


Abstract
Helicases, enzymes that unwind DNA or RNA structure, are present in the cell nucleus and in the mitochondrion. Although the majority of the helicases unwind DNA or RNA duplexes, some of these proteins are known to resolve unusual structures such as G-quadruplexes (G4) in vitro. G4 may form stable barrier to the progression of molecular motors tracking on DNA. Monitoring G4 unwinding by these enzymes may reveal the mechanisms of the enzymes and provides information about the stability of these structures. In the experiments presented herein, we developed a reliable, inexpensive and rapid fluorescence-based technique to monitor the activity of G4 helicases in real time in a 96-well plate format. This system was used to screen a series of G4 structures and G4 binders for their effect on the Pif1 enzyme, a 5′ to 3′ DNA helicase. This simple assay should be adaptable to analysis of other helicases and G4 structures.
INTRODUCTION
Helicases are a central class of enzymes found in all known organisms. By a combination of biochemical and genetic approaches, their activity has been shown to affect most metabolic processes that rely on nucleic acid unwinding in the cell, including DNA replication, transcription, translation, DNA repair or recombination. At the molecular level, helicases are motor proteins that move directionally along a nucleic acid phosphodiester backbone and are able to separate annealed nucleic acid strands using energy derived from adenosine triphosphate (ATP) hydrolysis.
Although the majority of the known helicase enzymes are involved in the process of unwinding duplex DNA or RNA, some of these proteins have been known to act on less canonical substrates like protein-nucleic acids complexes or secondary DNA structures, in particular G-quadruplexes (G4) (1–4). In the G4 field, helicase studies have been at the forefront for demonstrating the formation of these structures in vivo, through various biochemical and genetic approaches (5–10). Some of the outstanding questions in the field are now how specific helicases are to various G-quadruplex structures, or what is the degree of redundancy between different helicases in their capacity to process these structures.
It is well documented that G4 can adopt multiple structures and strand arrangements, which differ thermodynamically (11). How these affect the helicase activity is currently poorly assessed. Owing to technical limitations and to the relative youth of this field, G-quadruplex unwinding has been monitored for a limited number of helicases, including members of the RecQ family (12,13), the XPD family (FANCJ) (3) and the Pif1 family (2,14,15) and for a limited number of substrates.
The most common assay for measuring helicase activity in vitro employs gel electrophoresis (16). Electrophoresis has the obvious advantage to allow the determination of the diversity and relative abundance of the molecular species present in the reaction. This is however a relatively cumbersome and low-throughput technique (17,18). Several additional methods based on fluorescence assays have been developed to overcome some of the limitations of electrophoresis, and to tackle real-time kinetics aspects of helicase action (3,19). To improve tools aiming at characterizing the activity of DNA helicases towards G4 structures, we developed a fluorescence-based helicase assay that allows testing the unwinding of different G4 structures by a helicase in high throughput and in real time. In this work, we demonstrate its application to study the activity of the DNA helicase Pif1p, a prototypal member of the PIF1 family of DNA helicase, and for which substantial evidences have been accumulated suggesting its activity on G4 in vitro and in vivo (15,19,20). This assay is also used to assess the inhibitory effect of pharmacological G4 binders on the activity of Pif1p. To our knowledge this is the first real-time helicase assay developed that allows, in a single experiment, the screening of series G4 sequences under several conditions and in the presence of G4 ligands. Moreover, this assay should be adaptable to the specific requirements of other helicase enzymes.
MATERIALS AND METHODS
Oligonucleotides and compounds
All oligonucleotides used in this research were purchased from Eurogentec and stored at −20°C as 100–200 μM stock solutions. Oligonucleotide strand concentrations were determined by absorbance at 260 nm using the extinction coefficients provided by the manufacturer. Sequences are provided in Table 1.
Table 1. Oligonucleotide sequences used in the helicase assay and its validation.
DNA systems were annealed at 1 μM concentration by preparing a mixture of 1 μM Dabcyl-labelled oligonucleotide and 0.85 μM FAM-labelled oligonucleotide in 20 mM Tris-HCl buffer (pH 7.2, 5 mM MgCl2 1 mM KCl and 99 mM NaCl). The 1.2-fold excess of dabcyl-labelled strand was used in order to assure the total hybridisation of the FAM-labelled strand and therefore to achieve the maximum quenching of the fluorescent signal. Solutions were denatured at 90°C for 5 min and cooled slowly to room temperature. Samples were then aliquoted and stored at −20°C. Braco-19 was purchased from Sigma-Aldrich. PhenDC3, Pyridostatin and TrisQ were provided by Marie-Paule Teulade-Fichou (Institut Curie, France)
Pif1 helicase
The recombinant nuclear isoform of budding yeast Pif1p was overexpressed in bacteria and purified to homogeneity as described previously (21). The purified Pif1 enzyme was stored at 2 μM concentration in 25 mM HEPES buffer (pH 7.2, 25 mM (NH4)2SO4, 25 mM MgAc2, 100 mM NaCl, 1 mM DTT and 50% glycerol) at −20°C. For helicase assays, Pif1 was further diluted to 0.2 μM in the same buffer and directly added to the DNA solution.
