

Abstract
Poloxamer 188 (P188), a nonionic block copolymer chemical surfactant known to have cytoprotective, rheologic, anti-inflammatory, and anti-thrombotic activity, has shown promise in the management of selected trauma patients. We studied human PMN oxidative burst and adhesion molecule expression when exposed to P188.
After RBC lysis of whole blood samples, WBC components were primed with PAF, primed and activated with fMLP, primed and activated with PMA, or left unstimulated. Each group was treated with vehicle or P188 (0.005 – 15 mg/ml concentrations). Flow cytometry quantified: 1) PMN superoxide anion production and 2) PMN marker expression of CD11b and L-selectin.
Among non-PMA activated PMNs, P188 increased superoxide anion production. PMA-activated PMNs decreased superoxide anion production, proportional to P188 dose. Among fMLP-activated PMNs, the highest P188 dose increased the expression of CD11b. Among PMA-activated PMNs, decreased CD11b expression was seen for the mid-range doses.
PMN’s altered their oxidative burst and marker expression after exposure to P188. When used at lower doses, P188 may increase the oxidative burst response and, when used at very high doses, increase CD11b expression. However, if PMNs are in a maximally activated state, a higher dose of P188 may decrease the oxidative burst response and decrease CD11b expression.
Introduction
Traumatically injured patients develop pathophysiologic responses including inflammation, coagulopathy, and acidosis which lead to cell membrane breakdown, hypoperfusion, and microvascular disease. Subsequent to the morbidity related to acute trauma, these pathophysiologic immune responses lead to a cascade of events including the systemic inflammatory response syndrome, ischemia/reperfusion injury, and multiple organ failure (MOF).1, 2
Polymorphonuclear cells (PMNs) mediate the global response to major trauma via activation, microvascular adherence, migration, and oxidative burst. While some of the proinflammatory response and PMN activity is protective, reducing the risk of sepsis, PMN adherence and oxidative burst is also known to damage endothelial cells, leading to vascular permeability, edema, and, ultimately, MOF.3, 4
Poloxamer 188 (P188) is a nonionic block copolymer chemical surfactant known to have cytoprotective, rheologic, anti-inflammatory, and anti-thrombotic activity combined with minimal toxicity. P188 derives its wide range of therapeutic applications via adherence to hydrophobic surfaces exposed after cell damage, restoring normal hydrated surfaces. P188 has shown promise in the management of many diseases, including sickle cell disease, vascular disease, stroke, and cancer, and clinical trials, involving more than 4,000 patients, have shown it to be safe when administered intravenously.5–7 P188 has shown the ability to improve the microcirculation 8, minimize platelet aggregation 9, and improve survival after hemorrhagic shock.10 P188 has also shown the ability to mitigate reperfusion injury after myocardial infarction 11, 12 and improve hemodynamics, and subsequent oxygen delivery, after hemorrhagic shock in humans.13 Preliminary in vitro and animal model work has shown that P188 limits PMN migration14, 15, impairs PMN delivery to an inflammatory locus16, and attenuates enzyme release from PMNs.17 Although these beneficial effects of P188 on PMNs have been previously seen, the direct effects of P188 on human PMNs are unknown. Therefore, we designed experiments to specifically examine whether P188 influenced human PMN oxidative burst or cell surface marker expression and hypothesized that it would.
Materials and Methods
Materials
Phosphotidylcholine (PAF), N-Formyl-Met-Leu-Phe (fMLP), Phorbol 12-Myristate 13-Acetate (PMA) were purchased from Sigma-Aldrich (St. Louis, MO). Phosphate buffered saline (PBS) with glucose (7mM) (‘PBSG’) was used to re-suspend the cells, along with the additional priming and activating agents, to a final volume of 100 μl. Dihydroethidium (DHE), phycoerythrin (PE) conjugated CD11b antibody, and fluorescein isothiocyanate (FITC) conjugated CD62L (L-selectin) antibody were purchased from Invitrogen™ (Invitrogen Corporation, Carlsbad, CA). Purified poloxamer 188 formulated for clinical use was provided by SynthRx Corporation (Bellaire, TX).
