

Abstract
Arginine methylation is a posttranslational modification that impacts wide-ranging cellular functions, including transcription, mRNA splicing and translation. RNA binding proteins (RBPs) represent one of the largest classes of arginine methylated proteins in both mammals and the early diverging parasitic protozoan, Trypanosoma brucei. Here, we report the effects of arginine methylation on the functions of the essential and previously uncharacterized T. brucei RBP, DRBD18. RNAseq analysis shows that DRBD18 depletion causes extensive rearrangement of the T. brucei transcriptome, with increases and decreases in hundreds of mRNAs. DRBD18 contains three methylated arginines, and we used complementation of DRBD18 knockdown cells with methylmimic or hypomethylated DRBD18 to assess the functions of these methylmarks. Methylmimic and hypomethylated DRBD18 associate with different ribonucleoprotein complexes. These altered macromolecular interactions translate into differential impacts on the T. brucei transcriptome. Methylmimic DRBD18 preferentially stabilizes target RNAs, while hypomethylated DRBD18 is more efficient at destabilizing RNA. The protein arginine methyltransferase, TbPRMT1, interacts with DRBD18 and knockdown of TbPRMT1 recapitulates the effects of hypomethylated DRBD18 on mRNA levels. Together, these data support a model in which arginine methylation acts as a switch that regulates T. brucei gene expression.
INTRODUCTION
Trypanosoma brucei, T. cruzi and Leishmania spp. are protozoan parasites of the Order Kinetoplastida, which are together responsible for 20 million infections worldwide. T. brucei infection causes African Sleeping Sickness in humans and the related disease, nagana, in livestock, thereby imposing a large human health and economic burden on sub-Saharan Africa. Transmission of T. brucei occurs through its insect vector, the tsetse fly. The parasite's complex life cycle necessitates that it adapts to varied environments. For example, bloodstream form (BF) parasites in the mammalian host are subject to 37°C, whereas procyclic form (PF) cells in the tsetse fly midgut live at variable ambient temperatures around 27°C. Parasite metabolism also dramatically changes in response to the environment in which the organism finds itself. In BF, the major pathway for energy generation is glycolysis; however, upon entry into the tsetse midgut, the parasite elaborates its single mitochondrion and relies primarily on oxidative phosphorylation for energy (1,2). The surface coat of the parasite is extensively remodeled upon transition from one host to another. Moreover, within both its mammalian and insect hosts, the parasite transitions from a proliferative form to a non-proliferative transmission-competent form. Thus, the gene expression profile of T. brucei is dramatically regulated to permit adaptation to its changing environment.
Regulation of gene expression in kinetoplastids occurs almost entirely at the posttranscriptional level; thus, RNA binding proteins (RBPs) are key determinants of cell fate (2,3). Transcribed RNAs form long polycistrons that are processed through 5′ trans-splicing with the addition of spliced leader RNA and 3′ polyadenylation (4,5). Elements generally located in the RNA 3′ untranslated regions (UTRs) modulate RNA abundance through coordinated interactions with trans-acting factors. Abundances can vary widely among RNAs, and these are controlled through numerous processes mediated largely by RBPs. Indeed, a recent genome-wide tethering screen identified over 300 proteins that, when bound to RNA, were capable of modulating gene expression, both positively and negatively (6). Additionally, RBPs are major regulators of T. brucei differentiation (3,7,8). Many RBPs regulate gene expression by modulating the stability of target RNAs (9–11). For example, the zinc finger protein, ZC3H11, actively stabilizes the heat-shock protein 70 transcript in BF cells through binding to elements in its 3′ UTR (9). Expression of an RNA can also be regulated through interactions with translational machinery, which again is aided by RBPs. For example, the Alba proteins in T. brucei stimulate translation of a major surface coat protein while having little to no affect on the overall mRNA abundance (12). Due to the extraordinary reliance on RBPs to modulate gene expression, it follows that the RBPs themselves must be regulated as well. However, to date, little is known regarding the mechanisms by which posttranslational modifications expand and regulate the functions of RBPs in kinetoplastid parasites.