G4 substrate screening in helicase assay
Helicase reactions were carried out in triplicate in 96-well plates (Greiner Bio-one; 96-well, black, flat bottom) at 25°C and fluorescence monitored in a microplate reader (Tecan Infinite M1000 PRO). Every replicate contained a 50-μl solution of 40 nM FAM-dabcyl system (S-mut, S-dx, S-cmyc, S-htelo, S-ckit1, S-ckit2, S-CTA, S-TBA, S-CEB25 or S-Kras) previously annealed, 20 nM of Pif1 enzyme and 200 nM of Trap oligonucleotide (unlabelled, complementary to the FAM-labelled strand). Next, 5 μl of a 50 mM ATP solution was added to every well, the 96-well plate was stirred for 10 s and the fluorescence emission was recorded every 10 s (the excitation wavelength was set at 492 nm and the emission wavelength at 520 nm). Once the maximum emission was reached and the signal was stable (30–45 min), 5 μl (2 μM) of a strand complementary to the dabcyl-labelled sequence (C-mut, C-cmyc, C-htelo, C-ckit1, C-ckit2, C-CTA, C-TBA, C-CEB or C-Kras; the prefix ‘C’ stands for complementary) was added to every well, the plates were stirred for 10 s, and emission was monitored every 10 s.
G4 screening in the presence of a G4-ligand in helicase assay
The same procedure as used for G4 substrate screening was applied for the G4-ligand screening. A 50-μl solution of G4 system (40 nM), Pif1p protein (20 nM), Trap single-strand (200 nM) and selected ligand (1 μM) was prepared in 20 mM Tris-HCl buffer (pH 7.2, 10 mM MgCl2, 1 mM KCl and 99 mM NaCl). ATP and the single-stranded complementary sequence were added following the same procedure as above. The emission was monitored every 20 s.
RESULTS
Real-time helicase assay
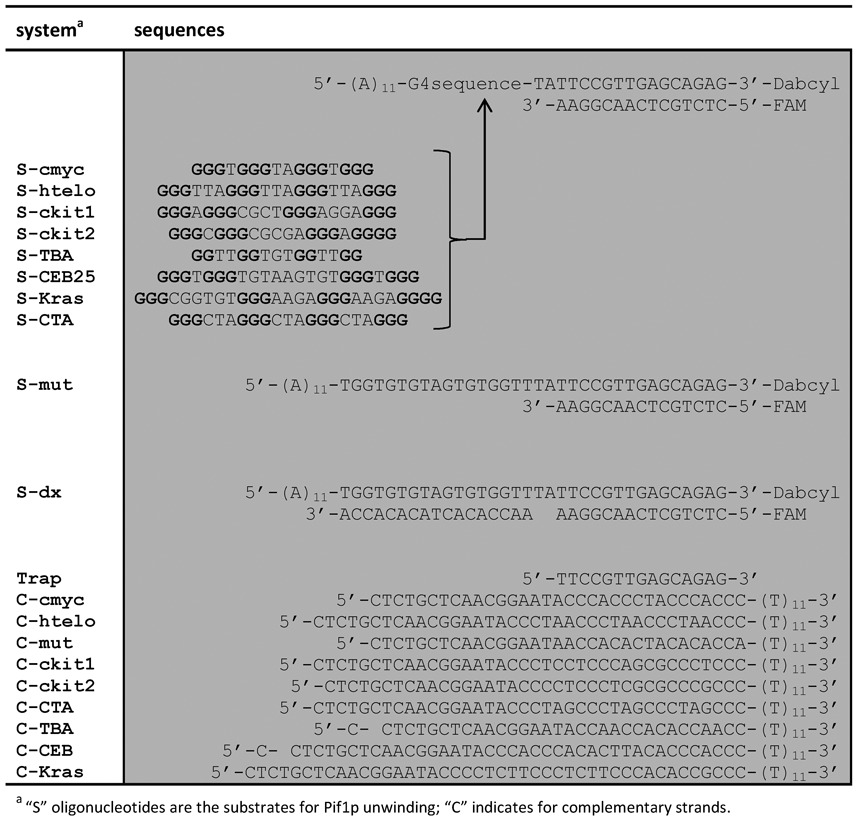
Fluorescence is a sensitive technique that has the clear advantage of monitoring a reaction process in real time, at low concentration and at high throughput. For this reason, the helicase assay was designed to monitor the unwinding process of a fluorophore-labelled DNA system. Pif1 helicases translocate in a 5′-to-3′ direction and require a 5′-tail to load on their substrate (2,22). Thus, we designed the DNA system to contain this recognition element (Figure 1). A 5′ single-stranded region of 11 deoxyadenines was used to create the required Pif1p docking site. Based on previous in vitro studies of Pif1, 11 nucleotides are sufficient for Pif1p loading (23). The substrate also contains a G4-forming region (Table 1) and a 3′ tail of 17 nucleotides covalently attached on its 3′ end to a dabcyl residue. A complementary oligonucleotide labelled at its 5′ end with the FAM fluorophore is annealed to 15 nucleotides on the 3′ side of the dabcyl-labelled oligonucleotide, forming a double-stranded substrate. In this design, two nucleotides separate the G4-forming sequence from the duplex moiety. These spacer nucleotides are necessary to avoid destabilisation of the G4 structure upon formation of the duplex structure but are not expected to provide a docking site for Pif1. In addition, in this system, the dabcyl quenches fluorescence of FAM when the duplex is formed. The helicase first loads on the 5′ tail and, after ATP addition, unwinds the G4 structure and then the fluorescently-labelled duplex DNA. This releases the FAM-labelled probe from the dabcyl-labelled strand, resulting in a measurable fluorescence increase of the FAM probe.
Figure 1.
