

Abstract
Pmi1525, an enzyme of unknown function from Proteus Mirabilis HI4320 and the amidohydrolase superfamily, was cloned, purified to homogeneity, and functionally characterized. The three-dimensional structure of Pmi1525 was determined with zinc and cacodylate bound in the active site (PDB id: 3RHG). The structure was also determined with manganese and butyrate in the active site (PDB id: 4QSF). Pmi1525 folds as a distorted (β/α)8-barrel that is typical for members of the amidohydrolase superfamily and cog1735. The substrate profile for Pmi1525 was determined via a strategy that marshaled the utilization of bioinformatics, structural characterization and focused library screening. The protein was found to efficiently catalyze the hydrolysis of organophosphonate and carboxylate esters. The best substrates identified for Pmi1525 are ethyl 4-nitrophenylmethyl phosphonate (kcat and kcat/Km values of 580 s−1 and 1.2 × 105 M−1 s−1, respectively) and 4-nitrophenyl butyrate (kcat and kcat/Km values of 140 s−1 and 1.4 × 105 M−1 s−1, respectively). Pmi1525 is stereoselective for the hydrolysis of chiral methylphosphonate esters. The enzyme hydrolyzes the (SP)-enantiomer of isobutyl 4-nitrophenyl methylphosphonate 14 times faster than the corresponding (RP)-enantiomer. The catalytic properties of this enzyme make it an attractive template for the evolution of novel enzymes for the detection, destruction, and detoxification of organophosphonate nerve agents.
The determination of the catalytic activity and substrate profile for uncharacterized enzymes is a significant experimental challenge as the availability of completely sequenced genomes has increased substantially in recent years (1). The rate at which new genes of uncertain function are being sequenced has greatly exceeded our ability to functionally annotate the catalytic properties of the corresponding enzymes. Annotations based primarily on sequence identity can be quite misleading in many instances because closely related sequences often have different substrate profiles, while highly divergent proteins can have identical functions (2–4). Recently, we have successfully annotated a broad spectrum of novel enzymes of previously unknown function using a comprehensive strategy of bioinformatics, computational docking, structural biology, and library screening (5–14).
Holm and Sander first identified the amidohydrolase superfamily (AHS) on the basis of similarities in the three-dimensional structures of phosphotriesterase, adenosine deaminase, and urease (15). This superfamily possesses a remarkable functional diversity with enzymes capable of hydrolyzing amide, or ester functional groups at carbon and phosphorus centers (15–16). The NCBI has partitioned the AHS into 24 Clusters of Orthologous Groups (COG) and enzymes from cog1735 catalyze the hydrolysis of organophosphate esters, lactones, and carboxylate esters (17–31). Sequence similarity networks for cog1735 at a BLAST E-value cutoffs of 10−80 and 10−40 are presented in Figure 1 (32). At an E-value cutoff of 10−80, cog1735 segregates into 18 subgroups that have been arbitrarily color-coded and numbered. The substrate profiles for subgroups 3–9 have been determined (18–31).
Figure 1.

Sequence similarity network for proteins of cog1735 from the amidohydrolase superfamily at BLAST E-value cutoffs of 10−80 (top) and 10−40 (bottom) created using Cytoscape (http://www.cytoscape.org). For these proteins an E-value of 10−80 corresponds to a sequence identity of approximately 42%, whereas an E-value of 10−40 corresponds to a sequence identity of approximately 30%. All protein sequences available through NCBI that are designated as belonging to cog1735 were used to create the network. Each node in the network represents a single sequence, and each edge (depicted as lines) represents the pairwise connection between two sequences at the given BLAST E-value (32). Lengths of edges are not significant except that sequences in tightly clustered groups are relatively more similar to each other than sequences with few connections. The triangular nodes represent proteins that have been functionally and/or structurally characterized. Additional information is available in the text.
The phosphotriesterase homology protein (PHP) from Escherichia coli and subgroup 1 has been structurally characterized (PDB id: 1BF6) but its catalytic function remains undetermined (24). Proteins from subgroup 3 catalyze the hydrolysis of N-acyl homoserine lactones (21–23). Recently, we determined that Rsp3690 from Rhodobacter sphaeroides 2.4.1 and subgroup 4 (PDB id: 3K2G) is a non-specific carboxylate esterase with the ability to hydrolyze methylphosphonate esters (29). Lmo2620 from Listeria monocytogenes (PDB id: 3PNZ) and Bh0225 from Bacillus halodurans and subgroup 5 catalyze the hydrolysis of D-lyxono-1, 4-lactone-5-phosphate and L-ribono-1, 4-lactone-5-phosphate (30). MS53_0025 (PDB id: 3OVG) from Mycoplasma synoviae and MAG_6390 from Mycoplasma agalactiae PG2 and subgroup 6 catalyze the hydrolysis of D-xylono-1, 4-lactone-5-phosphate and L-arabino-1,4-lactone-5-phosphate (31). Proteins from subgroup 7 are γ- and δ-lactonases (26–27). Proteins from subgroup 8 are phosphotriesterases (PTE), which catalyze the hydrolysis of organophosphate esters, including the insecticide paraoxon and the chemical warfare agents GB, GD, and VX (18–19). Proteins from subgroup 9 have weak phosphotriesterase activity but exhibit substantially faster rates toward the hydrolysis of lactones (20).
In this study, we have focused on the characterization of Pmi1525 from Proteus Mirabilis HI4320, an enzyme from subgroup 2 of cog1735. The three-dimensional structure of Pmi1525 was determined and the enzyme was shown to catalyze the hydrolysis of organophosphate and carboxylate esters. Pmi1525 is particularly active towards the hydrolysis of the (SP)-enantiomers of methylphosphonate esters that resemble the substructures contained within the G- and V-series of organophosphonate nerve agents. Therefore, this enzyme, and other enzymes from subgroup 2 of cog1735 may serve as templates for the evolution of novel enzyme catalysts for the detection, destruction, and detoxification of organophosphate nerve agents.
Materials and Methods
Materials
LB broth was purchased from Tpi Research Products International Corp. The HisTrap™ HP and gel filtration chromatographic columns were purchased from GE Healthcare. E. coli BL21(DE3) competent cells were obtained from Stratagene. The standards for ICP-MS determination of the metal content of the isolated proteins were purchased from Inorganic Ventures Inc. Organophosphate compounds 1-10 were supplied by Sigma/Aldrich, or synthesized by modifications of published procedures (33). All other chemicals, buffers, and purification reagents used in this work were purchased from Sigma/Aldrich, unless otherwise specified.