Arginine methylation is a widespread posttranslational modification that involves the transfer of a methyl group from the methyl donor S-adenosylmethionine (AdoMet) to the terminal nitrogen of a peptidyl arginine. The reaction is catalyzed by a family of enzymes termed protein arginine methyltransferases (PRMTs). PRMTs are divided into types based on the end product they catalyze, with all types being able to catalyze ω-NG-monomethylarginine (MMA). Type I PRMTs subsequently yield asymmetric ω-NG,NG-dimethylarginine (ADMA) as a final product, while type II PRMTs produce symmetric ω-NG,NG-dimethylarginine (SDMA) (13,14). Methylarginines are most commonly found in RGG, RG, or RXR motifs contained within unstructured regions (15–17). The addition of a methyl group(s) affects the residue's mass, hydrophobicity and hydrogen bonding capabilities, thereby modulating macromolecular interactions (13,14,18). Because arginine residues are overrepresented at protein–protein and protein–RNA interfaces, the potential for arginine to act as a key regulator of interactome dynamics is vast. While the majority of arginine methylation research has focused on histones and epigenetics, it is becoming increasingly clear that arginine methylation affects many critical cellular functions (13,14,19). Interestingly, RNA binding and processing proteins represent one of the most abundant methylated classes of protein in both mammals and trypanosomes (15,16,20). While the impact of arginine methylation on the functions of these substrates is not known in most cases, arginine methylation of RBPs in higher eukaryotes reportedly affects processes such as pre-mRNA splicing (21,22), RNA trafficking (23), RNA stability (24) and translation (25,26). Thus, arginine methylation can be a critical regulator of cellular function through its modulation of RBPs.
In addition to its medical importance, T. brucei is an excellent model organism in which to study the role of arginine methylation in RNA biology both due to its reliance on RBPs for gene regulation and its genetic tractability. T. brucei contains four characterized PRMTs which, together, catalyze each type of methylation (27–31). In a global screen aimed at identifying the arginine methylome of T. brucei, we identified over 800 PRMT substrates (15,16). These methylproteins represent a number of different cellular compartments and are implicated in a variety of cellular processes, suggesting that arginine methylation has widespread impacts on trypanosome biology. Of the 844 methylated proteins, 100 are either predicted or known to be involved in RNA metabolism, thereby implicating arginine methylation as an important regulator of parasite gene expression. We previously showed that arginine methylation affects T. brucei mitochondrial gene expression through at least one effector protein, RBP16 (32). TbPRMT1-catalyzed arginine methylation disrupted RBP16's protein–protein and protein–RNA interactions, which led to a destabilization of specific mitochondrial RNAs (33). However, the role of arginine methylation in regulating non-mitochondrial RBPs in T. brucei has yet to be examined.
To gain insight into how arginine methylation regulates RBPs in T. brucei, we set out to characterize the previously unstudied RBP, DRBD18 (Tb927.11.14090), and to define the impact of arginine methylation on its function. DRBD18 was named according to convention (34), as it is the eighteenth described protein containing two (Double) RNA Binding Domains, or RNA recognition motifs (RRMs). Here, we demonstrate that DRBD18 is an abundant and essential cytoplasmic protein in PF T. brucei, whose RNAi-meditated depletion results in significant increases and decreases in hundreds of mRNAs. Our global methylome studies revealed three methylarginine residues on DRBD18 (15). Utilizing knockdown cells complemented with mutant DRBD18 that is either hypomethylated or methylmimic, we demonstrate that DRBD18 methylation at these three arginine residues promotes the stabilization of the cohort of mRNAs that are normally stabilized by DRBD18. Conversely, methylation can adversely affect DRBD18-mediated mRNA destabilization. These effects on gene expression are likely due to the dramatic effects of arginine methylation on DRBD18 macromolecular interactions. As predicted by its domain structure, DRBD18 interacts with RNA in vivo; however, hypomethylated and methylmimic DRBD18 exhibit different RNA binding specificities in vivo consistent with their effects on RNA stability. Hypomethylated and methylmimic DRBD18 also engage in substantially different protein–protein interactions, some of which entail proteins with known functions in gene regulation. Finally, we implicate TbPRMT1 as an arginine methyltransferase that modulates DRBD18 function. DRBD18 interacts with TbPRMT1 in vivo, and TbPRMT1 knockdown recapitulates the effects of DRBD18 hypomethylation on gene expression. Collectively, our data support a model in which arginine methylation acts as a switch that changes the function of DRBD18 from an mRNA destabilizing to an mRNA stabilizing protein.