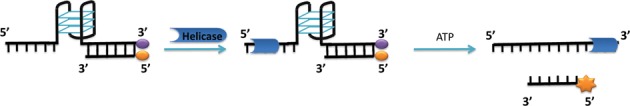
Illustration of the helicase reaction with G4/duplex DNA system. The substrate has a single-stranded region, a G4 region and a duplex region. When duplex is formed, the fluorescence of the FAM (orange symbol) labelling the single strand is quenched. In the first step of the reaction, the Pif1p helicase binds to the 5′ single-strand end. After ATP addition, the helicase proceeds in a 5′ to 3′ direction unwinding first the G-quadruplex structure and then the duplex DNA. Unwinding of the duplex releases the labelled single-stranded probe, which emits a strong fluorescence as it is no longer quenched by dabcyl (purple symbol) on the complementary strand.
Two different substrates were tested in order to validate the assay and to optimise the amount of DNA and enzyme required. In a first construction of the cmyc G4, a 16-mer sequence designed from a mutated version of the 3′ part of the NHEIII promoter of c-MYC was included in the DNA assembly (S-cmyc design, Table 1). This mutated sequence folds into a less polymorphic and more homogenous G4 structure than the natural c-MYC sequence. It adopts a parallel G4 structure in the presence of monovalent cations, which was maintained in the S-cmyc system (Supplementary Figure S1). In a second construction several guanines were mutated in the c-myc sequence in order to prevent the formation of a stable G4 structure. Thus, in this system a long single-strand region is followed by the short DNA duplex (S-mut design, Table 1).
Pif1 is active in the presence of either Na+ or K+; the optimal concentration of monovalent cations is 50–60 mM, but Pif1 is still highly active at higher ionic strength (100 mM) (23). Several concentrations of Pif1 were evaluated in a buffer containing 1 mM KCl and 99 mM NaCl using the S-cmyc system. Addition of ATP to this solution activated the helicase enzyme resulting in the release of the FAM-labelled single-strand (Figure 2A). The maximum emission observed and the kinetics of the reaction were directly dependent on the enzyme concentration with a maximum enzyme activity reached at a Pif1 concentration of 12 nM (Figure 2B). Similar experiments were carried out in a buffer containing 100 mM KCl. At this KCl concentration, we did not observe any fluorescence enhancement, suggesting either that the helicase is inactive in KCl at this ionic strength or that the cmyc G4 is too stable to be unwound by Pif1. To distinguish between these two possibilities, a similar experiment was carried out with the S-mut system, which cannot form a G4 structure. We found almost similar fluorescence emission enhancements in both buffers (1 mM KCl/99 mM NaCl and 100 mM KCl, Supplementary Figure S2), indicating that Pif1 helicase activity on the duplex part was independent of the nature of the monovalent cation at this ionic strength. Thus, the difference in activity was the result of a difference in stability of the G4 substrate to be unfolded. This is the first demonstration that a stable intramolecular G4 can resist unfolding by the Pif1 helicase.
Figure 2.
(A) Real-time emission enhancement of S-cmyc system as a function of Pif1p concentration. Pif1p helicase was incubated at several concentrations (3, 6, 12, 25, 50 and 100 nM) with the quadruplex-duplex S-cmyc system (40 nM) in a 1 mM KCl/99 mM NaCl buffer. Emission was monitored over time after ATP addition. (B) Maximum emission enhancement achieved for S-cmyc system after ATP addition as a function of Pif1 concentration. Error bars indicate standard deviation of three independent experiments.
The arbitrary 1 mM KCl/99 mM NaCl buffer solution was selected for further studies in this research. This K/Na mixture was found to be a good compromise to allow optimal activity of the protein while the quadruplex structure is properly folded. Increasing the concentration of K+ to physiological concentration (∼150 mM) would significantly stabilise the G4 structure requiring larger amounts of enzyme. In addition, high concentration of monocations would reduce the in vitro activity of the helicase (which is optimal at 50–60 mM of monocations).
In our system, the unwinding reaction is in competition with the re-hybridisation of the two separated strands, which is either spontaneous or promoted by the helicase itself (24). The reformation of the duplex DNA is slow but not negligible, even at low strand concentration (40 nM). It was observed in the unwinding traces after a few minutes (Supplementary Figure S3). The helicase activity of Pif1 reaches its maximum immediately after ATP addition; however, after a few minutes, the activity of the enzyme decreases and duplex formation results in emission quenching of the FAM fluorophore, a phenomenon that can be observed in real time. To overcome this limitation, we reasoned that addition of a trapping oligonucleotide to the reaction mixture would prevent duplex reannealing (25). A short single-stranded oligonucleotide (Trap) fully complementary to the FAM-labelled strand was added to the reaction mixture. Once the enzyme unwinds the duplex structure, the single stranded FAM-sequence is expected to be trapped by the non-labelled complementary strand. This is what we observed, since a small excess of this trapper oligonucleotide resulted in stable fluorescence even after 30 min (Supplementary Figure S3).
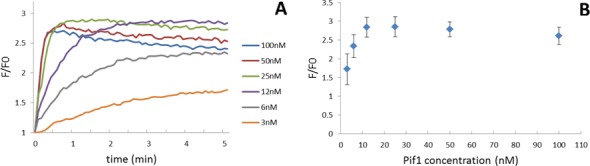
The next step in this research was to quantify the enzymatic activity. To measure the maximum level of fluorescence emission of the unwound substrate, complete separation of the FAM-dabcyl labelled duplex must be achieved. To this end, a second trapping step was introduced. Once the helicase reaction was completed and the fluorescence emission reached a stable plateau, an oligonucleotide complementary to the full-length dabcyl-labelled sequence (C-mut) was introduced into the reaction. This DNA strand displaces the FAM-labelled sequence in any unreacted G4-duplex system, leading to a highly stable duplex DNA (44 bp-long) in a quasi-irreversible process (see Supplementary Figure S4). Figure 3 shows an illustration of this two-step reaction for the S-cmyc system. The first step involves helicase-promoted separation of the G4/duplex and is ATP-dependent. Once the reaction reaches a stable plateau, the addition of the complementary sequence C-cmyc drives the reaction by displacing any unreacted G4-dx system; this second step being ATP-independent. Thus, the Pif1 helicase activity on the S-cmyc substrate can be calculated by comparing the fluorescence emission in the first and second stages of the reaction.