Purification of Pmi1525 from Proteus Mirabilis
The gene encoding the protein with the locus tag Pmi1525 from P. Mirabilis HI4320 (gi|197285384) was cloned. The plasmid encoding the gene for Pmi1525 was transformed into E. coli BL21 (DE3) competent cells (Invitrogen) and plated on LB agar. A single colony was used to inoculate a 5 mL culture of LB and allowed to grow overnight (13–15 hours) at 37 °C. A 5-mL overnight culture was subsequently used to inoculate one liter of LB medium supplemented with 50 μg/mL kanamycin. The inoculated culture was grown with agitation (200 rpm) at 30 °C to an OD600 of 0.1 – 0.2 before the addition of 100 μM 2, 2′-bipyridyl to reduce the iron content of the protein (34). The culture was allowed to grow to an OD600 of 0.6 and then 1.0 mM ZnCl2 (or MnCl2) and 0.5 mM isopropyl thio-β-D-galactopyranoside (IPTG) were added. After growing for an additional 16 hours, the cells were harvested by centrifugation and stored at −70 °C. Approximately 10 grams of cells were obtained from 3 L of liquid culture. The frozen cell pellet was thawed, re-suspended in binding buffer (20 mM HEPES, pH 7.9, 0.50 M NaCl, and 5.0 mM imidazole) and then disrupted by sonication. The cellular extract was clarified by centrifugation, filtered through a 0.2 μm syringe filter (VWR) and loaded onto a 5-mL HisTrap™ HP column (GE Healthcare), which was connected to an ÄKTA purifier (Amersham Pharmacia Biotech). The column was washed thoroughly with binding buffer and the target protein was eluted with a gradient of elution buffer (20 mM HEPES, pH 7.9, 0.25 M NaCl, and 0.50 M imidazole). The crude protein thus obtained was further purified by size-exclusion chromatography using a GE Healthcare Hiload 16/60 Superdex 200 prep grade gel filtration column with 50 mM HEPES, pH 8.0. The isolated protein was >95% pure based upon SDS-PAGE analysis. The Y126F mutant of Pmi1525 was constructed using the QuikChange site-directed mutagenesis kit from Agilent according to the manufacturer’s instructions and purified to homogeneity using the same procedures as for the wild-type protein.
Metal Analysis
The protein concentration was determined spectrophotometrically at 280 nm using a 1-cm quartz cuvette using a SPECTRAmax-340 UV-Vis spectrophotometer. The extinction coefficient at 280 nm for determination of the protein concentration of Pmi1525 was calculated to be 39,795 M−1 cm−1 (http://ca.expasy.org/tools/protparam.html). The metal content of the purified protein was determined using inductively coupled plasma emission mass spectrometry (ICP-MS). Before ICP-MS measurements, the protein sample was digested with concentrated nitric acid by refluxing for ~30 minutes and then diluted with distilled H2O until the final concentration of nitric acid was 1% (v/v) and the protein concentration was ~1.0 μM.
Kinetic Measurements and Data Analysis
The activity screening and kinetic measurements were performed using a SPECTRAmax-340 plate reader. The hydrolysis of carboxylate esters was monitored using a pH-sensitive colorimetric assay. Protons released from the hydrolysis reaction were measured using the pH indicator cresol purple (29, 35). The reactions were performed in 2.5 mM BICINE, pH 8.3, containing 0.20 M NaCl, various concentrations of substrates, 0.1 mM cresol purple, and Pmi1525. The final concentration of DMSO in the reaction mixture was 2%. The change in absorbance at 577 nm was monitored in a 96-well UV-visible plate. The conversion factor for the rate determination (ΔOD/mol of H+) was obtained from an acetic acid titration at 577 nm at pH 8.3, 30 °C (ε = 1.76 × 103 M−1 cm−1, 2% DMSO). The purified protein was stored in 50 mM HEPES, pH 8.0, and the buffer was exchanged with 10 mM BICINE, pH 8.3, using a PD-10 desalting column immediately before the kinetic measurements. The carboxylate ester substrates were dissolved in DMSO and then diluted into the reaction mixture. A background rate was observed that was independent of the initial substrate concentration and subtracted from the initial rate of the enzyme-catalyzed reactions (26, 29).
For substrates with 4-nitrophenol as a hydrolysis product, activity screening and kinetic parameters measurements were monitored at 400 nm (ε = 1.7 × 104 M−1 cm−1, pH 8.0). For substrates with 4-acetylphenol as the hydrolysis product, the kinetic constants were determined by monitoring the formation of 4-acetylphenol at 294 nm (ε = 7710 M−1 cm−1, pH 8.0). The reaction mixtures contained 50 mM HEPES, pH 8.0, 1.0 mM MnCl2, and various concentrations of substrate. When indoxyl acetate (15) was used as a substrate, the catalytic reaction was monitored at 678 nm in 50 mM HEPES, pH 8.0, 1% ethanol (ε = 7.0 × 103 M−1 cm−1) (36). The reactions were initiated by addition of enzyme.
Kinetic parameters were determined by fitting the initial rates to equation 1 using the nonlinear least-squares fitting program in SigmaPlot 9.0, where ν is the initial velocity of the reaction, Et is the enzyme concentration, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant.
| (1) |
Crystallization, X-ray Data Collection, and Structure Determination
Pmi1525 was crystallized by the sitting-drop vapor diffusion method. The concentrated (~30 mg/mL) protein solutions (0.5 μL) were mixed with an equal volume of precipitant and equilibrated at room temperature against the same precipitant solution in clear tape-sealed 96-well INTELLI-plates (Art Robbins Instruments, Sunnyvale, CA). Crystallization was performed using either a TECAN crystallization robot (TECAN US, Research Triangle Park, NC) or a PHENIX crystallization robot (Art Robbins Instruments) and four types of commercial crystallization screens: the WIZARD I & II screen (Emerald BioSystems, Bainbridge Island, WA); the INDEX HT and the CRYSTAL SCREEN HT (Hampton Research, Aliso Viejo, CA); and the MCSG screen (Microlytic, Woburn, MA). The appearance of protein crystals was monitored by visual inspection or using a Rock Imager 1000 (Formulatrix, Waltham, MA) starting within 24 hours of incubation and again at weeks 1, 2, 3, 5 8, and 12. Where necessary, the crystallization conditions were optimized manually using 24-well Cryschem sitting drop plates (Hampton Research). The zinc-containing protein was crystallized in the presence of 0.10 M sodium cacodylate (pH 6.5), 0.20 M ammonium sulfate and 30% PEG8000 as the precipitant. Crystals were flashed-cooled and stored in liquid nitrogen prior to data collection. The manganese-containing protein was crystallized in the presence of 0.10 M Bis-Tris (pH 5.5), 0.20 M lithium sulfate and 20% PEG 3350. The crystals were incubated in the precipitant solution supplemented with 50 mM 4-nitrophenylbutyrate (12) for 30 minutes and then flashed-cooled in liquid nitrogen.
The X-ray diffraction data were collected at 100 K on the beamline X29A (National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY) using a wavelength of 1.075 Å. The diffraction data were processed and scaled with HKL2000 software (37). The crystal structures were determined by molecular replacement using coordinates of PDB id: 3HTW as a starting model and PHASER software (the CCP4 program package suite (38). The initial models were built and refined by ARP/WARP (39) on the basis of the molecular replacement solutions. The models were further refined using REFMAC (40) and manually adjusted using COOT visualization and refinement software (41). The structures were deposited to the Protein Data Bank (PDB id: 3RHG and 4QSF). All figures were produced using PYMOL (42). The data collection and refinement statistics for the crystal structures are listed in the Table 1.
Table 1.
