

Abstract
Neurogenin3 (NEUROG3) is a basic helix-loop-helix transcription factor required for development of the endocrine pancreas in mice. In contrast, humans with NEUROG3 mutations are born with endocrine pancreas function, calling into question whether NEUROG3 is required for human endocrine pancreas development. To test this directly, we generated human embryonic stem cell (hESC) lines where both alleles of NEUROG3 were disrupted using CRISPR/Cas9-mediated gene targeting. NEUROG3−/− hESC lines efficiently formed pancreatic progenitors but lacked detectible NEUROG3 protein and did not form endocrine cells in vitro. Moreover, NEUROG3−/− hESC lines were unable to form mature pancreatic endocrine cells after engraftment of PDX1+/NKX6.1+ pancreatic progenitors into mice. In contrast, a 75–90% knockdown of NEUROG3 caused a reduction, but not a loss, of pancreatic endocrine cell development. We conclude that NEUROG3 is essential for endocrine pancreas development in humans and that as little as 10% NEUROG3 is sufficient for formation of pancreatic endocrine cells.
Introduction
In mice, pancreatic progenitor cells give rise to functional endocrine cells through an endocrine progenitor intermediate that expresses the basic helix-loop-helix (bHLH) transcription factor Neurogenin3 (Neurog3) (1–4). Neurog3 is required for development of all pancreatic endocrine cell types in mice (5–7) and does this through direct and indirect regulation of downstream targets, including the transcription factors NeuroD1 (8), Rfx6 (9), Pax4 (10), Nkx6.1 (11,12), Arx (13), and others. Neurog3+ cells are first observed during the primary transition in mouse between e9 and e12.5. Although some of these primary transition endocrine cells may contribute to adult islets (3), the majority of endocrine cell mass forms during a second wave of endocrine cell development between e12.5 and e16.5.
Neurog3 is also required for development of intestinal (enteric) enteroendocrine cells in mice (14–17). Similarly, patients with biallelic mutations in NEUROG3 are born with intractable malabsorptive diarrhea due to loss of enteroendocrine cells, also known as enteric anendocrinosis (18–20). Most mutations occur in, or result in a truncation of, the well-conserved bHLH domain of NEUROG3, which has been previously reported to render the protein transcriptionally inactive. However, all these patients were born with circulating C-peptide, suggesting that unlike in mice, NEUROG3 may not be required for the development of the human endocrine pancreas (21).
We sought to unambiguously determine whether NEUROG3 is functionally required for human pancreatic endocrine cell development using pancreatic differentiation of human embryonic stem cells (hESCs) as a model system. We used two methods to disrupt NEUROG3 function: short hairpin RNA (shRNA) knockdown and direct modification of the NEUROG3 locus with CRISPR/Cas-mediated gene editing. All hESC lines generated pancreatic progenitor cells with equal efficiency, but NEUROG3−/− hESC lines were deficient in endocrine cell development in vitro and after engraftment into mice. In contrast, knockdown of NEUROG3 transcripts by up to 90% using shRNAs had only a marginal effect on the production of hormone-expressing cells in vitro. These data are consistent with the idea that the published NEUROG3 mutations are hypomorphic and not complete loss of function, thus allowing these patients to be born with a functional endocrine pancreas.
Research Design and Methods
Cell Culture and Differentiation
The hESC line H1 (WiCell) was maintained in mTeSR (Stemcell Technologies) on Matrigel-coated plates. Before differentiation, cells were dispersed with Accutase (Stemcell Technologies), washed, collected, resuspended in mTeSR containing 10 μmol/L ROCK (Rho-associated, coiled-coil containing protein kinase) inhibitor (Y-27632; Tocris Bioscience), and plated at a concentration of 1 × 105 cells/cm2 onto Matrigel-coated, 24-well Nunclon plates (Delta treated). Differentiation was initiated when cells reached ∼75% confluence ∼48 h after plating. At the start of differentiation (day 0), cells were switched to RPMI 1640 supplemented with nonessential amino acids, 100 ng/mL activin A (Cell Guidance Systems), and 50 ng/mL BMP4 (R&D Systems). Days 1–2 media included 0.2% FBS (HyClone) and did not have BMP4. On day 3, media were changed to RPMI 1640 containing 2% FBS, 50 ng/mL FGF7 (R&D Systems), and 50 ng/mL Noggin (R&D Systems). On days 5 and 7, media were switched to high-glucose (HG) DMEM (Gibco) containing 50 ng/mL Noggin, 2 μmol/L all-trans retinoic acid (Stemgent), and 1% (0.5×) B27 without vitamin A (Gibco). Finally, days 9–11 media were prepared using HG-DMEM supplemented with 1% B27 and 25 ng/mL Noggin.