Sample collection and RBC lysis
Venous blood was collected from six healthy, human volunteers through a sterile 21-gauge butterfly needle into a heparinized collection tube (143 USP units of sodium heparin/tube; ~8 ml blood/tube). Red blood cell (RBC) lysis solution (Qiagen Sciences, Valencia, CA) was mixed with the collected blood (3:1 ratio) for ~5 min on a shaker at room temperature. After centrifugation at 2000 x g for 2 minutes, 100 μl of PBSG was added per every 100 μl of blood (~7.5 × 105 total WBC’s). For each experimental sample, 50 μl of the PBSG+cells solution was used. The final cell population was ~60% PMNs versus other WBC types by flow cytometry and >99% viable by trypan blue exclusion.
Superoxide anion production assay
This assay was performed to assess the production of superoxide anion, a key component of the oxidative burst by PMNs in response to a stimulus. Superoxide anion production was measured by hydroethidine (HE) conversion to ethidium bromide (EB), detected by flow cytometry (BD LSR II; BD Biosciences, San Jose, CA). The PMN + PBSG solution (100 μl) was incubated with DHE (10 μM final concentration) for 15 minutes at 37° C. Either the vehicle (sodium chloride, sodium citrate, and citric acid) or P188 was added to unprimed and unactivated experimental wells at final concentrations of 0.005 mg/ml (ultra low dose), 0.05 mg/ml (very low dose), 0.5 mg/ml (low dose), 2.0 mg/ml (medium dose*), 6.0 mg/ml (high dose), or 15 mg/ml (very high dose) and allowed to incubate for 5 minutes at 37° C. The ‘priming’ agent, PAF (10 μM final concentration), was added to certain control and experimental wells and allowed to incubate for 5 minutes at 37° C. P188 was added in the above concentrations to the appropriate experimental wells and allowed to incubate for 5 minutes at 37° C. The NADPH-dependent activating agent, fMLP (1 μM final concentration), or the NADPH-independent activating agent, PMA (10 μM final concentration), was added and allowed to incubate for 20 minutes at 37° C. The 96-well plate was placed on ice for 5 minutes to stop the reactions and immediately run on the high throughput system of the BD LSR II flow cytometer. The PMN population was gated on the FSC/SSC plot and the mean fluorescent intensity (MFI) was recorded. All samples were run in duplicate.
To ensure that decreased superoxide anion production was not the result of PMN cell death, the samples were checked for cell viability using the BD™ Cell Viability Kit (BD Biosciences, San Jose, CA) and viewed using Trypan blue exclusion.
CD11b or L-selectin surface marker expression
The adhesion molecules L-selectin and CD11b are important for PMN rolling, adhesion to the endothelium, and transmigration to an insult. Surface marker expression was measured by conjugated antibodies binding to the PMN cell surface, detected by flow cytometry. P188 was added to unstimulated (unprimed and unactivated) experimental wells at final concentrations of 0.5 mg/ml (low dose), 2.0 mg/ml (medium dose), 6.0 mg/ml (high dose), or 15 mg/ml (very high dose) and allowed to incubate for 5 minutes at 37° C. The ‘priming’ agent, PAF (10 μM final concentration), was added to certain control and experimental wells and allowed to incubate for 5 minutes at 37° C. P-188 was added in the above concentrations to the appropriate experimental wells and allowed to incubate for 5 minutes at 37° C. The NADPH-dependent activating agent, fMLP (1 μM final concentration), or the NADPH-independent activating agent, PMA (10 μM final concentration), was added and allowed to incubate for 20 minutes at 37° C. The conjugated antibodies (CD11b and L-selectin, 2 μl each antibody) were added to all wells and allowed to incubate for 30 minutes at 37° C. The 96-well plate was immediately run on the high throughput system of the BD LSR II flow cytometer. The PMN population was gated on the FSC/SSC plot and the MFI (mean receptor density) was recorded. All samples were run in duplicate.