Data collection and refinement statistics
| PDB identifier | 3RHG | 4QSF |
|---|---|---|
| Space group | P 32 2 1 | P 32 2 1 |
| Unit cell dimension (Å) | - | - |
| -a | 101.203 | 101.269 |
| -b | 101.203 | 101.269 |
| -c | 65.614 | 65.493 |
| Cell angles (degrees) | - | - |
| -alpha | 90.00 | 90.00 |
| -beta | 90.00 | 90.00 |
| -gamma | 120.00 | 120.00 |
| Molecules per ASU | 1 | 1 |
| Solvent content | 48.66 | 48.51 |
| Matthew’s Coefficient | 2.39 | 2.41 |
| Ligands | Zn2+, benzoic acid, cacodylate anion, unknown ligand | butanoic acid, Mn2+ |
| X-ray source | NSLS X29A | NSLS X29A |
| Wavelength | 1.075 | 1.075 |
| Method of structure solution | Molecular replacement | Molecular Replacement |
| Resolution | 90.00–1.53 (1.56–1.53) | 50.00–1.65 (1.68–1.65) |
| Resolution/refinement | 50.00–1.53 | 36.46–1.65 |
| Completeness (%) | 99.7 (95.0) | 99.8 (100.0) |
| I/sigma (I) | 19.90 (2.20) | 34.60 (3.10) |
| Rsym | 0.060 (0.80) | 0.050 (0.66) |
| Rwork (Rfree) | 12.1 (15.6) | 15.5 (18.1) |
| Rfree reflections (%) | 1795 (3.1%) | 1450 (3.1%) |
| Average B factor | 28.57 | 27.04 |
| RMSD | - | - |
| -bonds lengths | 0.010 | 0.013 |
| -bond angles | 1.227 | 1.567 |
| Number of solvent molecules | 453 | 385 |
| Ramachandran plot statistics | - | - |
| Most favored regions | 96.9% | 95.6% |
| Allowed regions | 2.6% | 4.11% |
| Outliers | 0.5% (F173, D264) | 0.29% (F173) |
Molecular Docking
Computational docking of potential substrates and intermediates to Pmi1525 was performed using Autodock Vina (43). The crystal structure of Pmi1525 (PDB id: 3RHG) was used in all docking calculations and the metal ions were retained in the active site. A pdbqt file format of the protein was generated by adding polar hydrogens and a grid box was centered at the active site of Pmi1525 with dimensions 26 × 26 × 26 Å with grid points spaced every 1 Å. Initial structures of the ligands were generated and charges were added using MGL tools 1.5.4 version software. High-energy hydrolysis intermediates of stereoisomers 9 and 10 were generated with the negative charge residing on one of the oxygen atoms attached to the penta-coordinated phosphorus atom. The zinc metals were assigned a net charge of +1.4 each, as described previously (44). The docking calculations were conducted with an exhaustiveness of 15. The output structures with the lowest binding energies were considered and analyzed using Discovery Studio Client 3.5
Results
Cloning, Expression and Purification of Pmi1525
The gene encoding Pmi1525 was cloned with a C-terminal His-tag. Cells were grown in the presence of added ZnCl2 or MnCl2 and the protein purified to homogeneity. The Zn- and Mn-containing proteins were >95% pure, based on SDS-PAGE analysis. ICP-MS analysis demonstrated that Pmi1525 expressed in the presence of added zinc contained 1.5 equivalents of Zn and 0.4 equivalents of Fe, whereas the protein expressed in the presence of added manganese contained 1.3 equivalents of Mn, 0.3 equivalents of Zn, and 0.4 equivalents of Fe. The Y126F mutant was purified in the presence of 1.0 mM MnCl2 and the purified enzyme contained 1.2 equivalents of Mn, 0.3 equivalents of Zn, and 0.05 equivalents of Fe.
Three-Dimensional Structure of Zn-Pmi1525
The three-dimensional structure of Zn-Pmi1525 was determined to a resolution of 1.53 Å (PDB id: 3RHG) with a single protein subunit in the asymmetric unit. The protein folds as a distorted (β/α)8-barrel that is typical for other structurally characterized enzymes from the amidohydrolase superfamily (16). A ribbon representation of the zinc-bound form of the Pmi1525 monomer is presented in Figure 2. The active site is located at the C-terminal end of the β-barrel and is partially open to bulk solvent. Two zinc ions are bound in the active site, and the coordination scheme is illustrated in Figure 3A. The more buried α-Zn is coordinated by His-23 and His-25 of β-strand 1, and Asp-294 of β-strand 8 at distances of 2.1, 2.0, and 2.4 Å, respectively. The β-Zn is coordinated by His-199 from β-stand 5, and His-229 from β-stand 6 at distances of 2.0, and 2.0 Å, respectively. Glu-166 from β-strand 4 acts as a bridging ligand and is coordinated to both α-Zn and β-Zn at distances of 2.1 and 1.9 Å, respectively. A bound cacodylate ion acts as the second ligand to bridge both Zn atoms at distances of 1.9 and 1.9 Å. The two Zn ions are 3.9 Å apart. A probable benzoate ion is found near the active site at a distance of 5.4 Å to β-Zn. A sulfate ion is found on the surface of the structure and an unidentified ligand (UNL) was found at ~6 Å from β-Zn.
Figure 2.

Cartoon representation of the Pmi1525 (PDB id: 3RHG) with bound zinc (colored in magenta) and cacodylate ion (CAC, carbon atoms are green, oxygen is red). Zinc and cacodylate are shown as vdw spheres. The protein subunit is rainbow colored with the N-terminus as dark blue and the C-terminus as red. The active site residues, an unknown ligand (UNL) and benzoic acid are drawn as stick models with carbon atoms gray colored, oxygen is red and nitrogen is blue.
Figure 3.

Metal and ligand coordination in the active site of Pmi1525. All residues are drawn as stick models, metals are shown as non-bonded spheres. (A) The structure of the zinc bound enzyme with cacodylate (CAC) (PDB id: 3RHG). (B) The structure the manganese bound enzyme with bound butyrate (BUT) (PDB id: 4QSF). Carbon atoms are gray colored, oxygen is red, nitrogen is blue. The coordination and selected hydrogen bonds are presented as dashed gray lines.
Three-Dimensional Structure of Mn-Pmi1525
The crystal structure of Mn-containing Pmi1525 was determined with butyrate bound in the active site. The overall fold and the active site scaffold of the Mn-Pmi1525 structure are nearly identical to the structure with bound zinc ions. Superimposition of atomic coordinates between the two Pmi1525 structures yields an RMSD of 0.13 Å. The difference between the active sites in these two structures is related to the coordination geometry of manganese and zinc ions. Zinc typically maintains tetrahedral coordination with bond distances of around 2 Å, although in bimetal centers the coordination geometry may be affected by the protein architecture. In the Zn-Pmi1525 structure the β-Zn is 4-coordinate and the α-Zn is “loosely” 5-coordinated because the distance between α-Zn and Asp-294 is only 2.4 Å. This organization indicates a relatively weak bond while other distances between α-Zn and protein atoms are around 2 Å. By contrast, the distances between the bound manganese and the coordinated protein atoms in the Mn-Pmi1525 structure are ~2.15 – 2.2 Å, which is typical for distances to this metal. Both Mn ions in the active site are 5-coordinated (Figure 3B).