Figure 3.
(Top) Illustration of the two-step helicase reaction assumed to occur in the S-cmyc system. In the first step, upon addition of ATP, the Pif1p helicase unwinds the G4/duplex releasing the labelled single-stranded probe. In the second step, the dabcyl-complementary strand C-cmyc was added; this displaces any remaining duplex leading to the maximum emission signal possible. (Bottom) Unwinding process monitored in real time by recording the emission enhancement of the FAM-labelled system.
The simplicity and rapidity of this test allows tuning buffer conditions as well as oligonucleotide and helicase concentrations to optimise the assay for a different helicase or different G4 substrates. The helicase reaction for the S-cmyc system was carried out in a buffer containing 1 mM KCl and 99 mM NaCl. The presence of 100 mM KCl, which is physiologically relevant, stabilised the c-myc G4 motif in the S-cmyc system and resulted in an obstacle for the enzyme. In order to prove the versatility of this test, the helicase assay conditions were re-optimised for the S-cmyc system at 100 mM KCl. As shown in Figure 4, increasing the Pif1 concentration from 20 nM (which was a saturating enzyme concentration in a 1 mM KCl/99 mM NaCl buffer) to 100 nM was sufficient to restore the helicase activity in this system.
Figure 4.
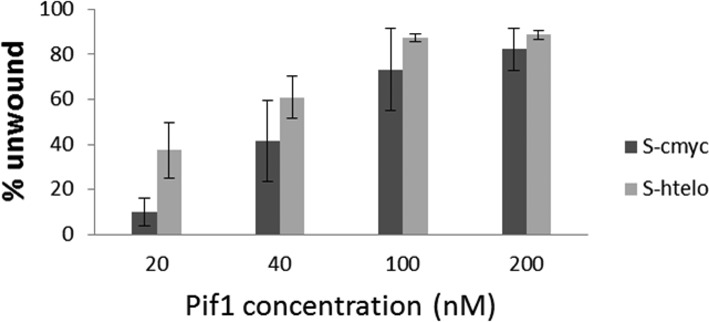
Helicase activity against the S-cmyc DNA system in buffer containing 100 mM KCl at 20, 40, 100 and 200 nM Pif1. Error bars indicate mean deviation between two independent experiments.
The unwinding process of a duplex motif immediately after a quadruplex structure by the action of the Pif1 helicase has been recently investigated by (19). In a single molecule FRET assay they found that Pif1 helicase was unable to resolve a duplex structure when preceded by a quadruplex motif. As this helicase is able to resolve the S-cmyc system, which is relatively similar to Zhou's substrate, a new design was proposed in order to verify the particular differences between the Zhou's design and the design proposed in the present helicase assay. The Pif1 helicase activity in a construction formed by a duplex motif followed by a single strand, a quadruplex motif and a second duplex (Supplementary Figure S5) was investigated under same condition as previously explained for the helicase assay. Interestingly, Pif1 was able to unwind both the quadruplex and the duplex motif that follows, thus showing a different behaviour than in Zhou's study. This difference in behaviour is therefore due to differences between the two experimental designs (single molecule versus bulk reaction with trapping) and not due to a sequence artefact (see Discussion).
G-quadruplex structure screening
Using the two-step assay, a series of G4 structures were screened for their ability to inhibit Pif1. Two non-G4 containing systems were also included as controls (Table 1): the first was the previously described single-stranded S-mut system and the second was the S-dx system, which is formed by the S-mut system to which is added an unlabelled 17-nucleotide oligonucleotide complementary to the mutated G-rich region. The first control substrate is single stranded, whereas the second involves a duplex. The series of G4 sequences screened included a number of well-characterised G4 structures (Table 1): S-htelo, the human telomeric DNA h-telo sequence (26); S-cmyc, S-ckit1, S-ckit2 and S-Kras systems, these contain G4 sequences located in the promoter regions of well-known oncogenes (c-myc (27), c-kit1 (28), c-kit2 (29) and 27Kras (30), respectively); S-CEB, containing the G-rich region from the minisatellite CEB25 (31); S-TBA, the thrombin binding aptamer TBA sequence (32); S-CTA, containing the human telomeric variant CTA (33). With the exception of S-TBA substrate, all the proposed systems exhibited a quadruplex structure under the buffer conditions considered for this test (Supplementary Figure S1). The system S-TBA however failed to fold into a stable quadruplex structure. TBA quadruplex is a quadruplex formed by only two G-tetrads and exhibits poor stability; hence the presence of a long poly(dA) sequence at the 5′ termination and a duplex motif at the 3′-end (S-TBA design) may be detrimental to G4 formation.
Figure 5A shows data from a representative two-step helicase assay monitoring the effect of Pif1 on G4 sequences in real time in 1 mM KCl and 99 mM NaCl. The addition of ATP to the solution of enzyme and DNA activated Pif1 and resulted in unwinding of the DNA duplex, leading to an increase in fluorescence emission. After 30 min, a stable plateau of fluorescence emission was reached in all the cases. At 30 min, the complementary strand of each dabcyl-labelled sequence was added. This led to the complete unfolding of all G4 structures and the displacement of the FAM-labelled sequence. The complete release of the FAM-labelled strands resulted in the maximum emission possible with each system. Raw data for each individual system are provided in Supplementary Figure S6.
Figure 5.