Cell viability/osmolality assays
To ensure that there was not significant PMN cell death in the presence of P188, we performed two experiments to ensure cell viability. First, we placed the same number of cells in increasing doses of P188 (same doses as above) and identified cell death via trypan blue exclusion. Second, we ran a flow cytometric cell viability assay (BD cell viability kit) which uses thiazole orange (TO) to stain live cells and propidium iodide (PI) to stain dead cells. Finally, we determined the osmolality of the cell media, with the increasing doses of P188, to be sure that alterations in the osmolality of the cell suspension solution were not responsible for observed effects.
Statistical analysis
Data were compared using a repeated measures analysis of variance (ANOVA) with a Dunnett’s test versus a control for post-hoc comparisons. Data are reported as the mean ± SEM for each group. p < 0.05 was considered statistically significant. All groups contain six different human donors.
Results
Superoxide anion production
Among control groups (no P188), PAF alone led to an insignificant increase in superoxide anion production (172 ± 38 vs 214 ± 44 MFI). When the cells were subsequently activated with fMLP or PMA, the MFI increased to 474 ± 80 (p<0.05 vs. baseline and primed only) and 2446 ± 132 (p<0.05 vs. other 3 groups), respectively.
Exposure to the ultra low doses of P188 did not increase the superoxide anion production among the unstimulated cells (no priming/activating agents; hereafter unstimulated), primed cells (PAF), and primed and NADPH-dependently activated (PAF+fMLP) cells (Figure 1). When the PMNs were exposed to the very low dose, cells stimulated by PAF and PAF+fMLP began to increase their superoxide anion production. Exposure to the low dose of P188 led to a significant increase in superoxide anion production (Figure 1) among the unstimulated cells and cells stimulated by PAF and PAF+fMLP, but not among the primed and NADPH-independently activated (PAF+PMA) cells. Exposure to increasing doses of P188 caused an increased response proportional to the low doses. The response peaked with the low dose and an inversely proportional superoxide anion production response was noted for the higher doses (Figure 1). The low, medium, and high P188 dose exposures led to statistically significant increases in superoxide anion production, compared to the baseline controls among unstimulated cells and cells stimulated with PAF or PAF+fMLP (p< 0.001 for all). The difference between superoxide anion production of cells exposed to the very high P188 dose and the baseline controls was not statistically significant among the unstimulated and PAF-stimulated PMN groups.
Figure 1. Superoxide anion production by PMNs in the presence of P188.
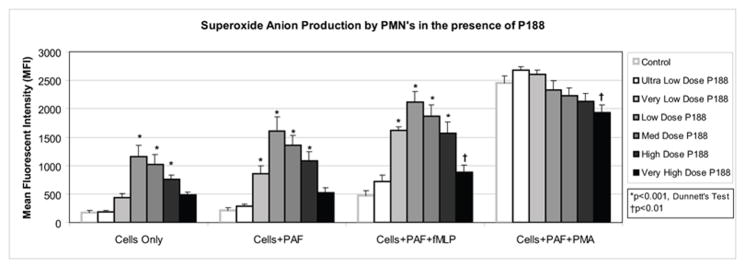
After incubation with DHE and priming (PAF), P188 (in the following final doses: Ultra low dose: 0.005 mg/ml, very low dose: 0.05 mg/ml, low dose: 0.5 mg/ml, medium dose: 2.0 mg/ml, high dose: 6.0 mg/ml, very high dose: 15 mg/ml) was added to the human PMNs. Cells were then activated (fMLP or PMA). Mean fluorescent intensity (MFI), a surrogate for superoxide anion production, was measured by flow cytometry. Unstimulated, primed, and activated (with fMLP) PMNs exhibited increased superoxide anion production in the presence of P188 in a dose-dependent fashion. Among the maximally activated PMNs, addition of very high dose P188 reduced superoxide anion production.
Among the cells stimulated with PAF+PMA, exposure to the ultra low, very low, low, medium, and high doses of P188 did not significantly change the superoxide anion production. Interestingly, exposure to the very high dose of P188 led to a statistically significant decrease in superoxide anion production (p<0.01) (Figure 1). No difference in PMN cell death between the control and experimental groups was observed using the flow cytometric cell viability kit or via Trypan blue exclusion (>95% viability for all groups by both methods). The osmolality of the cell suspension media, with increasing doses of P188 ranged from 377–388 mmol/kg, with no differences observed between different concentrations of P188.