CRISPR Design and Targeted Mutagenesis
The plasmid encoding Cas9-2A-GFP (22) was acquired from Addgene (#44719). Guide RNAs (gRNAs) were designed to target downstream of the NEUROG3 start codon (gRNA1 5′-GTGGGCGCACCCGAGGGTTGAGG, gRNA2 5′-GGAAGGACCGCTCCGTCTCACGG). All gRNAs were synthesized as gBlocks by Integrated DNA Technologies and PCR cloned into the pENTR/D-TOPO vector (Life Technologies). H1 cells were transfected with 2.5 μg of each plasmid using the Amaxa P3 Primary Cell 4D-Nucleofector Kit (Lonza). Positively transfected H1 cells were then collected by FACS (using the 2A-GFP) and plated at a limiting dilution for subcloning. Individual colonies were isolated and clonally expanded. Genomic DNA was collected using the HotSHOT method (23). For genotyping, PCR products were amplified, column purified (QIAGEN), and Sanger sequenced. The mixed sequence reader (24) was used to screen resulting mixed traces for insertions and deletions (INDELs). Predicted genotypes were then confirmed by subcloning using the Zero Blunt TOPO PCR Cloning Kit (Life Technologies) followed by Sanger sequencing.
Generating shRNA NEUROG3 Knockdown and Reporter Lines
Lentiviral vectors for NEUROG3 shRNA were obtained from the Cincinnati Children’s Hospital Medical Center (CCHMC) Lenti-shRNA Library Core (TRCN0000020034, Mission Library; Sigma-Aldrich), and the mCherry reporter was generated using a 5.5-kb promoter region 5′ to the NEUROG3 transcriptional start site. Vectors were packaged into high-titer lentivirus by the CCHMC viral production core. The shRNA was designed for the NEUROG3 sequence 5′-CAGTCTGGCTTTCTCAGATTT. Low-passage H1 embryonic stem cells were dissociated into a single-cell suspension using Accutase and then replated in mTeSR + 10 μmol/L Y-27632. shNEUROG3 viral particles were added to the cells immediately before plating. Puromycin selection (2 μg/mL) was added 72 h after transduction, and lines were maintained under selection.
Aggregation of hESC-Derived Pancreatic Progenitors and Transplantation
Day 12 cultures were lifted off the plate by treatment with Dispase and gentle scraping, collected by centrifugation, dispersed into 100–500-μm-sized pieces, and aggregated for 24–48 h in ultra-low attachment six-well plates. Aggregates were then embedded into purified type I collagen (rat tail collagen; BD Biosciences) 12 h before surgery and then transplanted under the kidney capsule or directly into the splenic lobe of the pancreas of immunodeficient NOD-Scid IL-2Rγnull (NSG) mice. Grafts were harvested 6 weeks after engraftment.
Cell and Tissue Processing and Immunofluorescence
Monolayers were fixed for 20 min at room temperature in 4% paraformaldehyde. Transplants were fixed overnight in 4% paraformaldehyde at 4°C, cryopreserved overnight in 30% sucrose, frozen in optimal cutting temperature compound, and cryosectioned in 8–10-μm increments. Before staining, monolayers and sections were blocked for 30 min (5% donkey serum and 0.5% Triton X in PBS). Primary antibodies were diluted in PBS + 0.1% Tween and incubated with the samples overnight at 4°C. Secondary antibodies were incubated for 2 h at room temperature. Cells were stained with DAPI (5 μg/mL in PBS) for 5 min. Sections were mounted using Fluoromount-G. All antibodies are listed in Supplementary Table 1.
Image Acquisition and Analysis
Confocal images were captured using a Nikon A1R confocal microscope with photomultiplier-based detectors and motorized stage. The microscope has 405-, 488-, 561-, and 640-nm lasers with appropriate filters. All image analyses were performed using Bitplane Imaris software. Figures were assembled using Adobe Photoshop and Adobe Illustrator CS6.