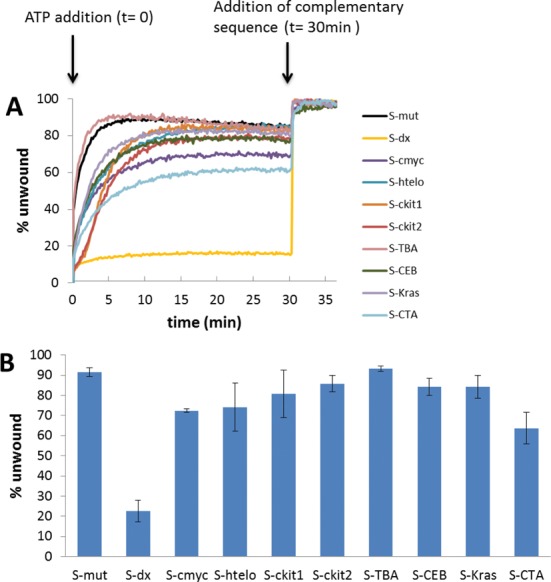
(A) Representative plots of emission as a function of time for unwinding of selected DNA systems (S-mut, S-dx, S-cmyc, S-htelo, S-ckit1, S-ckit2, S-TBA, S-CEB, S-Kras and S-CTA). ATP was added to begin the reaction (t = 0 min); the complementary strands (C-mut, C-cmyc, C-htelo, C-ckit1, C-ckit2, C-TBA, C-CEB, C-Kras and C-CTA, respectively) were added once the reactions reached a plateau (t = 30 min). (B) Quantitation of Pif1p helicase activity against selected DNA systems; error bars indicate standard deviation of three independent experiments.
Figure 5B shows the quantified Pif1 helicase activity in the presence of every structure studied. Under these conditions, none of the G4 structure was able to completely block the helicase activity of the Pif1 enzyme. Nevertheless, the presence of stable G4 structures in some assemblies was an obstacle to the enzyme, and, therefore, the unwinding activity was slightly reduced as determined by comparison of the emission intensities after the first and second steps of the reaction. Most of the kinetic profiles were hyperbolic except those obtained with the two c-kit sequences which were sigmoidal. These sequences formed higher order structures that did not enter a gel during electrophoresis (Supplementary Figure S7). The presence of these higher order structures, presumably intermolecular, could explain this behaviour. The curve observed for the S-Kras G4 substrate, which also displays the formation of higher order structures (Supplementary Figure S7), was not sigmoidal, however.
Unsurprisingly, the single-stranded S-mut was the most reactive assembly and the unwinding was found to be nearly complete (90%). Similarly, the TBA substrate, which contains a relatively unstable G4 motif formed by the stacking of only two guanine-quartets, was found to be 90% unfolded. Pif1 unwound the other G4 structures with efficiencies of 65–85%. Interestingly, the 17-base pair duplex DNA in the S-dx design was the strongest obstacle to Pif1 protein of the structures tested at 1 mM K+ concentration. This confirms earlier reports using intermolecular G4 structures that showed that Pif1 is able to resolve G4 structures more efficiently than duplex DNA structures in vitro (10,15).
The stability of DNA substrates against Pif1 enzyme was compared with the thermal stability of the quadruplex moiety structure. For this purpose the Tm of the quadruplex core motif was determined for each system. A precise Tm determination of the G4 structure embedded in the complete system was difficult as the single strand and duplex overhangs contributed to the absorbance and polluted the signal. The Tm of the quadruplex cores structures revealed a similar trend in stability as the one observed in the helicase assay (Supplementary Figure S8). The Tm of the S-cmyc, S-htelo and S-CTA quadruplex core motifs, which correspond to the DNA systems that presented a higher barrier to the enzyme, were found to show the highest Tm values (59.9°C, 56.8°C and 57.9°C respectively). On the contrary, the TBA quadruplex core was found to be the least thermodynamically stable (29.5°C).
To probe the versatility of this test, S-htelo system was also studied in more physiological [K+] conditions. As previously shown for S-cmyc (Figure 4), the presence of a higher amount of potassium cations prevent Pif1p unwinding, which can be overcome by increasing the concentration of the enzyme in the reaction. Again, this suggests that a high concentration of K+ stabilises the quadruplex, thus the optimal enzyme concentration to unwind the quadruplex is higher in this buffer (Figure 4).
Quadruplex ligand screening
The unwinding of G4 structures by Pif1 was further studied by increasing the stability of the G4 motif using known G4 ligands. Four well-characterised G4 ligands were evaluated: the trisubstituted acridine compound Braco-19 (34), the bisquinolinium PhenDC3 (35), the bis(quinolinyl)pyridine Pyridostatin (36) and triazoniatrinaphthylene TrisQ (37) (Figure 6). The G4-containing DNA systems, the S-mut system (as an example of substrate containing unstructured single-stranded DNA in place of a quadruplex motif), and the S-dx design (as an example of a duplex structure) were tested in the high-throughput format in the presence and absence of each ligand using the two-step assay strategy. Figure 6A and B show real-time unwinding curves of S-mut and S-cmyc systems, respectively, by Pif1 in the presence and absence of ligand. With the exception of Braco-19 which slowed the activity of the enzyme, the presence of G4 ligands in the reaction media did not significantly affect the unwinding activity of Pif1 on the S-mut substrate. This was expected as the selected ligands bind G4 structures but not duplex or single-stranded DNA (Supplementary Figure S9). Braco-19 showed a weak inhibition of Pif1 activity in these two systems, and this decrease was attributed to an alteration in the kinetics of the reaction. Pif1 activity was considerably reduced on the S-cmyc substrate when any of the G4 binders were present, indicating that the formation of a G4-ligand complex inhibits unwinding by the enzyme.