No water molecules are directly bound to either zinc or manganese as the potential binding sites for water/hydroxide are occupied by butyrate (PDB id: 4QSF) or cacodylate (PDB id: 3RHG). The complex of Mn-Pmi1525 and the product butyrate was obtained by soaking the protein crystals with the substrate 4-nitrophenyl butyrate (12). The hydrolysis product butyrate functions as a monodentate ligand bound to both Mn ions in the active site, unlike the cacodylate ion (PDB id: 3RHG), which bridges the two Zn ions as a bidentate ligand (2.15 – 2.20 Å). The hydrophobic segment of butyrate is found in a predominantly hydrophobic pocket within the active site that is formed by the side chains of Asn-28, Leu-30, Thr-96, Ile-101, Tyr-126, Phe-130, Phe-296, and Leu-297.
Metal Specificity of Pmi1525
The Zn-Pmi1525 was tested as a catalyst for the hydrolysis of compounds presented in Scheme 1, but catalytic activity was barely detectable. However, significant activity could be detected with the Mn-Pmi1525 when the assays were supplemented with the addition of 1.0 mM MnCl2. Higher concentrations of MnCl2 did not alter the rate of hydrolysis. These results are consistent with the observation that the manganese occupancy in the structure (PDB id: 4QSF) was less than 1.0 (it was refined with an occupancy of 0.8). All subsequent assays used to determine the catalytic activity of Pmi1525 utilized Mn-Pmi1525 in the presence of 1.0 mM of added MnCl2. It is unclear why this enzyme is not active with zinc in the active site since closely related enzymes, such as the phosphotriesterase from Pseudomonas diminuta are active with a variety of divalent cations (16).
Scheme 1.

Function Determination and Substrate Specificity of Pmi1525
Pmi1525 belongs to cog1735 from the amidohydrolase superfamily. Other members of this COG have been found to hydrolyze organophosphate triesters, lactones, and carboxylate esters (18–31). Pmi1525 was screened with a small library of those compounds that have previously been identified as substrates for other enzymes in cog1735. These experiments demonstrated that Pmi1525 is unable to hydrolyze γ- and δ-lactones, which have been shown to be substrates from subgroups 3 and 7 of cog1735 (Figure 1). However, Pmi1525 is capable of catalyzing the hydrolysis of organophosphates, organophosphonates, and carboxylate esters. The best substrates identified for Pmi1525 are ethyl 4-nitrophenyl methylphosphonate (4) and 4-nitrophenyl butyrate (12) with values of kcat/Km of 1.2 × 105 and 1.4 × 105 M−1 s−1, respectively. The structures of the best substrates are presented in Scheme 1 and the kinetic constants are provided in Table 2. The hydroxyl group of Tyr-126 in the Pmi1525 structures (PDB id: 3RHG and 4QSF) is hydrogen bonded to one of oxygen atoms of cacodylate or butyrate. It is therefore possible that Tyr-126 may be directly involved in catalysis. To address this proposal, we constructed the Y126F mutant of Pmi1525 and tested this protein with selected substrates (Table 3). With the phosphate esters, the mutant enzyme is nearly as active as the wild-type enzyme. However, with the carboxylate esters 12 and 13, the activity is reduced approximately 100-fold.
Table 2.
Kinetic Parameters for Pmi1525a
| Substrate | compound | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| 1 | dimethyl 4-nitrophenyl phosphate | (1.7 ± 0.1) × 102 | ||
| 2 | diethyl 4-nitrophenyl phosphate | 3.5 ± 0.2 | 5.1 ± 0.4 | (6.9 ± 0.7) × 102 |
| 3 | dibutyl 4-nitrophenylphosphate | (2.8 ± 0.1) × 103 | ||
| 4 | ethyl 4-nitrophenyl methylphosphonate | 583 ± 15 | 4.8 ± 0.23 | (1.2 ± 0.1) × 105 |
| 5 | isopropyl 4-nitrophenyl methylphosphonate | (9.6 ± 0.2) × 102 | ||
| 6 | isobutyl 4-nitrophenyl methylphosphonate | 61 ± 2 | 2.9 ± 0.2 | (2.1 ± 0.2) × 104 |
| 7 | cyclohexyl 4-nitrophenyl methylphosphonate | (1.3 ± 0.1) × 102 | ||
| 8 | ethyl 4-acetylphenyl methylphosphonate | (8.5 ± 0.3) × 103 | ||
| 9 | (RP)-isobutyl 4-acetylphenyl methylphosphonate | (1.9 ± 0.1) × 102 | ||
| 10 | (SP)-isobutyl 4-acetylphenyl methylphosphonate | 5.4 ± 0.3 | 2.7 ± 0.3 | (2.0 ± 0.2) × 103 |
| 11 | 4-nitrophenyl acetate | (8.3 ± 0.3) × 102 | ||
| 12 | 4-nitrophenyl butyrate | 137 ± 6 | 1.0 ± 0.1 | (1.4 ± 0.2) × 105 |
| 13 | 4-nitrophenyl hexanoate | 7.6 ± 0.1 | 0.074 ± 0.004 | (1.0 ± 0.1) × 105 |
| 14 | 4-nitrophenyl octanoate | 0.20 ± 0.01 | 0.015 ± 0.002 | (1.3 ± 0.2) × 104 |
| 15 | indoxyl acetate | (4.5 ± 0.4) × 100 | ||
| 16 | 2-naphthyl acetate | (4.2 ± 0.3) × 101 | ||
| 17 | 2-naphthyl propionate | (2.5 ± 0.3) × 102 | ||
| 18 | 4-methylumbelliferyl butyrate | 0.92 ± 0.02 | 0.21 ± 0.02 | (4.3 ± 0.4) × 103 |
Assays for compounds 1-10 were conducted at pH 8.0 and 30 °C; Assays for compounds 11-18 were conducted at pH 8.3 and 30 °C; Compounds 4-8 were used as a racemic mixture.
Table 3.
Kinetic Parameters for Mutant Y126F of Pmi1525a
| substrate | compound | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| 2 | diethyl 4-nitrophenyl phosphate | 0.64 ± 0.03 | 1.3 ± 0.1 | (5.1 ± 0.5) × 102 |
| 3 | dibutyl 4-nitrophenylphosphate | (1.2 ± 0.1) × 103 | ||
| 4 | ethyl 4-nitrophenyl methylphosphonate | 570 ± 30 | 3.1 ± 0.4 | (1.8 ± 0.2) × 105 |
| 12 | 4-nitrophenyl butyrate | 1.1 ± 0.1 | 1.1 ± 0.1 | (1.0 ± 0.1) × 103 |
| 13 | 4-nitrophenyl hexanoate | 0.32 ± 0.01 | 0.07 ± 0.006 | (4.3 ± 0.1) × 102 |
Assays for compounds 1, 3, and 4 conducted at pH 8.0 and 30 °C; Assays for compounds 12-13 were conducted at pH 8.3 and 30 °C.
Compound 4 was used as a racemic mixture.
The isolated wild-type protein contained a mixture of Zn, Mn, and Fe when expressed in a medium that was supplemented with added Zn or Mn. At high concentrations the purified protein is pink in color with an adsorption maximum at 500 nm (data not shown). This property may be attributed to a charge transfer complex between Tyr-126 and iron bound to the β-metal position within the binuclear metal center (26). The Y126F mutant is colorless at 500 nm.