Quantitative Real-Time PCR
All RNAs were column purified using a NucleoSpin RNA kit (Macherey-Nagel), including an on-column DNAse digestion. cDNA was produced with the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Five nanograms cDNA were amplified per reaction with QuantiTect SYBR Green (QIAGEN) then amplified using a CFX96 Real-Time PCR Detection System (Bio-Rad). All primers are listed in Supplementary Table 2.
Statistical Analysis
All results are expressed as mean ± SEM unless otherwise noted. Statistical significance between two groups was tested using a two-tailed unpaired t test. P < 0.05 was considered significant.
Results
Targeted Mutagenesis of the NEUROG3 Locus in hESCs
To investigate a role for human NEUROG3 during endocrine pancreas development, we generated hESC lines with targeted disruption of NEUROG3 using CRISPR-Cas9 technology. In this approach, we used gRNAs to target the Cas9 nuclease to sequences just downstream of the NEUROG3 start codon (Fig. 1A). Potential gRNAs were screened and ranked for specificity using BLAST (Basic Local Alignment Search Tool) algorithms and the CRISPR Design Tool (http://crispr.mit.edu) to minimize the risk of off-target effects (25) (Supplementary Fig. 1A and F). Moreover, two separate gRNAs that recognize different target sequences in NEUROG3 were used to generate independent NEUROG3+/− and NEUROG3−/− lines with the rationale that a similar phenotype caused by different target sequences is exceedingly unlikely to be due to off-target effects. Clonal lines were generated and mutations in NEUROG3 detected by Sanger sequencing (Fig. 1B and Supplementary Fig. 1B). Approximately 25% of clones had no INDELs in NEUROG3 (NEUROG3+/+), ∼50% of clones had INDELs in one allele (NEUROG3+/−), and ∼25% of clones had INDELs in both alleles (NEUROG3−/−). All lines exhibited a characteristic pluripotent stem cell (PSC) morphology, expressed pluripotency markers OCT4 and NANOG (Supplementary Fig. 1C and D), grew at a rate similar to that of the parental H1 line, and were karyotypically normal (Supplementary Fig. 1E). Importantly, the NEUROG3 paralogs NEUROG1 and NEUROG2 were sequenced and confirmed to be normal in all NEUROG3−/− lines (data not shown).
Figure 1.

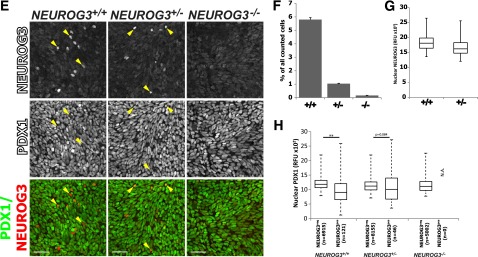
CRISPR/Cas9-mediated mutagenesis disrupts expression of NEUROG3 in differentiated pancreatic precursors. A: Adapted image from UCSC Genome Browser illustrating the full NEUROG3 gene with aligned sites targeted by the designed gRNAs (gRNA1 and gRNA2) and the primers used for sequencing. Vertebrate conservation is illustrated by the histogram. B: Sequenced genotypes of NEUROG3 wild-type (WT), heterozygous (+/−), and knock-out (−/−) clones were generated independently using either gRNA1 or gRNA2. The NEUROG3 start codon is indicated in green. The targeted mutation in each NEUROG3 allele (Al-1 and Al-2) is indicated. The Cas9 endonuclease cut sites are indicated by the scissors and the protospacer adjacent motif (PAM) is indicated in red. The insertions (red) or deletions (−) in NEUROG3 are indicated on the right side. C: Schematic summarizing the four-stage directed differentiation of human PSCs to pancreatic precursors. The y-axis lists the reagents and growth factors used, and the x-axis shows the time and stage that each factor was used. D: Representative time course of H1 NEUROG3+/+, NEUROG3+/−, and NEUROG3−/− hESCs differentiated to pancreatic precursors. mRNA for markers of several developmental stages indicated in C were assessed by quantitative PCR (n = 3, representative of four separate experiments). E: NEUROG3 protein expression in NEUROG3+/+, NEUROG3+/−, and NEUROG3−/− pancreatic precursors (differentiation day 12). NEUROG3pos cells (examples illustrated with yellow arrowheads) were counted (F), and nuclear expression was quantified (G) by immunofluorescence and high-content analysis. H: The nuclear levels of PDX1 protein in either NEUROG3pos or NEUROG3neg cells were compared across cell lines (NEUROG3+/+, NEUROG3+/−, NEUROG3−/−). Data are displayed with a box-and-whisker plot, and the whiskers represent the minimum and maximum values. Scale bars = 50 μm. Data are mean ± SEM. **P < 0.01. D, day; fwd, forward; n, number of total nuclei counted; NA, not applicable; RFU, relative fluorescence unit; rvs, reverse.