Figure 6.
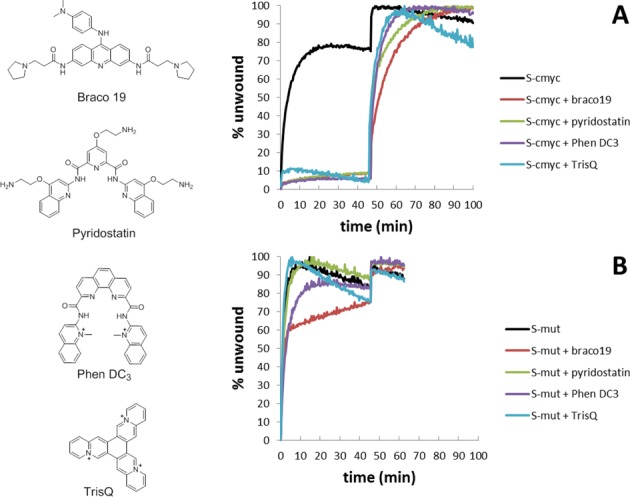
Typical real-time unfolding of (A) S-cmyc and (B) S-mut systems in absence or presence of selected ligands (Braco19 (34), Pyridostatin (36), Phen DC3 (35) and TrisQ (37)). ATP was added at t = 0; the complementary strand (C-mut and C-cmyc, respectively) was added at t = 45 min.
Similar results were found when the series of G4 structures were screened in the presence of these G4 ligands (Figure 7). As observed in the initial experiment, unwinding of S-mut or S-dx was not significantly influenced by the presence of a G4 binder. On the contrary, all the G4 ligands inhibited unwinding of the G4 substrates. Interestingly, TrisQ inhibition was selective: TrisQ blocked the enzyme activity by more than 50% on S-cmyc, S-ckit1, S-ckit2 and S-CEB substrates, but failed to affect the unwinding of other systems (S-htelo, S-Kras and S-CTA). S-htelo substrate, which contains the human telomeric sequence, was found to be the system least influenced by the addition of ligands. S-TBA system presented an interesting behaviour. As mentioned before, this substrate did not fold into a quadruplex structure under the assay buffered conditions. However, it was found to be an obstacle for the Pif1 helicase in the presence of G4 ligands. This suggests that, although the S-TBA substrate does contain a stable quadruplex motif, G4 ligands may act as chaperones promoting its formation and therefore acting as a barrier for the enzyme.
Figure 7.
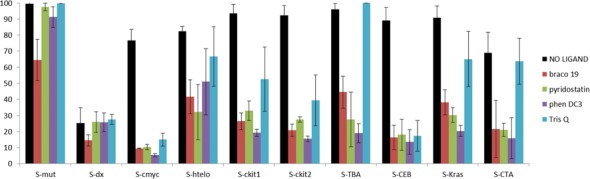
Pif1 helicase activity against DNA systems S-mut, S-dx, S-cmyc, S-htelo, S-ckit1, S-ckit2, S-TBA, S-CEB, S-KRas and S-CTA in the absence or presence of selected G4 ligands (Braco-19, Pyridostatin, PhenDC3 and TrisQ). Error bars indicate standard deviation of three independent experiments.
In overall, these results show that the activity of Pif1 can be inhibited by the presence of G4 binders, and that these G4 ligands appear to be true inhibitors of the G4-resolving activity of Pif1p.
DISCUSSION
Using a simple DNA system consisting of a G4-forming sequence, the interaction between the Pif1 helicase and G4 DNA structures was studied. Oligonucleotides in the DNA system were modified with the FAM and dabcyl quenchers to allow monitoring the unfolding of the G4 substrate by fluorescence spectroscopy. Thus, this new helicase assay is very sensitive. The assay required both low sample concentrations and reaction volumes, allowing the development of a high-throughput approach. With this technique, the kinetic behaviour of the enzyme can be followed in real time, which is a clear addition to more conventional techniques based on gel electrophoresis.
The system we designed is composed of three oligodeoxynucleotides: the FAM-labelled single strand, the G4 motif forming strand modified with dabcyl, and the trap oligonucleotide. In the annealed DNA systems, two nucleotides separated the duplex and quadruplex moieties. For the DNA substrates considered in this study, this separation was found to be insufficient to become a new binding site for the protein while preventing any stabilisation/destabilisation effect between these two DNA motifs. However, a potential destabilisation impact in other G4 by the duplex motif could be expected.
The DNA design allowed the rapid screening of several G4 structures. The comparison of the helicase activity against each structure was possible by simply changing the G4-forming sequence domain. In addition, although the present study was focused on the Pif1 helicase, the proposed system can also be applied for analysis of other helicases, provided that they show sufficient stability and activity in buffers compatible with G4 formation in vitro.
Pif1 was selected due to its stability in the buffer condition used; this enzyme proved stable in a 96- well plate for the time frame of the assay. The activity of this protein depends on the concentration of cations in the buffer and on the DNA and the enzyme concentrations. In this study, some factors were fixed (such as the DNA substrate concentration and the buffer), whereas others were optimised (such as the helicase concentration). The simplicity of this test permits re-optimisation when parameters are modified. For example, we evaluated an increase of potassium concentration from 1 mM to a more physiological condition of 100 mM. This increase in K+ inhibited Pif1p activity against the highly stable c-myc quadruplex structure under the conditions optimised in the presence of 1 mM KCl. We evaluated a range of concentrations of enzyme and were able to identify a concentration in which the G4 structure was unfolded by the Pif1 protein.
