SOD activity was assayed as described in “methods section”. Activity was expressed as percentage of inhibition of autoxidation of pyrogallol. Data are expressed as the mean±SEM of three independent experiments performed in duplicate. *P≤0.05: control vs 2 µM NE; ***P≤0.001: 100 µM NE; ###P≤0.001: 2 µM NE vs 100 µM NE.
Discussion
Heart failure is a multi-factorial condition by which cardiac myocytes are progressively lost due to apoptosis. While limited adrenergic stimulation induces myocyte hypertrophy, adrenergic overdrive is the primary contributor towards myocyte loss. Studies done with ex vivo myocytes suggest that both hypertrophic and apoptotic responses induced by NE are a relatively slow process (~24–48 h) wherein ROS is the common determinant. The precise mechanism of such differential response elicited by ROS is unknown [24,25].
Role of ROS in inducing apoptosis has long been documented. Based on the cell type and stimuli, ROS might induce apoptosis through multiple mechanisms [59]. Interestingly, increasing evidences suggest that a large number of redox enzymes previously thought to be involved in energy metabolism also contribute to the intracellular redox homeostasis and therefore might be associated with apoptotic responses [59].
In the present study, we examined the nature and the pattern of ROS generation that leads to the proliferation vis-à-vis death of cardiac myoblasts when treated with two different doses of NE (2 and 100 µM). Analysis of ROS generation by DCFH probe suggested that immediately after NE treatment, low but comparable levels of ROS is generated in both treatment sets. At later time points, cells treated with only the higher dose showed a second phase of ROS generation, also at a lower intensity. Thus, throughout the course of progression towards hypertrophic and apoptotic responses, level of ROS generation in between the two sets did not appear to be differential enough to accept it as “oxidative stress” in one setup compared to the other. It thus reiterates our earlier conclusion that at the onset, low intensity generation of ROS initiate signaling processes that lead to hypertrophy and apoptosis after 24–48 h [24]. Interestingly, upon treatment with various quenchers of O2•− and H2O2, there were subtle differences in the repertoire of ROS in two treatment sets; which presumably contribute to the differential outcome further downstream. This observation gains significance in the context that reactive species are involved in modulating a number of kinase signaling pathways. Based upon the site of generation and the kinetics of their attenuation by various cellular antioxidant systems, they modify specific thiol residues of certain signaling proteins [26–29,60]. It is thus expected that future identification of those targets of NE induced ROS will further enhance our understanding of differential adrenergic signaling in cardiac myocytes.
Since the primary objective of this study was to identify the reactive species, if any, directly responsible for the oxidative damage followed by apoptosis; we further examined whether peroxynitrite or hydroxyl radicals could be a candidate. Amongst various reactive nitrogen species, peroxynitrite is a highly reactive and has been widely attributed to apoptosis [42]. Available literature suggests that HPF is oxidized only by highly reactive species such as the peroxynitrite, hydroxyl radical and those generated from a peroxidase/H2O2 system [44]. We observed comparable levels of sustained HPF fluorescence in cells treated with either doses of NE, albeit with differential kinetics. However, peroxynitrite specific probe PF1 showed no peroxynitrite generation in both treatment sets, while both treated and untreated cells showed baseline fluorescence. Paradoxically though, the HPF fluorescence were partially susceptible to the scavenger of nitric oxide, suggesting the presence of some other yet to be identified reactive nitrogen species. Independent estimation of hydroxyl radical also showed an increased generation under both treatment sets, hence suggesting its contribution in HPF fluorescence as well. However, regardless of the nature of ROS/RNS involved, HPF fluorescence could not explain the differential response by the two doses of NE. It would be of future interest to see how NE generates those highly reactive species in both treatment conditions leading to two different responses i.e., proliferation and apoptosis.
To further substantiate our hypothesis that low but distinctive repertoire of moderate as well as highly reactive species leads to the deleterious effects only in 100 µM NE treated cells, we measured the extent of DNA damage and apoptosis, if any, under both treatment conditions. Phosphorylation of the histone variant H2AX, concurrent with the PARP cleavage; the hallmarks of oxidant induced apoptosis was seen only in 100 µM NE treated cells. Such observation was then reinforced by demonstrating that DNA base modification (8-OH-dG) occurs only in apoptotic cells. Formation of 8-OH-dG has long been studied in the contexts of superoxide, H2O2, hydroxyl radical and peroxynitrite generation [53,61]. Significant increase in 8-OH-dG content only in 100 µM NE treated cells strongly suggested the selective damaging effects of those radicals which were not detrimental in case of a lower dose of NE treatment. Finally, although we observed that in cells undergoing apoptosis; the level of catalase declined over time, it might not cause the collapse of antioxidant defense, as there was also a robust increase in SOD levels in those cells. Taken together, these data unequivocally suggests that rather than a generalized surge in ROS, discrete redox dynamics induces hypertrophy and apoptosis in cardiac myocytes under adrenergic stress. Further experimentation will be required for establishing the biochemical/proteomic basis of such differential effects of ROS/RNS axis in eliciting those responses.