Human NEUROG3 Is Essential for Formation of Pancreatic Endocrine Cells In Vitro
For differentiation of hESCs into pancreatic progenitors and endocrine cells, we used a four-step protocol similar to several previous methods (26–29) (summarized in Fig. 1C): differentiation of hESC monolayers (marked by OCT4) into definitive endoderm (DE) (marked by SOX17 and FOXA2) and then into posterior foregut (marked by PDX1, >95%) and, finally, formation of pancreatic progenitor cells (marked by NKX6.1, 61%) (Fig. 1D and Supplementary Fig. 2A, B, and D). Differentiation into DE, posterior foregut, and pancreatic progenitors was comparable in all lines tested (NEUROG3+/+, NEUROG3+/−, NEUROG3−/−) (Fig. 1D). In NEUROG3+/+ hESCs, NEUROG3 transcripts were detectable starting around day 9 (Fig. 4A) and ∼6% of cells expressed NEUROG3 protein by day 12 (Fig. 1E and F). In contrast, NEUROG3−/− hESCs had virtually no NEUROG3 protein (Fig. 1E) or mRNA (Supplementary Fig. 3A and B). We observed an 80% decrease in NEUROG3-expressing cells in NEUROG3+/− heterozygous lines compared with NEUROG3+/+ wild-type controls, consistent with a published report that NEUROG3 haploinsufficiency in mice causes a reduction in pancreatic endocrine cell mass and impaired glucose regulation (30). Of note, quantitative analysis of NEUROG3 protein levels in each cell indicated that NEUROG3 levels were the same in NEUROG3+/− and NEUROG3+/+ cells (Fig. 1G). During pancreas development, PDX1 and NKX6.1 expression is initiated before endocrine specification, and early expression of these genes was similar across genotypes (Fig. 1D). To investigate whether expression of PDX1 and NKX6.1 in endocrine cells is altered by NEUROG3 haploinsufficiency, we quantified the level of nuclear PDX1 and NKX6.1 in cells either positive or negative for NEUROG3 protein (NEUROG3pos, NEUROG3neg) across genotypes. NEUROG3 mutations had no impact on PDX1 levels (Fig. 1H), but the range of PDX1 expression was much broader in NEUROG3pos cells than in NEUROG3neg cells. However, there was a small reduction in NKX6.1 protein levels in NEUROG3+/− and NEUROG3−/− lines (Supplementary Fig. 2C).
Figure 4.
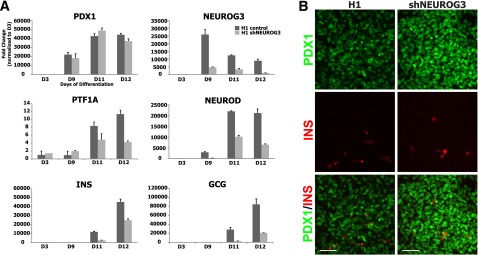
Efficient shRNA-based knockdown of NEUROG3 reduces but does not abolish hormone expression in differentiated hESCs. A: Human PSCs constitutively expressing a NEUROG3-silencing mRNA were differentiated into pancreatic precursors. Markers of pancreas and endocrine differentiation (PDX1, PTF1A, NEUROD, INS, GCG) were assessed by quantitative PCR (n = 2, representative of three separate experiments). B: Day 12 cultures were analyzed by immunofluorescence for PDX1 and INS. Scale bars = 50 μm. Data are mean ± SEM. D, day.
On day 12, we observed differentiated endocrine cells in NEUROG3+/+ and NEUROG3+/− cultures that expressed the panendocrine marker chromagranin A (CHGA) (Fig. 2A and Supplementary Fig. 2A) and the hormones insulin (INS), glucagon (GCG), and somatostatin (SST) (Fig. 2B). In contrast, NEUROG−/− cultures showed no evidence of endocrine differentiation: CHGA protein and mRNA were completely absent (Fig. 2A and C), and there were no hormone-expressing cells (Fig. 2B), demonstrating that NEUROG3 is required for endocrine specification in vitro. Quantification of INS, GCG, and SST cells in day 12 cultures demonstrated that many cells were polyhormonal. NEUROG3+/− cultures had ∼75% fewer hormone-expressing cells overall (Supplementary Fig. 2H), and there were slight changes in the relative proportions of the hormone-expressing cell types.