CD11b expression
Among control groups (no P188), PAF alone led to a mild increase in CD11b expression (1140 ± 254 vs 1468 ± 92 MFI). When the cells were subsequently activated with fMLP or PMA, the MFI slightly increased to 1673 ± 99 and 1779 ± 114, respectively (Figure 2).
Figure 2. CD11b expression on PMNs in the presence of P188.
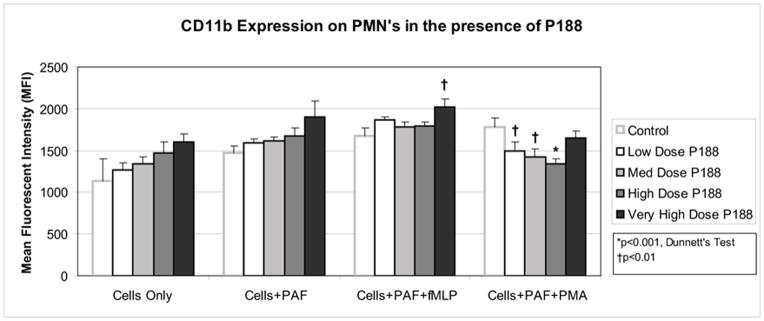
After priming (PAF), P188 (in the following final doses: low dose: 0.5 mg/ml, medium dose: 2.0 mg/ml, high dose: 6.0 mg/ml, very high dose: 15 mg/ml) was added to the human PMNs. Cells were then activated (fMLP or PMA) and PE conjugated antibodies specific for human CD11b antibodies were added. Mean fluorescent intensity (MFI), a surrogate for CD11b marker expression, was measured by flow cytometry. Among unstimulated or primed PMNs, P188 did not alter CD11b marker expression. Primed and activated (fMLP) PMNs increased CD11b expression in the presence of very high dose P188. Primed and activated (PMA) PMNs decreased CD11b expression in the presence of low, medium, and high dose P188.
Incubation of the cells with P188 did not lead to a statistically significant change in the CD11b expression of unstimulated cells or PAF-stimulated cells. After PMN stimulation with PAF+fMLP, the very high dose of P188 increased the expression of CD11b (1673 ± 99 vs 2022 ± 92 MFI, p<0.05). Among PMNs stimulated with PAF+PMA, the low, medium, and high doses of P188 decreased the CD11b expression relative to the control (p<0.05) (Figure 2).
L-selectin expression
Among control groups (no P188), PAF did not alter L-selectin expression (81 ± 19 vs 75 ± 22 MFI). When the cells were subsequently activated with fMLP or PMA, the MFI decreased significantly to 28 ± 3 and 27 ± 2, respectively (Figure 3).
Figure 3. L-selectin expression on PMNs in the presence of P188.
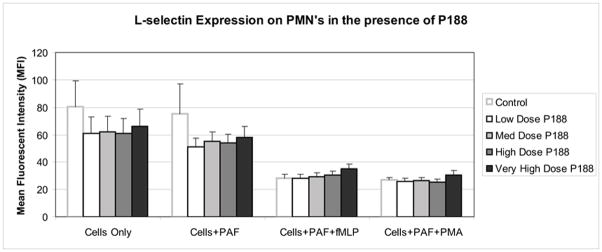
Incubation of the PMN’s with P188 did not lead to any significant change in L-selectin expression, irrespective of dose used or stimulation status of the cells.
Incubation of the PMNs with P188 did not lead to any significant change in L-selectin expression, irrespective of dose used or stimulation status of the cells (Figure 3).
Discussion
We have shown that PMNs alter their oxidative burst and surface marker expression when exposed to P188. When unstimulated, primed, or primed and NADPH dependently activated, PMNs exposed to low, medium, and high doses of P188 have significantly increased superoxide anion production in an inversely proportional dose-dependent fashion. When PMNs are primed and NADPH independently activated, however, exposure to very high dose P188 leads to a significant decrease in superoxide anion production. Expression of CD11b among unstimulated and primed PMNs was unchanged in the presence of P188. Very high dose P188 increased CD11b expression among primed and NADPH dependently activated PMNs. After priming and NADPH independent activation of PMNs, low, medium, and high dose P188 significantly decreased CD11b expression. The expression of L-selectin on PMNs was unaffected by exposure to P188.