Acknowledgments
The authors thankfully acknowledge funding support from the Council of Scientific and Industrial Research, Government of India (No. 37 (1479)/11), and the Department of Biotechnology, Government of India (BT/PR4268/BRB/10/1016/2011) awarded to SKG. We also thank Dr. Pritam Mukhopadhyaya, School of Physical Sciences, Jawaharlal Nehru University for helping synthesize the PF1 probe in his laboratory. AT was a recipient of a JR/SR fellowship from CSIR, Government of India and MJA is a recipient of JR/SR Fellowship from DBT, Government of India.
Appendix A. Supplementary information
References
- 1.Edrey Y.H., Salmon A.B. Revisiting an age-old question regarding oxidative stress. Free Radical Biology & Medicine. 2014;71:368–378. doi: 10.1016/j.freeradbiomed.2014.03.038. 24704971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho E., Karimi Galougahi K., Liu C.-C., Bhindi R., Figtree G.A. Biological markers of oxidative stress: applications to cardiovascular research and practice. Redox Biology. 2013;1:483–491. doi: 10.1016/j.redox.2013.07.006. 24251116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray T.V.A., Ahmad A., Brewer A.C. Reactive oxygen at the heart of metabolism. Trends in Cardiovascular Medicine. 2014;24(3):113–120. doi: 10.1016/j.tcm.2013.09.003. 24183795 [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Tocchetti C.G., Krieg T., Moens A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radical Biology & Medicine. 2012;53(8):1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010. 22819981 [DOI] [PubMed] [Google Scholar]
- 5.Sawyer D.B. Oxidative stress in heart failure: what are we missing? American Journal of the Medical Sciences. 2011;342(2):120–124. doi: 10.1097/MAJ.0b013e3182249fcd. 21747279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sag C.M., Santos C.X.C., Shah A.M. Redox regulation of cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2014;73:103–111. doi: 10.1016/j.yjmcc.2014.02.002. 24530760 [DOI] [PubMed] [Google Scholar]
- 7.Octavia Y., Brunner-La Rocca H.P., Moens A.L. NADPH oxidase-dependent oxidative stress in the failing heart: from pathogenic roles to therapeutic approach. Free Radical Biology & Medicine. 2012;52(2):291–297. doi: 10.1016/j.freeradbiomed.2011.10.482. 22080085 [DOI] [PubMed] [Google Scholar]
- 8.Brandes R.P., Weissmann N., Schröder K. Redox-mediated signal transduction by cardiovascular Nox NADPH oxidases. Journal of Molecular and Cellular Cardiology. 2014;73:70–79. doi: 10.1016/j.yjmcc.2014.02.006. 24560815 [DOI] [PubMed] [Google Scholar]
- 9.Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biology. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. 24634836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stangherlin A., Reddy A.B. Regulation of circadian clocks by redox homeostasis. Journal of Biological Chemistry. 2013;288(37):26505–26511. doi: 10.1074/jbc.R113.457564. 23861436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J.-G., Baek K., Soetandyo N., Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nature Communication. 2013;4:1568. doi: 10.1038/ncomms2532. 23463011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groitl B., Jakob U. Thiol-based redox switches. Biochimica Biophysica Acta. 2014;1844(8):1335–1343. doi: 10.1016/j.bbapap.2014.03.007. 24657586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forman H.J., Ursini F., Maiorino M. An overview of mechanisms of redox signaling. Journal of Molecular and Cellular Cardiology. 2014;73:2–9. doi: 10.1016/j.yjmcc.2014.01.018. 24512843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y.-R., Zweier J.L. Cardiac mitochondria and reactive oxygen species generation. Circulation Research. 2014;114(3):524–537. doi: 10.1161/CIRCRESAHA.114.300559. 24481843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madamanchi N.R., Runge M.S. Redox signaling in cardiovascular health and disease. Free Radical Biology & Medicine. 2013;61:473–501. doi: 10.1016/j.freeradbiomed.2013.04.001. 23583330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi M., Suzuki E., Takeda R., Oba S., Nishimatsu H., Kimura K. Angiotensin II and tumor necrosis factor-alpha synergistically promote monocyte chemoattractant protein-1 expression: roles of NF-kappaB, p38, and reactive oxygen species. American Journal of Physiology. Heart and Circulatory Physiology. 2008;294(6):H2879–H2888. doi: 10.1152/ajpheart.91406.2007. 18441197 [DOI] [PubMed] [Google Scholar]