Figure 2.

NEUROG3 is required for specification of human pancreatic endocrine cells in vitro. A: NEUROG3+/+, NEUROG3+/−, and NEUROG3−/− hESC lines were differentiated and then analyzed on day 12 for markers of pancreatic precursors (PDX1 and NKX6.1) and endocrine cells (CHGA) by immunofluorescence. Representative images show a complete loss of the panendocrine marker CHGA in NEUROG3−/− cells. B: Analysis of hormone-expressing cells in day 12 cultures (INS, GCG, and SST). B′: High-magnification image of highlighted box in B with separated channels to show expression of individual hormones in the same cells. C: Analysis of genes involved in endocrine lineage commitment and development by quantitative PCR. Genes were subdivided into NEUROG3-dependent, –partially dependent, or -independent expression categories (n = 3, representative of four separate experiments). NEUROG3+/− and NEUROG3−/− lines generated using gRNA1 were compared with parental H1 ESCs (NEUROG3+/+). D: NEUROG3 target genes have the same response to loss of NEUROG3 in hESC clones generated from a second NEUROG3 gRNA (gRNA2 shown in Fig. 1A). NEUROG3+/− and NEUROG3−/− lines show reduced and absent expression of NEUROD1 and CHGA compared with a NEUROG3+/+ control line. Scale bars = 50 μm. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
We next investigated the impact of NEUROG3 loss on the expression of several transcription factors involved in pancreatic endocrine development. The data suggest that expression of these factors falls into one of three profiles: expression independent of, partially dependent on, or completely dependent on NEUROG3 (Fig. 2C and Supplementary Fig. 3). NEUROG3 status had little effect on the levels of PDX1 and NKX6.1 at the pancreatic precursor stage, consistent with the protein data (Fig. 1H and Supplementary Fig. 3C). In contrast, expression of NEUROD1, PAX4, PAX6, and MYT1 completely depended on NEUROG3. Factors that partially depended on NEUROG3 were IA1 and ARX, which were decreased in NEUROG3+/− cells and further reduced in NEUROG3−/− cells. Surprisingly, PTF1A appeared to be partially dependent on NEUROG3 both in CRISPR-targeted lines and in shRNA knockdown lines (Fig. 4A).
To further investigate whether the expression of these transcription factors was elevated in NEUROG3-expressing cells, we generated a transgenic NEUROG3 reporter line using a 5.5-kb region 5′ to the NEUROG3 transcriptional start site to drive expression of mCherry fluorescent protein (NEUROG3mCherry) in NEUROG3-expressing cells (Supplementary Fig. 3E and F). Sorted NEUROG3mCherry-expressing cells had high levels of NEUROD1, NKX2.2, PAX4, RFX6, IA1, and the hormones INS and GCG compared with mCherry-negative cells (Supplementary Fig. 2G). Conversely, the levels of PDX1, NKX6.1, and PTF1A were not enriched in NEUROG3-expressing cells, suggesting that the impact of NEUROG3 loss of function on PTF1A expression is noncell autonomous.
It was surprising that we did not observe the NEUROG3-dependent expression of RFX6, IA1, and ARX previously observed in mice lacking Neurog3−/− (9,13,31). Also different from other mouse studies was that NKX2.2 appeared to depend on NEUROG3, as its expression is absent in NEUROG3−/− lines. This observation is consistent with NKX2.2 expression during human fetal pancreas development where NKX2.2 is first detected shortly after the onset of NEUROG3 expression (32). Together, these data indicate that endocrine cell development is a NEUROG3-dependent process in humans, but there are qualitative differences between mouse and humans regarding the transcription factors downstream of NEUROG3.