Stereoselectivity of Pmi1525
To address the stereoselectivity of Pmi1525 for the hydrolysis of chiral organophosphate substrates, racemic isobutyl 4-nitrophenyl methylphosphonate (6) was used as a substrate. The time courses for the hydrolysis of racemic 6 by wild-type phosphotriesterase (PTE) and Pmi1525 are presented in Figure 4. The addition of wild-type PTE results in the rapid hydrolysis of ~50% of the total substrate. The addition of Pmi1525 catalyzes the hydrolysis of the remaining material (Figure 4A). A similar time course was observed when the order of enzyme addition was reversed (data not shown). From the time course for the hydrolysis of racemic 6 by PTE alone (Figure 4B) and Pmi1525 alone (Figure 4C) it can be concluded that Pmi1525 and PTE preferentially hydrolyze the opposite enantiomers from one another. Since PTE hydrolyzes the (RP)-enantiomer of isobutyl 4-acetylphenyl methylphosphonate (9) 25 times faster than the (SP)-enantiomer (10), Pmi1525 prefers to hydrolyze the (SP)-enantiomer of this substrate (33). For wild-type PTE, the (RP)-enantiomer of compound 6 is hydrolyzed 75 times faster than the (Sp)-enantiomer based on a fit of the data in Figure 4B to the sum of two exponentials (29). For Pmi1525, the (SP)-enantiomer of compound 6 is hydrolyzed 14 times faster than the (RP)-enantiomer. For the hydrolysis of the isolated enantiomers of isobutyl 4-acetylphenyl methylphosphonate (9 and 10), the values of kcat/Km for Pmi1525 were determined to be 2.0 × 103 for the (SP)-enantiomer and 1.9 × 102 M−1 s−1 for the (Rp)-enantiomer.
Figure 4.

Time courses for the hydrolysis of 90 μM racemic substrate 6 using wild-type PTE and Pmi1525. (A) The reaction was initiated with the addition of 0.5 nM wild-type PTE. After 23 minutes, 50 nM of Pmi1525 was added to hydrolyze the remaining material. (B) Hydrolysis of 90 μM racemic 6 using 1.0 nM wild-type PTE. (C) Hydrolysis of 90 μM racemic 6 using 50 nM Pmi1525.
Computational Docking of Substrates to the Active Site of Pmi1525
The structural basis of the observed stereoselectivity of Pmi1525 was investigated using computational docking. The high-energy reaction intermediates for the hydrolysis of substrates 9 and 10 were generated with the leaving group (acetylphenyl group) and the attacking hydroxyl group in the axial position as shown in Scheme 2 (44). A productive docking pose is formed when the nucleophilic hydroxide ion is in the axial position of the proposed high-energy pentavalent intermediate and positioned between the two metal ions. The phosphorus oxy-anion in the equatorial position is expected to interact with the β-metal. Unfortunately, docking of the proposed high-energy intermediates 19 and 20 using Autodock Vina did not result in any productive conformations as described above. The phosphonate oxygen atoms in the top docking poses were at least 4 Å away from the zinc atoms with the dominant interaction being the hydrophobic contacts with other active site residues. Therefore, the penta-coordinated H-phosphonate ligands 21 and 22 were designed to reduce the non-specific hydrophobic contacts within the active-site.
Scheme 2.
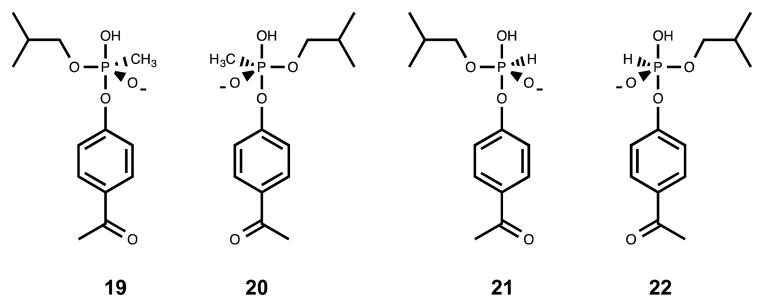
The top docking pose of ligand 22 (SP-enantiomer) resulted in a productive conformation. The ligand-bound active site structure is shown in Figure 5. The axial hydroxide group coordinates with the two zinc metals at a distance of 2.3 Å from the α-metal and 2.5 Å from the β-metal. The phosphoryl oxyanion at the equatorial position of 22 interacts with the β-Zn metal at a distance of 2.8 Å and with Tyr-126 at a distance of 2.7 Å. The interaction of the isobutyl side chain with Leu-30 and Ile-101 is in accord with the positioning of butyrate in the active site of Mn-Pmi1525 (PDB id: 4QSF). The leaving group in the axial position makes hydrophobic contacts with Trp-203 from loop 5.
Figure 5.

Active site of Pmi1525 computationally docked with compound 22. The axial OH group attached to the pentacoordinated phosphorus occupies the position of the nucleophilic hydroxide that is bridged by the two metal ions. The isobutyl side chain makes hydrophobic contacts with the residues (Leu-30, Ile-101, and Tyr-126) in the larger pocket of Pmi1525
The high-energy intermediate 21 (RP enantiomer) did not dock in the active-site of Pmi1525 in a productive conformation. The equatorial phosphoryl oxyanion of 21 preferentially coordinates the two Zn metal atoms. While there is no interaction between the axial hydroxyl group and either of the Zn metal atoms, the isobutyl side chain and the acetyl phenyl leaving group have similar interactions as that of the SP-enantiomer (22).
Discussion
Substrate Profile for Pmi1525
Pmi1525 is a promiscuous enzyme that catalyzes the hydrolysis of a variety of substrates. With organophosphonate esters, Pmi1525 efficiently catalyzes the hydrolysis of racemic ethyl 4-nitrophenyl methylphosphonate (4) with a kcat/Km value of 1.2 × 105 M−1 s−1. However, the catalytic activity decreases significantly when the ethyl group of this substrate is replaced by an isopropyl (5), isobutyl (6), or cyclohexyl (7) group. Pmi1525 also catalyzes the hydrolysis of the organophosphate esters methyl paraoxon (1), paraoxon (2) and dibutyl 4-nitrophenyl phosphate (3) but the values of kcat/Km are 2–3 orders of magnitude lower than those determined for ethyl 4-nitrophenyl methylphosphonate (4). Among the carboxylate esters, Pmi1525 efficiently hydrolyzes 4-nitrophenyl butyrate (12) with a value of kcat/Km 1.4 × 105 M−1 s−1. The value of kcat/Km decreases as the carboxylate moiety of the substrate is either shortened (11) or lengthened (13 and 14). Pmi1525 also catalyzes the hydrolysis of naphthyl, indole, and 4-methyllumbelliferyl esters. Those substrates with a longer carboxylate moiety are hydrolyzed more efficiently (17 and 18).
Pmi1525 preferentially hydrolyzes the (SP)-enantiomer of compounds such as isobutyl 4-nitrophenyl methylphosphonate (6) and isobutyl 4-acetylphenyl methylphosphonate (10). This stereoselectivity is opposite to that observed for wild-type PTE (33). The organophosphate and organophosphonate esters are toxic because of their ability to rapidly inactivate the enzyme acetyl cholinesterase. Since the corresponding (SP)-enantiomers of sarin (GB), soman (GD), and VX are considerably more toxic than the (RP)-enantiomers, the enzymes from subgroup 2 of cog1735 are promising candidates for the directed evolution of enzymes that can be enhanced in their ability to detect, destroy, and detoxify the most toxic enantiomers of chemical warfare agents and agricultural pesticides.