- 17.Pimentel D.R., Adachi T., Ido Y., Heibeck T., Jiang B., Lee Y. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. Journal of Molecular and Cellular Cardiology. 2006;41(4):613–622. doi: 10.1016/j.yjmcc.2006.05.009. 16806262 [DOI] [PubMed] [Google Scholar]
- 18.Sag C.M., Wagner S., Maier L.S. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radical Biology & Medicine. 2013;63:338–349. doi: 10.1016/j.freeradbiomed.2013.05.035. 23732518 [DOI] [PubMed] [Google Scholar]
- 19.Donoso P., Sanchez G., Bull R., Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Frontiers in Bioscience (Landmark Edition) 2011;16:553–567. doi: 10.2741/3705. 21196188 [DOI] [PubMed] [Google Scholar]
- 20.Zhu H., Shan L., Schiller P.W., Mai A., Peng T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. Journal of Biological Chemistry. 2010;285(13):9429–9436. doi: 10.1074/jbc.M109.071274. 20097764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrara N., Komici K., Corbi G., Pagano G., Furgi G., Rengo C. β-Adrenergic receptor responsiveness in aging heart and clinical implications. Frontiers in Physiology. 2014;4:396. doi: 10.3389/fphys.2013.00396. 24409150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciccarelli M., Santulli G., Pascale V., Trimarco B., Iaccarino G. Adrenergic receptors and metabolism: role in development of cardiovascular disease. Frontiers in Physiology. 2013;4:265. doi: 10.3389/fphys.2013.00265. 24106479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clerk A. The radical balance between life and death. Journal of Molecular and Cellular Cardiology. 2003;35(6):599–602. doi: 10.1016/s0022-2828(03)00121-4. 12788376 [DOI] [PubMed] [Google Scholar]
- 24.Gupta M.K., Neelakantan T.V., Sanghamitra M., Tyagi R.K., Dinda A., Maulik S. An assessment of the role of reactive oxygen species and redox signaling in norepinephrine-induced apoptosis and hypertrophy of H9c2 cardiac myoblasts. Antioxidants and Redox Signalling. 2006;8(5–6):1081–1093. doi: 10.1089/ars.2006.8.1081. 16771697 [DOI] [PubMed] [Google Scholar]
- 25.Fu Y.-C., Chi C.-S., Yin S.-C., Hwang B., Chiu Y.-T., Hsu S.-L. Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species-TNF alpha-caspase signaling pathway. Cardiovascular Research. 2004;62(3):558–567. doi: 10.1016/j.cardiores.2004.01.039. 15158148 [DOI] [PubMed] [Google Scholar]
- 26.Guo J., Gertsberg Z., Ozgen N., Steinberg S.F. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circulation Research. 2009;104(5):660–669. doi: 10.1161/CIRCRESAHA.108.186288. 19168439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong F., Xiao D., Zhang L. Norepinephrine causes epigenetic repression of PKCε gene in rodent hearts by activating Nox1-dependent reactive oxygen species production. FASEB Journal. 2012;26:2753–2763. doi: 10.1096/fj.11-199422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corbi G., Conti V., Russomanno G., Longobardi G., Furgi G., Filippelli A. Adrenergic signaling and oxidative stress: a role for sirtuins? Frontiers on Physiology. 2013;4:324. doi: 10.3389/fphys.2013.00324. 24265619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jindal E., Goswami S.K. In cardiac myoblasts, cellular redox regulates FosB and Fra-1 through multiple cis-regulatory modules. Free Radical Biology & Medicine. 2011;51(8):1512–1521. doi: 10.1016/j.freeradbiomed.2011.07.008. 21820506 [DOI] [PubMed] [Google Scholar]
- 30.Ueno S., Kashimoto T., Susa N., Shiho K., Seki T., Ito N. Estimation of hydroxyl radical generation by salicylate hydroxylation method in kidney of mice exposed to ferric nitrilotriacetate and potassium bromate. Free Radical Research. 2007;41(11):1246–1252. doi: 10.1080/10715760701644019. 17907000 [DOI] [PubMed] [Google Scholar]