Pif1p requires the presence of either Na+ or K+, which affect the helicase activity in a similar manner. This is remarkable considering that the nature the cation dramatically influences the stability of the quadruplex structures. Thus, the G4 stability can be tuned by simply adjusting the Na+ or K+ concentrations without modifying the activity of the enzyme over a limited range of ion concentration. Our system is flexible enough to allow the fine-tuning of these conditions, for example to adjust quadruplex structure stability, without major impact on enzyme activity.
The screening of several G4 DNA substrates showed that Pif1p was able to resolve each quadruplex motif tested with little preference for any specific G4 structure. Nevertheless, some unwinding profiles were more complex (e.g. S-ckit1, S-ckit2) than others. This could be due to structural heterogeneity of the G4 species formed in these substrates, leading to a complex kinetic behaviour. For example, dimers of c-kit2 quadruplexes have been observed by nuclear magnetic resonance spectroscopy (29). Duplex substrates were also considered in this study and, as previously reported (15), Pif1p was found to resolve quadruplex structures more efficiently than duplex motifs. A recent study reported that G-quadruplexes enhance Pif1p activity on adjacent double-stranded DNA by promoting its dimerisation (38). It is tempting to speculate that the structural heterogeneity of some of our substrates lead to activation of Pif1p with different kinetic rates (according to different dimerisation profiles), leading to more complex kinetic behaviours than the ones obtained with homogenous G4-structures. The observations made by (38) provide also a rationale to understand the difference between our results and the single molecule study by (19). In Zhou's study, Pif1p is maintained in a monomeric state by antibody pull-down, which might keep its activity on double stranded DNA low. In our study, it is likely that Pif1p also becomes dimeric, explaining the difference between our results and single molecule studies on monomeric Pif1p.
The comparison of enzyme activities toward different DNA or RNA substrates can provide an estimation of the efficiency of the enzyme to remove quadruplexes of various structures. Our results (Figure 5) show that Pif1p can unwind very diverse G4 structures with similar kinetics and therefore does not exhibit a clear preference for a given quadruplex fold. The assay was also very efficient for testing effect of G4-ligands on Pif1p activity. When a G4 structure was stabilised by a selective G4 ligand, the activity of the protein decreased and in most cases was completely blocked. TrisQ ligand was found to be a remarkable exception as this ligand did not impair Pif1p activity in all the cases; this indicates that TrisQ has a selective effect on a subset of G4 structures. This is the first report of TrisQ selectivity, and our data should be confirmed by other means. The activity on the S-htelo substrate was less effected than that on other substrates. This sequence is known to be highly polymorphic in solution. One possible interpretation for our data is that the ligands shift the equilibrium between conformations in solution. If the ligands tend to stabilise a minor species, the kinetic effect could be minimal after averaging the ensemble population.
In conclusion, the results reported here validate this helicase assay for the screening of G4 structures and G4 ligands as inhibitors of helicase activity. This assay can be rapidly implemented and easily optimised, and should be adaptable to other helicases, buffers or DNA substrates. Testing other helicases would be of great interest to evaluate if the lack of selectivity of Pif1p for a particular G4-fold is a common property within G4 resolvases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fondation ARC, Conseil Régional d'Aquitaine, Agence Nationale de la Recherche (OligoSwitch [ANR-12-IS07–0001], ‘Quarpdiem’ [ANR-12-BSV8–0008–01] & ‘VIBBnano’ [ANR-10-NANO-04–03]). Funding for open access charge: Institut national de la santé et de la recherche médicale (INSERM).
Conflict of interest statement. None declared.
REFERENCES
- 1.Wu Y., Shin-ya K., Brosh R.M. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders C.M. Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 2010;430:119–128. doi: 10.1042/BJ20100612. [DOI] [PubMed] [Google Scholar]
- 3.Liu J., Chen C., Xue Y., Hao Y., Tan Z. G-quadruplex hinders translocation of BLM helicase on DNA: a real-time fluorescence spectroscopic unwinding study and comparison with duplex substrates. J. Am. Chem. Soc. 2010;132:10521–10527. doi: 10.1021/ja1038165. [DOI] [PubMed] [Google Scholar]
- 4.Booy E.P., Meier M., Okun N., Novakowski S.K., Xiong S., Stetefeld J., McKenna S.A. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012;40:4110–4124. doi: 10.1093/nar/gkr1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaffitzel C., Berger I., Postberg J., Hanes J., Lipps H.J., Plückthun A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. 2001;98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biffi G., Tannahill D., McCafferty J., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin W., Sampathi S., Dai H., Liu C., Zhou M., Hu J., Huang Q., Campbell J., Shin‐Ya K., Zheng L., et al. Mammalian DNA2 helicase/nuclease cleaves G‐quadruplex DNA and is required for telomere integrity. EMBO J. 2013;32:1425–1439. doi: 10.1038/emboj.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen G.H., Tang W., Robles A.I., Beyer R.P., Gray L.T., Welsh J.A., Schetter A.J., Kumamoto K., Wang X.W., Hickson I.D., et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc. Natl. Acad. Sci. 2014;111:9905–9910. doi: 10.1073/pnas.1404807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postberg J., Tsytlonok M., Sparvoli D., Rhodes D., Lipps H.J. A telomerase-associated RecQ protein-like helicase resolves telomeric G-quadruplex structures during replication. Gene. 2012;497:147–154. doi: 10.1016/j.gene.2012.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paeschke K., Capra J.A., Zakian V.A. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stegle O., Payet L., Mergny J.-L., MacKay D.J.C., Huppert J.L. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009;25:i374–i1382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivessa A.S., Zhou J.-Q., Zakian V.A. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000;100:479–489. doi: 10.1016/s0092-8674(00)80683-2. [DOI] [PubMed] [Google Scholar]