Human NEUROG3 Is Essential for Formation of Mature Pancreatic Endocrine Cells
Forty percent of endocrine cells derived in vitro were polyhormonal (Figs. 2B′ and 3C) and did not coexpress β-cell transcription factors such as NKX6.1 and PDX1, suggesting that they were not definitive pancreatic endocrine cells. To investigate whether NEUROG3 is required for the development of mature endocrine cells that arise during the secondary transition, we engrafted human PSC–derived pancreatic progenitors into NSG mice, which are known to promote their development into more mature functional endocrine cells (27). Progenitors were engrafted either into the splenic lobe of the pancreas or under the kidney capsule and were matured for 6 weeks (Supplementary Fig. 4A and B). Of note, we observed that the pancreas seemed to support better growth and survival, with 8 of 11 grafts recovered from the pancreas compared with 6 of 19 grafts recovered from the kidney. NEUROG3+/+ cells transplanted into the pancreas contained an average of 16% endocrine cells expressing hormones INS, GCG, and SST. NEUROG3+/+ and NEUROG3+/− endocrine cells were 99% and 91% monohormonal, respectively (Fig. 3A and B). NEUROG3+/− transplants showed an ∼50% decrease in endocrine cell numbers and an increase in the number of polyhormonal cells compared with NEUROG3+/+ lines (Supplementary Fig. 4C). The only hormone-positive cells observed in NEUROG3−/− transplants expressed glucagon (7 of 57,393 counted cells from n = 3 transplants), similar to what was observed in Neurog3−/− mice (33). Unlike insulin-expressing cells derived in vitro, insulin-expressing cells matured in vivo coexpressed the definitive β-cell transcription factors NKX6.1 and PDX1, indicating that these are more mature β-cells (Fig. 3D). The protein levels of NKX6.1 and PDX1 were quantified by immunofluorescence in INSpos and INSneg cells. Both NKX6.1 and PDX1 were more highly expressed in INSpos cells than in INSneg cells. There were no major differences in NKX6.1 and PDX1 expression among NEUROG3+/+, NEUROG3+/−, and NEUROG3−/− transplants (Supplementary Fig. 4D and E).
Figure 3.

NEUROG3 is required for endocrine maturation in vivo. A: Human endocrine precursors wild type (+/+), heterozygous (+/−), and null (−/−) for NEUROG3 were transplanted into the pancreas of NSG mice, allowed to mature for 6 weeks, and then analyzed for expression of pancreatic hormones INS, GCG, and SST by immunofluorescence. Human tissue is distinguished from mouse by costaining for human nuclear antigen (HNUC). B: The total number of mono- and polyhormonal cells were quantified by high-content imaging (n = 3 transplants each for NEUROG3+/+, NEUROG3+/−, and NEUROG3−/− lines; data are the total number of endocrine cells as a percentage of all human cells counted). C: A similar analysis was performed on in vitro–derived hormone-expressing cells to compare the relative proportion of polyhormonal cells in vitro with in vivo matured cells. D: Immunofluorescence staining for cells coexpressing INS, NKX6.1, and PDX1. Coexpression of these markers indicates the presence of mature β-cells. Scale bars = 50 μm.
Endocrine Cell Development Is Not Severely Affected by an 89% Reduction in NEUROG3
The aforementioned data demonstrate that genetic ablation of the NEUROG3 locus results in a complete loss of specification of human pancreatic endocrine cells differentiated from PSCs. However, patients with homozygous or biallelic NEUROG3 mutations are all born with endocrine pancreas function. It is possible that the reported NEUROG3 mutations retain enough residual activity to allow for development of pancreatic endocrine cells. To investigate the impact of reduced NEUROG3 levels on endocrine pancreas development, we generated hESC lines expressing shRNA constructs for NEUROG3 (shNEUROG3) and differentiated these into pancreatic precursors. The shNEUROG3 hESCs formed pancreatic progenitors (marked by PDX1 and NKX6.1) with the same efficiency as control lines (Fig. 4A) and had up to an 89% reduction in NEUROG3 mRNA at the endocrine differentiation stages. However, despite this level of knockdown, there were still significant levels of NEUROG3 target genes, such as NEUROD1. INS and GCG mRNA were only reduced by 40% and 75%, respectively. Importantly, NEUROG3 knockdown had only a modest effect on the number of insulin-expressing cells in vitro as assessed by immunofluorescence (Fig. 4B). These data demonstrate that as little as 11% NEUROG3 was sufficient for relatively normal formation of insulin-expressing cells in vitro.