- 31.Bolin C., Cardozo-Pelaez F. Assessing biomarkers of oxidative stress: analysis of guanosine and oxidized guanosine nucleotide triphosphates by high performance liquid chromatography with electrochemical detection. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2007;856(1–2):121–130. doi: 10.1016/j.jchromb.2007.05.034. 17581804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sikora A., Zielonka J., Lopez M., Joseph J., Kalyanaraman B. DiRecT oxidation of boronates by peroxynitrite: mechanism and implications in fluorescence imaging of peroxynitrite. Free Radical Biology & Medicine. 2009;47(10):1401–1407. doi: 10.1016/j.freeradbiomed.2009.08.006. 19686842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Modak M.A., Parab P.B., Ghaskadbi S.S. Pancreatic islets are very poor in rectifying oxidative DNA damage. Pancreas. 2009;38(1):23–29. doi: 10.1097/MPA.0b013e318181da4e. 18695629 [DOI] [PubMed] [Google Scholar]
- 34.Beers R.F., Sizer I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. Journal of Biological Chemistry. 1952;195(1):133–140. 14938361 [PubMed] [Google Scholar]
- 35.Weydert C.J., Cullen J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nature Protocols. 2010;5(1):51–66. doi: 10.1038/nprot.2009.197. 20057381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marklund S., Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. European Journal of Biochemistry. 1974;47(3):469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. 4215654 [DOI] [PubMed] [Google Scholar]
- 37.Karlsson M., Kurz T., Brunk U.T., Nilsson S.E., Frennesson C.I. What does the commonly used DCF test for oxidative stress really show? Biochemistry Journal. 2010;428(2):183–190. doi: 10.1042/BJ20100208. 20331437 [DOI] [PubMed] [Google Scholar]
- 38.Chen X., Zhong Z., Xu Z., Chen L., Wang Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radical Research. 2010;44(6):587–604. doi: 10.3109/10715761003709802. 20370560 [DOI] [PubMed] [Google Scholar]
- 39.Gough D.R., Cotter T.G. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death & Disease. 2011;2:e213. doi: 10.1038/cddis.2011.96. 21975295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kliment C.R., Oury T.D. Extracellular superoxide dismutase protects cardiovascular syndecan-1 from oxidative shedding. Free Radical Biology & Medicine. 2011;50(9):1075–1080. doi: 10.1016/j.freeradbiomed.2011.02.014. 21334435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalyanaraman B., Dranka B.P., Hardy M., Michalski R., Zielonka J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes − the ultimate approach for intra- and extracellular superoxide detection. Biochimica Biophysica Acta. 2014;1840(2):739–744. doi: 10.1016/j.bbagen.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khalil H., Rosenblatt N., Liaudet L., Widmann C. The role of endogenous and exogenous RasGAP-derived fragment N in protecting cardiomyocytes from peroxynitrite-induced apoptosis. Free Radical Biology & Medicine. 2012;53(4):926–935. doi: 10.1016/j.freeradbiomed.2012.06.011. 22721922 [DOI] [PubMed] [Google Scholar]
- 43.Yin H., Zhu M. Free radical oxidation of cardiolipin: chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radical Research. 2012;46(8):959–974. doi: 10.3109/10715762.2012.676642. 22468920 [DOI] [PubMed] [Google Scholar]
- 44.Setsukinai K., Urano Y., Kakinuma K., Majima H.J., Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. Journal of Biological Chemistry. 2003;278(5):3170–3175. doi: 10.1074/jbc.M209264200. 12419811 [DOI] [PubMed] [Google Scholar]
- 45.Paulander W., Wang Y., Folkesson A., Charbon G., Løbner-Olesen A., Ingmer H. Bactericidal antibiotics increase hydroxyphenyl fluorescein signal by altering cell morphology. PLoS One. 2014;9(3):e92231. doi: 10.1371/journal.pone.0092231. 24647480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diez L., Livertoux M.H., Stark A.A., Wellman-Rousseau M., Leroy P. High-performance liquid chromatographic assay of hydroxyl free radical using salicylic acid hydroxylation during in vitro experiments involving thiols. Journal of Chromatography B: Biomedical Sciences and Applications. 2001;763(1–2):185–193. doi: 10.1016/s0378-4347(01)00396-6. 11710577 [DOI] [PubMed] [Google Scholar]