Lane and colleagues first identified P188 inhibition of neutrophil migration and adherence in vitro.15 Tan and coworkers subsequently showed that neutrophil migration was inhibited by synthetic polymers in an in vitro, three-dimensional collagen gel.14 Lane and colleagues followed their original work by assessing PMN migration in vivo after P188 treatment in a rodent model of bacterial sepsis.16 They found a significantly increased mortality among rodents treated with P188 and attributed this to impaired neutrophil delivery to the inflammatory locus and a subsequent increased rate of sepsis. Animals that received P188 alone (without the bacterial injection) did not have increased mortality. Additionally, the effects of P188 on neutrophil-mediated injury to endothelial cells in an in vitro sheep model of microvascular injury revealed that neutrophil incubation with P188 significantly attenuated neutrophil adhesion, cytotoxicity, and proteolytic enzyme release.17 Pretreatment of the endothelial cells with P188 did not inhibit neutrophil adherence, indicating that P188 may have direct effects on the cell membrane of the PMN.
Separating the effects of P188 on PMNs, in isolation from endothelium or activated endothelium interactions, required in vitro experimentation. Our study design is a simultaneous strength and recognized limitation, as in vivo study could elucidate further physiologic relevance. Additionally, identifying the direct effects of P188 on isolated PMNs, as opposed to the lymphocyte, monocyte, PMN mixture we used, would require gradient separation, an action that is known to prime/activate PMNs. Given this, we chose to use a non-separated cell mixture and gate on the PMN population. We recognize that the effects of P188 on PMNs may be indirect effects, through interactions with other cell types.
PMNs appear to respond to P188 by selectively increasing their expression of CD11b and generally increasing the release of oxygen free radicals. Under maximally activated conditions, however, PMNs appear to respond to high dose P188 by reducing the oxidative burst response and decreasing their CD11b expression. The mechanism of action behind the latter response is unknown. Low doses may prime/activate PMNs, similar to any other foreign substance, while higher doses may prevent activation. P188 has been previously shown to attenuate neutrophil adhesion and transendothelial migration in vivo14, 16, 17, prompting the hypothesis that P188 may interact with the cell membrane or adhesion molecule, acting as a competitive inhibitor of adhesion molecule interactions. The poloxamer may be acting in a similar fashion at very high doses in our study, preventing some level of NADPH independent activation by PMA and preventing interaction between the adhesion molecule and the antibody.
Our work has significant clinical implications. Among mild and moderately injured patients with unstimulated or primed PMNs, use of P188, particularly at low or moderate doses, may exacerbate the PMN oxidative burst response. This may have serious ramifications such as eventual development of multiple organ failure (MOF). On the other hand, severely injured trauma patients or patients developing ARDS and/or MOF, whose PMNs are likely to be maximally primed and activated, may benefit from P188 therapy through reduced superoxide anion production and/or decreased CD11b expression.
Human PMNs alter their oxidative burst and surface marker expression after exposure to P188. These findings should be taken into consideration when considering optimal dosing and patient selection for future trials involving P188.