Three-Dimensional Structure of Pmi1525
The structural fold of Pmi1525 is that of a distorted (β/α)8-barrel, similar to those structures previously determined from cog1735 and the amidohydrolase superfamily. The active site is located at the C-terminal end of the β-barrel and is highlighted by the binding of two divalent cations. The enzyme is most active with manganese. The two metal ions are coordinated to the protein via direct interactions with the side chains of two histidine residues from the end of β-strand 1, two additional histidine residues from the ends β-strands 5 and 6, and an aspartate from the end of β-strand 8. The two metal ions are bridged by the side chain carboxylate of a glutamate that originates from the end of β-strand 4. This metal coordination arrangement of a binuclear metal center is quite common in the active site of enzymes from cog1735 and the rest of the amidohydrolase superfamily (16). However, in many enzymes, such as phosphotriesterase, the bridging carboxylate from glutamate is replaced with a carboxylated lysine residue (45). We have determined the structures of two complexes of Pmi1525. The first structure has cacodylate in the active site and the second contained butyrate, formed from the enzymatic hydrolysis of the substrate p-nitrophenyl butyrate (12). The cacodylate bridges the two metal ions in a manner that represents a mimic of the tetrahedral intermediate formed from the hydrolysis of carboxylate esters.
Amino Acid Sequence Comparisons within cog1735
At an E-value cutoff of 10−80, Pmi1525 is localized to subgroup 2 of the sequence similarity network presented in Figure 1. A sequence alignment of Pmi1525 with four representative proteins from cog1735 is shown in Figure 6. Rsp3690, PHP (b3379), Dr0930, and PTE are examples from subgroups 4, 1, 7, and 8, respectively, of cog1735. The aspartate and four histidine residues that coordinate the two metal ions are highlighted in red; the residue bridging the two metal ions is colored in pink. The residues that form the hydrophobic pocket in the active site of Pmi1525 and Rsp3690 are colored in yellow and the corresponding residues in the other three proteins are highlighted in green. The positions of the eight loops that follow the eight β-strands are highlighted in gray. When the E-value cutoff of the sequence similarity network is changed from 10−80 to 10−40, subgroup 2 merges with subgroup 4 and this observation is consistent with the observed substrate specificity for enzymes from these two subgroups (Figure 1). Recently, we determined that Rsp3690 is a non-specific carboxylate esterase with a promiscuous ability to hydrolyze methylphosphonate esters (29). Pmi1525 has a similar substrate profile to that of Rsp3690 but the substrate specificities of these two proteins are not the same. Both of these proteins have an extended loop 1and shortened loops 7 and 8, relative to PTE. The importance of loops 7 and 8 to the substrate specificity of PTE has been well-documented (26, 29, 46). Wild-type Dr0930 has very weak phosphotriesterase activity but this activity has been significantly improved by directed evolution of active site residues (47).
Figure 6.

Amino acid sequence alignment of Pmi1525 and four other proteins from cog1735, including the non-specific carboxylesterase Rsp3690 from subgroup 4, phosphotriesterase homology protein (PHP, b3379) from subgroup 1, γ,δ-lactonase Dr0930 from subgroup 7, and phosphotriesterase PTE from subgroup 9. The four histidine and the aspartate residues coordinated to the two metal ions are highlighted in red. The residue bridging the two metal ions is colored in pink. The residues forming the hydrophobic pocket in the active site in Pmi1525 and Rsp3690 are colored yellow and the corresponding residues in the other four proteins are colored green.
Structural Comparison of Pmi1525, Rsp3630 and PTE
A three-dimensional structural comparison of Pmi1525, Rsp3630 and PTE is presented in Figure 7. The central β-barrel, the surrounding α-helices, and the two metal atoms of these three proteins superimpose well, but conformational differences in the loops that follow the eight β-strands are obvious. There are minor differences in the length and conformations of loops 2 to 6, but there are larger differences between loops 1, 7, and 8. Pmi1525 and Rsp3690 have a longer loop 1 but loops 7 and 8 are shorter, relative to PTE. Overall, Pmi1525 and Rsp3690 have hydrophobic active site pockets, but Pmi1525 has fewer hydrophobic residues compared to Rsp3630.
Figure 7.

Superposition of coordinates for structures Pmi1525 (PDB id: 3RHG), Rsp3690 (PDB id: 3k2g), and PTE (PDB id: 1dpm). The eight β/α loops from Pmi1525, Rsp23690, and PTE are depicted in magenta, green, and orange, respectively. The central β-barrel, the surrounding α-helices, and the two metal atoms are depicted in grey. The RMSD between grey colored parts of Pmi1525/Rsp3690, Pmi1525/PTE, and Rsp3690/PTE are 0.9, 1.0, and 1.1 Å, respectively.
Active site comparison of Pmi1525 and Rsp3690
Overall, Pmi1525 and Rsp3690 both have relatively hydrophobic active sites; however, the Pmi1525 active site is less hydrophobic than that of Rsp3630. A superposition of the active site binding pockets for Pmi1525 and Rsp3690 is presented in Figure 8. The residues that coordinate the two divalent cations are identical. The major differences include the replacement of a cysteine in Rsp3690 for Leu-30 in loop 1 in Pmi1525, substitution of a serine for Phe-130, and a valine for Leu-297. The hydrophobic residues that enclose butyrate in the active site of Pmi1525 and DTT in Rsp3690 are mostly conserved and occupy similar positions. The structural comparison supports the experimental finding that the substrate specificities of these two enzymes are similar, but not identical.
Figure 8.

Superimposition of active site residues coordinated to the two metal ions and residues around the butyrate ion in Pmi1525 and homologous residues near the DTT bound in the active site of Rsp3690. The residues of Pmi1525 are colored in light gray; the residues of Rsp3690 are colored in cyan and the residue labels are enclosed in brackets. The carbon chain of butyrate bound in the active site of Pmi1525 is shown in green and the carbon chain of DTT bound in Rsp3690 is shown in orange. Manganese atoms are colored pink and the zinc atoms are colored in dark grey. Oxygen is shown in red and sulfur in yellow.