- 47.Zielonka J., Sikora A., Joseph J., Kalyanaraman B. Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: direct reaction with boronate-based fluorescent probe. Journal of Biological Chemistry. 2010;285(19):14210–14216. doi: 10.1074/jbc.M110.110080. 20194496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwarzer C., Fu Z., Fischer H., Machen T.E. Redox-independent activation of NF-kappaB by Pseudomonas aeruginosa pyocyanin in a cystic fibrosis airway epithelial cell line. Journal of Biological Chemistry. 2008;283(40):27144–27153. doi: 10.1074/jbc.M709693200. 18682396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berenyiova A., Grman M., Mijuskovic A., Stasko A., Misak A., Nagy P. The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants. Nitric Oxide. 2015;46:123–130. doi: 10.1016/j.niox.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 50.Ivashkevich A., Redon C.E., Nakamura A.J., Martin R.F., Martin O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Letters. 2012;327(1–2):123–133. doi: 10.1016/j.canlet.2011.12.025. 22198208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mah L.-J., El-Osta A., Karagiannis T.C., Gamma H. 2AX as a molecular marker of aging and disease. Epigenetics. 2010;5:129–136. doi: 10.4161/epi.5.2.11080. [DOI] [PubMed] [Google Scholar]
- 52.Turinetto V., Giachino C. Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Research. 2015;43(5):2489–2498. doi: 10.1093/nar/gkv061. 25712102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergeron F., Auvré F., Radicella J.P., Ravanat J.L. HO* radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proceedings of the National Academy of Sciences USA. 2010;107(12):5528–5533. doi: 10.1073/pnas.1000193107. 20212167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki S., Shishido T., Ishino M., Katoh S., Sasaki T., Nishiyama S. 8-Hydroxy-2′-deoxyguanosine is a prognostic mediator for cardiac event. European Journal of Clinical Investigation. 2011;41(7):759–766. doi: 10.1111/j.1365-2362.2010.02465.x. 21261617 [DOI] [PubMed] [Google Scholar]
- 55.Susa T., Kobayashi S., Tanaka T., Murakami W., Akashi S., Kunitsugu I. Urinary 8-hydroxy-2′-deoxyguanosine as a novel biomarker for predicting cardiac events and evaluating the effectiveness of carvedilol treatment in patients with chronic systolic heart failure. Circulation Journal: Official Journal of Japanese Circulation Society. 2012;76(1):117–126. doi: 10.1253/circj.cj-11-0537. 22008315 [DOI] [PubMed] [Google Scholar]
- 56.Stedeford T., Cardozo-Pelaez F., Nemeth N., Song S., Harbison R.D., Sanchez-Ramos J. Comparison of base-excision repair capacity in proliferating and differentiated PC 12 cells following acute challenge with dieldrin. Free Radical Biology & Medicine. 2001;31(10):1272–1278. doi: 10.1016/s0891-5849(01)00715-8. 11705706 [DOI] [PubMed] [Google Scholar]
- 57.Srivastava S., Chandrasekar B., Gu Y., Luo J., Hamid T., Hill B.G. Downregulation of CuZn-superoxide dismutase contributes to beta-adrenergic receptor-mediated oxidative stress in the heart. Cardiovascular Research. 2007;74(3):445–455. doi: 10.1016/j.cardiores.2007.02.016. 17362897 [DOI] [PubMed] [Google Scholar]
- 58.Neri M., Cerretani D., Fiaschi A.I., Laghi P.F., Lazzerini P.E., Maffione A.B. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. Journal of Cellular and Molecular Medicine. 2007;11(1):156–170. doi: 10.1111/j.1582-4934.2007.00009.x. 17367510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Circu M.L., Aw T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology & Medicine. 2010;48(6):749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. 20045723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Billia F., Hauck L., Grothe D., Konecny F., Rao V., Kim R.H. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proceedings of the National Academy of Sciences USA. 2013;110(15):6085–6090. doi: 10.1073/pnas.1303444110. 23530187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Briedé J.J., van Delft J.M.H., de Kok T.M.C.M., van Herwijnen M.H.M., Maas L.M., Gottschalk R.W.H. Global gene expression analysis reveals differences in cellular responses to hydroxyl- and superoxide anion radical-induced oxidative stress in caco-2 cells. Toxicological Sciences: Official Journal of Society of Toxicology. 2010;114(2):193–203. doi: 10.1093/toxsci/kfp309. 20044591 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.