Discussion
Neurog3 is necessary for the development of pancreatic and gastrointestinal endocrine cells in mice. Patients with biallelic mutations in NEUROG3 present with an absence of intestinal enteroendocrine cells, thus phenocopying the mouse. In contrast to the mouse data, all patients are born with a functional endocrine pancreas (18–20,34,35). Here, we use genetically modified hESCs to provide definitive evidence that NEUROG3 is required for the development of human pancreatic endocrine cells. These data suggest that the NEUROG3 mutations identified in humans were not complete loss of function because they still supported some degree of endocrine pancreas development. Consistent with this, the current data show that as little as 11% NEUROG3 mRNA is sufficient to allow pancreatic endocrine cell development but is insufficient for intestinal enteroendocrine development (36).
The molecular basis for the differential requirement for NEUROG3 in pancreatic versus enteroendocrine development is unknown. It is possible that pancreatic endocrine cells express higher levels of NEUROG3 than enteroendocrine cells, where even a hypomorphic protein would be present at sufficient levels to specify a pancreatic endocrine fate. Another possibility may be context- dependent associations with transcriptional cofactors because bHLH transcription factors function as dimers. Consistent with this possibility, most of the point mutations identified in NEUROG3 in humans occur in the HLH dimerization domain. Finally, these mutations may affect posttranslational processing and/or stability of the protein, as is the case with neurogenin1 wherein the half-life is regulated by phosphorylation of a threonine residue (T188) in the loop region, which is highly conserved across neurogenin paralogs (37).
We also observed that loss of one allele of NEUROG3 resulted in a substantial reduction in NEUROG3 target genes and endocrine cell numbers. Furthermore, we found a substantial increase in polyhormonal cells after in vivo engraftment of NEUROG3+/− pancreatic precursors relative to NEUROG3+/+, consistent with the observation that timing and dose of NEUROG3 may affect specification of endocrine subtypes (38). The developmental phenotype associated with Neurog3 haploinsufficiency in mice is not as dramatic; however, postnatally these animals have reduced islet mass and are glucose intolerant (30). In contrast, NEUROG3 heterozygous parents have normal glucose tolerance (20), suggesting that either any reduced islet mass is not sufficient for loss of glucose regulation or mutations are only partial loss of function and are sufficient for normal endocrine pancreas development. Finally, the in vitro nature of the current model may render pancreatic cells more sensitive to reduced levels of NEUROG3. Another interesting observation is that loss of one allele of NEUROG3 caused a >50% reduction in mRNA consistent with mouse studies showing that Neurog3 participates in a feed-forward loop with both Foxa2 (39) and Myt1 (33) and that a certain threshold of Neurog3 is required to maintain this regulatory loop.
In conclusion, we used genetic targeting to demonstrate that NEUROG3 is required for development of human endocrine pancreatic cells. These studies suggest that the described human mutations in NEUROG3 are hypomorphic and that the residual function is sufficient for endocrine pancreas function in patients. Moreover, we demonstrated that this approach can be used to manipulate the human genome and study human embryonic organ development in a way that was previously only possible in model organisms. However, these studies also demonstrate that endocrine pancreas development is highly conserved between humans and mice and emphasizes the utility of the mouse as a model organism to study human development.
Article Information
Acknowledgments. The authors thank Drs. Aaron Zorn and Kyle McCracken for scientific discussion (Division of Developmental Biology, CCHMC, Cincinnati, OH). The authors also thank the CCHMC Pluripotent Stem Cell Facility, Confocal Imaging Core, Research Flow Cytometry Core, Mouse Cytogenetics Core, and Lenti-shRNA Library Core for support and services.
Funding. This study was supported by National Institutes of Health grants R01-DK-080823 and R01-DK-092456. The authors also acknowledge core support from a Cincinnati Digestive Health Center award (P30-DK-078392) and a Center for Clinical and Translational Science and Training award (U54-RR-025216).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. P.S.M. performed all experiments except the mouse transplantations and designed the study, interpreted results, and wrote the manuscript. C.L.W. carried out the mouse transplantations. C.I. contributed to the experiments, tissue processing, and image quantitation and read and provided input on the manuscript. M.A.H. read and provided input on the manuscript. J.M.W. designed the study, interpreted results, and wrote the manuscript. J.M.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1412/-/DC1.