Structural Superimposition of Pmi1525 and PTE
A structural comparison of the active sites for Pmi1525 and phosphotriesterase (PTE) from Pseudomonas diminuta is presented in Figure 9. The residues that are coordinated to the two divalent cations metals are omitted for clarity. The complex of PTE and the substrate analog diethyl 4-methylbenzylphosphate highlight substrate binding site composed of side chains from twelve residues which can be subdivided into three pockets that accommodate the small-, large-, and leaving-group moieties of the substrate (48–50). From Figure 9 it is apparent that those residues that facilitate the binding of the substrates in the active site of PTE structure are not conserved in the active site of Pmi1525. However, one can still discern three subpockets in the active site of Pmi1525. The primary difference occurs in the so-called large pocket, which is smaller in Pmi1525, relative to that found in PTE. The superimposition of these two proteins illustrates the clash of Phe-296 from Pmi1525 with the benzyl ring of the substrate analog in the PTE structure. In addition, Met-300 of Pmi1525 is very close to the methyl group of the benzyl ring of the inhibitor in PTE. This may partially explain why Pmi1525 has low catalytic activity with substrates such as isopropyl 4-nitrophenyl methylphosphonate (5), isobutyl 4-nitrophenyl methylphosphonate (6), and cyclohexyl 4-nitrophenyl methylphosphonate (7) relative to ethyl 4-nitrophenyl methylphosphonate (4). The binding subpocket of Pmi1525 that corresponds to the small pocket of PTE is bigger. Space filling renditions of the three substrate binding subpockets in these two proteins are shown in Figure 10. It is apparent that the large pocket in PTE superimposes with the small pocket in Pmi1525 and the small pocket in PTE corresponds to the large subpocket in Pmi1525. This difference in active site geometry helps explain the difference in the substrate stereoselectivity for PTE and Pmi1525.
Figure 9.

Superimposition of the residues of the three binding pockets in the active site of PTE (PDB id: 1dpm) and the corresponding residues in Mn-containing structure Pmi1525. For PTE, the large pocket residues are colored in purple, the small pocket residues are colored in blue, and leaving group pocket residues are colored in green. The corresponding residues of Pmi1525 are colored in light grey. The labels of the residues of PTE are colored the same as the residues themselves. The residue labels of Pmi1525 are colored in black.
Figure 10.

Substrate binding pocket comparison of wild-type PTE with the bound substrate analog, diethyl 4-methylbenzylphosphonate (top) and Pmi1525 with the bound product butyrate (bottom). The color schemes are the same as shown in Figure 9 (purple, blue and green represents the large, small, and leaving group residues, respectively.
Annotation Strategy
Proteins of unknown function from different subgroups in cog1735 were functionally identified using bioinformatics, three-dimensional structure determination, computational docking, and physical compound library screening (26, 29–31). From our previous study of nonspecific carboxylate esterase Rsp3690 from subgroup 2 of cog1735(29), protein Pmi1525 from subgroup 4 was predicted to catalyze the hydrolysis of hydrophobic carboxylate esters similar to the substrates of Rsp3690. The present study confirms this prediction, as Pmi1525 efficiently catalyzes the hydrolysis of hydrophobic carboxylate esters and the hydrolysis of methylphosphonate esters.
Acknowledgments
This work was supported in part by the National Institutes of Health (GM 71790) and Defense Threat Reduction Agency (HDTRA1-14-1-0004). Use of National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Contract No. DE-AC02-98CH10886.
Abbreviations
- COG
cluster of orthologous groups
- AHS
amidohydrolase superfamily
- IPTG
isopropyl thio-β-D-galactopyranoside
- DTT
dithiothreitol
- PTE
phosphotriesterase
- ICP-MS
inductively coupled plasma mass spectrometry
References
- 1.Benson DA, Karsch-Mizrachi I, Lipman DJ, Sayers EW. GenBank. Nucleic Acids Res. 2009;37:D26–31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public database: Misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seffernick JL, de Souza ML, Sadowsky MJ, Wackett LP. Melamine deaminase and atrazine chlorohydrolase: 98 percent identical but functionally different. J Bacterial. 2001;183:2405–2410. doi: 10.1128/JB.183.8.2405-2410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odokonyero D, Ragumani S, Lopez MS, Bonanno JB, Ozerova ND, Woodard DR, Machala BW, Swaminathan S, Burley SK, Almo SC, Glasner ME. Divergent evolution of ligand binding in the o-succinylbenzoate synthase family. Biochemistry. 2013;52:7512–7521. doi: 10.1021/bi401176d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall RS, Fedorov AA, Marti-Arbona R, Fedorov EV, Kolb P, Sander JM, Burley SK, Shoichet BK, Almo SC, Raushel FM. The hunt for 8-oxoguanine deaminase. J Am Chem Soc. 2010;113:1762–1763. doi: 10.1021/ja909817d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448:775–779. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hermann JC, Ghanem E, Li Y, Raushel FM, Irwin JJ, Shoichet BK. Predicting substrates by docking high-energy intermediates to enzyme structures. J Am Chem Soc. 2006;128:15882–15891. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- 8.Ghodge SV, Cummings JA, Williams HJ, Raushel FM. Discovery of a cyclic phosphodiesterase that catalyzes the sequential hydrolysis of both ester bonds to phosphorus. J Am Chem Soc. 2013;135:16360–16363. doi: 10.1021/ja409376k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hitchcock DS, Fan H, Kim J, Vetting M, Hillerich B, Seidel R, Almo SC, Shoichet BK, Sali A, Raushel FM. Structure-guided discovery of new deaminase enzymes. J Am Chem Soc. 2013;135:13927–13933. doi: 10.1021/ja4066078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goble AM, Feng Y, Raushel FM, Cronan JE. Discovery of a cAMP deaminase that quenches cyclic AMP-dependent regulation. ACS Chem Biol. 2013;8:2622–2629. doi: 10.1021/cb4004628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hobbs ME, Vetting M, Williams HJ, Narindoshvili T, Kebodeaux DM, Hillerich B, Seidel RD, Almo SC, Raushel FM. Discovery of an L-fucono-1, 5-lactonase from cog3618 of the amidohydrolase superfamily. Biochemistry. 2013;52:239–253. doi: 10.1021/bi3015554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ornelas A, Korczynska M, Ragumani S, Kumaran D, Narindoshvili T, Shoichet BK, Swaminathan S, Raushel FM. Functional annotation and three-dimensional structure of an incorrectly annotated dihydroorotase from cog3964 in the amidohydrolase superfamily. Biochemistry. 2013;52:228–238. doi: 10.1021/bi301483z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goble AM, Zhang Z, Sauder JM, Burley SK, Swaminathan S, Raushel FM. Pa0148 from Pseudomonas aeruginosa catalyzes the deamination of adenine. Biochemistry. 2011;50:6589–6597. doi: 10.1021/bi200868u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goble AM, Fan H, Sali A, Raushel FM. Discovery of a cytokinin deaminase. ACS Chem Biol. 2011;6:1036–1040. doi: 10.1021/cb200198c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydolase related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- 16.Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 17.Tatusor RL, Loonin EV, Lipman DJ. A Genomic Perspective on Protein Families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 18.Gnanem E, Raushel FM. Detoxification of organophosphate nerve agents by bacterial phosphotriesterase. Toxicol Appl Pharmacol. 2005;207:459–470. doi: 10.1016/j.taap.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 19.Ely F, Pedroso MM, Gahan LR, Ollis DL, Guddat LW, Schenk G. Phosphate-bound structure of an organophosphate-degrading enzyme from Agrobacterium radiobacter. J Inorg Biochem. 2012;106:19–22. doi: 10.1016/j.jinorgbio.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Merone L, Mandrich L, Rossi M, Manco G. A thermostable phosphotriesterase from the archeon Sulfolous solfataricus: Cloning, overexpression and properties. Extremophiles. 2005;9:297–305. doi: 10.1007/s00792-005-0445-4. [DOI] [PubMed] [Google Scholar]