- 13.Schulz V.P., Zakian V.A. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–155. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 14.Ribeyre C., Lopes J., Boulé J.-B., Piazza A., Guédin A., Zakian V.A., Mergny J.-L., Nicolas A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLOS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013;497:458–462. doi: 10.1038/nature12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J.-H., Seo Y.-S. In vitro assays for studying helicaseactivities. Methods Mol Biol. 2009;521:361–379. doi: 10.1007/978-1-60327-815-7_20. [DOI] [PubMed] [Google Scholar]
- 17.Bharti S.K., Sommers J.A., George F., Kuper J., Hamon F., Shin-ya K., Teulade-Fichou M.-P., Kisker C., Brosh R.M. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013;288:28217–28229. doi: 10.1074/jbc.M113.496463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharti S.K., Sommers J.A., Zhou J., Kaplan D.L., Spelbrink J.N., Mergny J.-L., Brosh R.M. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014;289:29975–29993. doi: 10.1074/jbc.M114.567073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou R., Zhang J., Bochman M.L., Zakian V.A., Ha T. Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife. 2014;3:e02190. doi: 10.7554/eLife.02190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piazza A., Serero A., Boulé J.-B., Legoix-Né P., Lopes J., Nicolas A. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulé J.-B., Vega L.R., Zakian V.A. The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature. 2005;438:57–61. doi: 10.1038/nature04091. [DOI] [PubMed] [Google Scholar]
- 22.Boulé J.-B., Zakian V.A. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006;34:4147–4153. doi: 10.1093/nar/gkl561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulé J.-B., Zakian V.A. The yeast Pif1p DNA helicase preferentially unwinds RNA–DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramanagoudr-Bhojappa R., Byrd A.K., Dahl C., Raney K.D. Yeast Pif1 accelerates annealing of complementary DNA strands. Biochemistry. 2014;53:7659–7669. doi: 10.1021/bi500746v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tani H., Fujita O., Furuta A., Matsuda Y., Miyata R., Akimitsu N., Tanaka J., Tsuneda S., Sekiguchi Y., Noda N. Real-time monitoring of RNA helicase activity using fluorescence resonance energy transfer in vitro. Biochem. Biophys. Res. Commun. 2010;393:131–136. doi: 10.1016/j.bbrc.2010.01.100. [DOI] [PubMed] [Google Scholar]
- 26.Phan A.T., Mergny J.-L. Human telomeric DNA: G‐quadruplex, i‐motif and Watson–Crick double helix. Nucleic Acids Res. 2002;30:4618–4625. doi: 10.1093/nar/gkf597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phan A.T., Modi Y.S., Patel D.J. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei D., Parkinson G.N., Reszka A.P., Neidle S. Crystal structure of a c-kit promoter quadruplex reveals the structural role of metal ions and water molecules in maintaining loop conformation. Nucleic Acids Res. 2012;40:4691–4700. doi: 10.1093/nar/gks023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuryavyi V., Phan A.T., Patel D.J. Solution structures of all parallel-stranded monomeric and dimeric G-quadruplex scaffolds of the human c-kit2 promoter. Nucleic Acids Res. 2010;38:6757–6773. doi: 10.1093/nar/gkq558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cogoi S., Xodo L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amrane S., Adrian M., Heddi B., Serero A., Nicolas A., Mergny J.-L., Phan A.T. Formation of pearl-necklace monomorphic G-quadruplexes in the human CEB25 minisatellite. J. Am. Chem. Soc. 2012;134:5807–5816. doi: 10.1021/ja208993r. [DOI] [PubMed] [Google Scholar]
- 32.Macaya R.F., Schultze P., Smith F.W., Roe J.A., Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim K.W., Alberti P., Guédin A., Lacroix L., Riou J.-F., Royle N.J., Mergny J.-L., Phan A.T. Sequence variant (CTAGGG)n in the human telomere favors a G-quadruplex structure containing a G·C·G·C tetrad. Nucleic Acids Res. 2009;37:6239–6248. doi: 10.1093/nar/gkp630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burger A.M., Dai F., Schultes C.M., Reszka A.P., Moore M.J., Double J.A., Neidle S. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005;65:1489–1496. doi: 10.1158/0008-5472.CAN-04-2910. [DOI] [PubMed] [Google Scholar]
- 35.De Cian A., DeLemos E., Mergny J.-L., Teulade-Fichou M.-P., Monchaud D. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 2007;129:1856–1857. doi: 10.1021/ja067352b. [DOI] [PubMed] [Google Scholar]
- 36.Muller S., Sanders D.A., Di Antonio M., Matsis S., Riou J.-F., Rodriguez R., Balasubramanian S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012;10:6537–6546. doi: 10.1039/c2ob25830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertrand H., Granzhan A., Monchaud D., Saettel N., Guillot R., Clifford S., Guédin A., Mergny J.-L., Teulade-Fichou M.-P. Recognition of G-quadruplex DNA by triangular star-shaped compounds: with or without side chains? Chem. – A Eur. J. 2011;17:4529–4539. doi: 10.1002/chem.201002810. [DOI] [PubMed] [Google Scholar]
- 38.Duan X.-L., Liu N.-N., Yang Y.-T., Li H.-H., Li M., Dou S.-X., Xi X.-G. G-Quadruplexes Significantly Stimulate Pif1 Helicase-catalyzed Duplex DNA Unwinding. J. Biol. Chem. 2015 doi: 10.1074/jbc.M114.628008. doi:10.1074/jbc.M114.628008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.