References
- 1.Schwitzgebel VM, Scheel DW, Conners JR, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 2000;127:3533–3542 [DOI] [PubMed] [Google Scholar]
- 2.Apelqvist A, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature 1999;400:877–881 [DOI] [PubMed] [Google Scholar]
- 3.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002;129:2447–2457 [DOI] [PubMed] [Google Scholar]
- 4.Miettinen PJ, Huotari M, Koivisto T, et al. Impaired migration and delayed differentiation of pancreatic islet cells in mice lacking EGF-receptors. Development 2000;127:2617–2627 [DOI] [PubMed] [Google Scholar]
- 5.Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 2000;97:1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JC, Smith SB, Watada H, et al. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 2001;50:928–936 [DOI] [PubMed] [Google Scholar]
- 7.Xu X, D’Hoker J, Stangé G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008;132:197–207 [DOI] [PubMed] [Google Scholar]
- 8.Huang H-PH, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai M-J. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by Neurogenin 3. Mol Cell Biol 2000;20:3292–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soyer J, Flasse L, Raffelsberger W, et al. Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 2010;137:203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 1997;386:399–402 [DOI] [PubMed] [Google Scholar]
- 11.Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, Sander M. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development 2005;132:3139–3149 [DOI] [PubMed] [Google Scholar]
- 12.Sander M, Sussel L, Conners J, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000;127:5533–5540 [DOI] [PubMed] [Google Scholar]
- 13.Collombat P, Mansouri A, Hecksher-Sorensen J, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 2003;17:2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenny M, Uhl C, Roche C, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J 2002;21:6338–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev 2002;16:1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.López-Díaz L, Jain RN, Keeley TM, et al. Intestinal Neurogenin 3 directs differentiation of a bipotential secretory progenitor to endocrine cell rather than goblet cell fate. Dev Biol 2007;309:298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med 2009;15:701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Galen C, Wu V, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med 2006;356:1781–1782; author reply 1782 [DOI] [PubMed] [Google Scholar]
- 19.Pinney SE, Oliver-Krasinski J, Ernst L, et al. Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin-3 gene coding sequence. J Clin Endocrinol Metab 2011;96:1960–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubio-Cabezas O, Jensen JN, Hodgson MI, et al. Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes 2011;60:1349–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubio-Cabezas O, Codner E, Flanagan SE, Gómez JL, Ellard S, Hattersley AT. Neurogenin 3 is important but not essential for pancreatic islet development in humans. Diabetologia 2014;57:2421–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013;12:393–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000;29:52, 54 [DOI] [PubMed] [Google Scholar]
- 24.Chang C-T, Tsai C-N, Tang CY, et al. Mixed sequence reader: a program for analyzing DNA sequences with heterozygous base calling. ScientificWorldJournal 2012;2012:365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 2005;23:1534–1541 [DOI] [PubMed] [Google Scholar]
- 27.Kroon E, Martinson LA, Kadoya K, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008;26:443–452 [DOI] [PubMed] [Google Scholar]
- 28.Rezania A, Bruin JE, Riedel MJ, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012;61:2016–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagliuca FW, Melton DA. How to make a functional β-cell. Development 2013;140:2472–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Yan J, Anderson DA, et al. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol 2010;339:26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellitzer G, Bonné S, Luco RF, et al. IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. EMBO J 2006;25:1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jennings RE, Berry AA, Kirkwood-Wilson R, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013;62:3514–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Hecksher-Sorensen J, Xu Y, et al. Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol 2008;317:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohsie S, Gerney G, Gui D, Kahana D, Martín MG, Cortina G. A paucity of colonic enteroendocrine and/or enterochromaffin cells characterizes a subset of patients with chronic unexplained diarrhea/malabsorption. Hum Pathol 2009;40:1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayar E, Islek A, Yilmaz A, Akcam M, Flanagan SE, Artan R. Extremely rare cause of congenital diarrhea: enteric anendocrinosis. Pediatr Int 2013;55:661–663 [DOI] [PubMed] [Google Scholar]
- 36.Spence JR, Mayhew CN, Rankin SA, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011;470:105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vosper JMD, Fiore-Heriche CS, Horan I, Wilson K, Wise H, Philpott A. Regulation of neurogenin stability by ubiquitin-mediated proteolysis. Biochem J 2007;407:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desgraz R, Herrera PL. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 2009;136:3567–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ejarque M, Cervantes S, Pujadas G, Tutusaus A, Sanchez L, Gasa R. Neurogenin3 cooperates with Foxa2 to autoactivate its own expression. J Biol Chem 2013;288:11705–11717 [DOI] [PMC free article] [PubMed] [Google Scholar]