- 21.Afriat L, Roodveldt C, Manco G, Tawfik DS. The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry. 2006;45:13677–13686. doi: 10.1021/bi061268r. [DOI] [PubMed] [Google Scholar]
- 22.Uroz S, Oger PM, Chapelle E, Adeline M-T, Faure D, Dessaux Y. A Rhodococcus qsdA-encoded enzyme defines a novel class of large-spectrum quorum-quenching lactonases. Appl Environ Microbiol. 2008;74:1357–1366. doi: 10.1128/AEM.02014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow JY, Wu L, Yew WS. Directed evolution of a quorum-quenching lactonase from Mycobacterium avium subsp. Paratuberculosis K-10 in the amidohydrolase superfamily. Biochemistry. 2009;48:4344–4553. doi: 10.1021/bi9004045. [DOI] [PubMed] [Google Scholar]
- 24.Buchbinder JL, Stephenson RC, Dresser MJ, Pitera JW, Scanlan TS, Fletterick RJ. Biochemical characterization and crystallographic structure of an Escherichia coli protein from the phosphotriestease gene family. Biochemistry. 1998;37:5096–5106. doi: 10.1021/bi971707+. [DOI] [PubMed] [Google Scholar]
- 25.Hawwa R, Aikens J, Turner RJ, Santarsiero BD, Mesecar AD. Structural basis for thermostability revealed through the identification and characterization of a highly thermostable phosohotriestserase-like lactonase from Geobacillus stearothermophilus. Arch Biochem Biophys. 2009;488:109–120. doi: 10.1016/j.abb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiang DF, Kolb P, Fedorov AA, Meier MM, Fedorov IV, Nguyen TT, Sterner R, Almo SC, Shoichet BK, Raushel FM. Funtional annotation and three-dimensional structure of Dr0930 from Deinococcus radiodurans, a close relative of phosphotriesterase in the amidohydrolase superfamily. Biochemistry. 2009;48:2237–2247. doi: 10.1021/bi802274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawwa R, Larsen SD, Ratia K, Mesecar AD. Structure-based and random mutagenesis approaches increase the organophosphate-degrading activity of a phosphotriesterase homologue from Deinococcus radiodurans. J Mol Biol. 2009;393:36–57. doi: 10.1016/j.jmb.2009.06.083. [DOI] [PubMed] [Google Scholar]
- 28.Mandrich L, Manco G. Evolution in the amidohydrolase superfamily: Substrate-assisted gain of function in the E183K mutant of a phosphotriesterase-like metal-carboxylesterase. Biochemistry. 2009;48:5602–5612. doi: 10.1021/bi801932x. [DOI] [PubMed] [Google Scholar]
- 29.Xiang DF, Kumaran D, Swaminathan S, Raushel FM. Structural characterization and function determination of a nonspecific carboxylate esterase from the amidohydrolase superfamily with a promiscuous ability To hydrolyze methylphosphonate esters. Biochemistry. 2014;53:3476–3485. doi: 10.1021/bi5004266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang DF, Kolb P, Fedorov AA, Xu CC, Fedorov EV, Narindoshivili T, Williams WJ, Shoichet BK, Almo SC, Raushel FM. Structure-based function discovery of an enzyme for the hydrolysis of phosphorylated sugar lactones. Biochemistry. 2012;51:1762–1773. doi: 10.1021/bi201838b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korczynska M, Xiang DF, Zhang Z, Xu C, Narindoshvili T, Kamat SS, Williams HJ, Chang SS, Kolb P, Hillerich B, Sauder JM, Burley SK, Almo SC, Swaminathan S, Shoichet BK, Raushel FM. Functional annotation and structural characterization of a novel lactonase hydrolyzing D-xylono-1,4-lactone-5-phosphate and L-arabino-1,4-lactone-5-phosphate. Biochemistry. 2014;53:4727–4738. doi: 10.1021/bi500595c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity network for visualization of relationships across diverse protein superfamilies. PloS One. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai PC, Bigley AN, Li Y, Ghanem E, Cadieux CL, Kasten SA, Reeves TE, Cerasoli DM, Raushel FM. Stereoselective hydrolysis of organophosphate nerve agents by the bacterial phosphotriesterase. Biochemistry. 2010;49:7978–7987. doi: 10.1021/bi101056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamat SS, Bagaria A, Kumaran D, Holmes-Hampton GP, Fan H, Sali A, Sauder JM, Burley SK, Lindahl PA, Swaminathan S, Raushel FM. Catalytic mechanism and three-dimensional structure of adenine deaminase. Biochemistry. 2011;50:1917–1927. doi: 10.1021/bi101788n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman E, Wong CH. A pH sensitive colorimetric assay for the high-throughput screening of enzyme inhibitors and substrates: a case study using kinases. Bioorg Med Chem. 2002;10:551–555. doi: 10.1016/s0968-0896(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 36.Mills CK, Gherna RL. Hydrolysis of indoxyl acetate by Campylobacter species. J Clin Microbiol. 1987;25:1560–1561. doi: 10.1128/jcm.25.8.1560-1561.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CWJ, Sweet RM, Abelson JN, Simon MI, editors. Methods in Enzymology. Academic Pres; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project Number 4. The CCP4 Suit: Programs for Protein Crystallography. Acta Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D. 1997;53(3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA: 2002. [Google Scholar]
- 43.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 45.Benning MM, Shim H, Raushel FM, Holden HM. High resolution X-ray structures of different metal-substituted forms of phosphotriesterase from Pseudomonas diminuta. Biochemistry. 2001;40:2712–2722. doi: 10.1021/bi002661e. [DOI] [PubMed] [Google Scholar]
- 46.Afriat-Jurnou L, Jackson CJ, Tawfik DS. Reconstructing a missing link in the evolution of a recently diverged phosphotriesterase by active-site loop remodeling. Biochemistry. 2012;51:6047–6055. doi: 10.1021/bi300694t. [DOI] [PubMed] [Google Scholar]
- 47.Meier MM, Rajendran C, Malisi C, Fox NG, Xu C, Schlee S, Barondeau DP, Höcker B, Sterner R, Raushel FM. Molecular engineering of organophosphate hydrolysis activity from a weak promiscuous lactonase template. J Am Soc Chem. 2013;135:11670–11677. doi: 10.1021/ja405911h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanhooke JL, Benning MM, Raushel FM, Holden HM. Three-dimensional structure of the zinc-containing phosphotriesterase with the bound substrate analog diethyl 4-methylbenzylphosphonate. Biochemistry. 1996;35:6020–6025. doi: 10.1021/bi960325l. [DOI] [PubMed] [Google Scholar]
- 49.Tsai PC, Fan Y, Kim J, Yang L, Almo SC, Gao YQ, Raushel FM. Structural determinants for the stereoselective hydrolysis of chiral substrates by phosphotriesterase. Biochemistry. 2010;49:7988–7997. doi: 10.1021/bi101058z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bigley AN, Xu C, Henderson TJ, Harvey SP, Raushel FM. Enzymatic neutralization of the chemical warfare agent VX: Evolution of phosphotriesterase for phosphorothiolate hydrolysis. J Am Chem Soc. 2013;135:10426–10432. doi: 10.1021/ja402832z. [DOI] [PMC free article] [PubMed] [Google Scholar]