Acknowledgments
Supported by NIH grants: T32 GM008792-06, P50 GM38529, M01 RR02558, Texas Higher Education Coordinating Board
Footnotes
2 mg/ml was chosen as the mid-range dose based on an approximate average ~120 mg/kg (range: 50–200 mg/kg) in published pre-clinical and clinical work.6, 10 For a 120 mg/kg dose, in 250g rats, with 60 ml/kg blood per rat, the average dose is approximately 2 mg/ml.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. The Surgical Clinics of North America. 1995;75(2):257–77. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 2.Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38(12):1336–45. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Botha AJ, Moore FA, Moore EE, Fontes B, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation states: therapeutic challenges. Shock. 1995;3(3):157–66. doi: 10.1097/00024382-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation: an early vulnerable window. Surgery. 1995;118(2):358–64. doi: 10.1016/s0039-6060(05)80345-9. discussion 64–5. [DOI] [PubMed] [Google Scholar]
- 5.Jewell RC, Khor SP, Kisor DF, LaCroix KA, Wargin WA. Pharmacokinetics of RheothRx injection in healthy male volunteers. Journal of Pharmaceutical Sciences. 1997;86(7):808–12. doi: 10.1021/js960491e. [DOI] [PubMed] [Google Scholar]
- 6.Orringer EP, Casella JF, Ataga KI, et al. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: A randomized controlled trial. JAMA. 2001;286(17):2099–106. doi: 10.1001/jama.286.17.2099. [DOI] [PubMed] [Google Scholar]
- 7.Grindel JM, Jaworski T, Emanuele RM, Culbreth P. Pharmacokinetics of a novel surface-active agent, purified poloxamer 188, in rat, rabbit, dog and man. Biopharmaceutics & Drug Disposition. 2002;23(3):87–103. doi: 10.1002/bdd.297. [DOI] [PubMed] [Google Scholar]
- 8.Hymes AC, Safavian MH, Gunther T. The influence of an industrial surfactant Pluronic F-68, in the treatment of hemorrhagic shock. The Journal of Surgical Research. 1971;11(4):191–7. doi: 10.1016/0022-4804(71)90060-6. [DOI] [PubMed] [Google Scholar]
- 9.Edwards CM, May JA, Heptinstall S, Lowe KC. Effects of pluronic F-68 (poloxamer 188) on platelet aggregation in human whole blood. Thrombosis Research. 1996;81(4):511–2. doi: 10.1016/0049-3848(96)00025-4. [DOI] [PubMed] [Google Scholar]
- 10.Mayer DC, Strada SJ, Hoff C, Hunter RL, Artman M. Effects of poloxamer 188 in a rabbit model of hemorrhagic shock. Annals of Clinical and Laboratory Science. 1994;24(4):302–11. [PubMed] [Google Scholar]
- 11.Schaer GL, Hursey TL, Abrahams SL, et al. Reduction in reperfusion-induced myocardial necrosis in dogs by RheothRx injection (poloxamer 188 N.F), a hemorheological agent that alters neutrophil function. Circulation. 1994;90(6):2964–75. doi: 10.1161/01.cir.90.6.2964. [DOI] [PubMed] [Google Scholar]
- 12.Schaer GL, Spaccavento LJ, Browne KF, et al. Beneficial effects of RheothRx injection in patients receiving thrombolytic therapy for acute myocardial infarction. Results of a randomized, double-blind, placebo-controlled trial. Circulation. 1996;94(3):298–307. doi: 10.1161/01.cir.94.3.298. [DOI] [PubMed] [Google Scholar]
- 13.Haneda KHK, Kanno M, Murakami S, Ohta M. Use of Fluosol DA (FDA) in hemorrhagic shock: Effects on oxygen carrying capacity and peripheral perfusion. New York: Alan R. Liss; 1983. [Google Scholar]
- 14.Tan J, Saltzman WM. Influence of synthetic polymers on neutrophil migration in three-dimensional collagen gels. Journal of Biomedical Materials Research. 1999;46(4):465–74. doi: 10.1002/(sici)1097-4636(19990915)46:4<465::aid-jbm4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 15.Lane TA, Lamkin GE. Paralysis of phagocyte migration due to an artificial blood substitute. Blood. 1984;64(2):400–5. [PubMed] [Google Scholar]
- 16.Lane TA, Lamkin GE. Increased infection mortality and decreased neutrophil migration due to a component of an artificial blood substitute. Blood. 1986;68(2):351–4. [PubMed] [Google Scholar]
- 17.Babbitt DG, Forman MB, Jones R, Bajaj AK, Hoover RL. Prevention of neutrophil-mediated injury to endothelial cells by perfluorochemical. The American Journal of Pathology. 1990;136(2):451–9. [PMC free article] [PubMed] [Google Scholar]