MATERIALS AND METHODS
T. brucei cell culture and generation of cell lines
PF T. brucei strain 29–13 (35) and all cell lines derived from this strain were grown in SM media supplemented with 10% fetal bovine serum. To create tetracycline inducible RNAi against DRBD18 (Tb927.11.14090), 520 bps of its 3′ UTR was amplified using DRBD18 5′ BamHI primer (5′-GAGGATCCAATACCTGAGCATTGGGTATATGC-3′) and DRBD18 3′ XhoI primer (5′- GGCTCGAGAGCGGTAGGGCGTTCAATACCAAC-3′) and ligated into the BamHI-XhoI sites of RNAi vector p2T7–177 creating p2T7–177-DRBD18. Tet inducible MHT-DRBD18 cell lines were generated by amplifying DRBD18 ORF using DRBD18 5′ HindIII (5′-TAAAATTCACAAGCTTATGCAAGGCGCATACGGAGG-3′) and DRBD18 3′ BamHI (5′-TCTGTTCCATGGATCCTGCTGAACCATTTTCCCCAGCAC-3′) primers and ligated into the HindIII/BamHI sites of pLEW100 (35) containing an MHT C-terminal tag using Infusion cloning. Constructs expressing an untagged version of DRBD18 were produced by insertion of a stop codon directly upstream of the MHT tag. Triple R to K and triple R to F mutant constructs were generated through QuikChange (Stratagene) mutagenesis and verified by DNA sequencing.
TAP purification
TAP purification of MHT-DRBD18(WT) and mutants was carried out essentially as in (15). Briefly, cells were harvested 2 days post-induction with 4 μg/ml of tetracycline and lysed in 50 mM HEPES (pH 7.5), 150 mM KCl, 150 mM sucrose and 3 mM MgCl2. Lysates were then incubated with IgG Sepharose resin (GE Healthcare Life Sciences), and washed in buffer containing 20 mM Tris (pH 7.7), 150 mM KCl and 3 mM MgCl2 before cleavage with AcTEV protease (Invitrogen). TEV eluates were then subjected to mass spectrometry.
Sample preparation for LC/MS
The protein mixture was spiked with 1% SDS followed by reduction and alkylation, as described previously (36,37). The mixture was precipitated by stepwise addition of 6 volumes of cholorform:acetone(1:2) mixture with continuous vortexing and then incubated overnight at −20°C. After centrifugation at 20 000 g for 30 min at 4°C, the supernatant was removed and then washed with cold acetone, then the pellet was allowed to air-dry. Two consecutive digestion steps were employed to achieve a complete proteolysis. In step 1 (digestion-aided pellet dissolution), Tris buffer (50 mM, pH 8.5) containing trypisn at an enzyme/substrate ratio of 1:40 (w/w) was added and incubated at 37°C for 6 h with vortexing at 500 rpm in a ThermoMixer shaking incubator; in step 2 (complete cleavage), another portion of trypsin solution was added at an enzyme/substrate ratio of 1:40 (w/w), the final volume was controlled at 50 μl. The mixture was incubated at 37°C overnight (12 h). The digestion was terminated by adding 1% (v:v) formic acid and centrifuged at 20 000 g for 30 min at 4°C; the supernatant was used for mass spectrometry anaylsis.
Nano-LC/high-resolution-MS
A nano-LC system, including an ultra-high pressure Eksigent ekspert NanoLC 425 system (Eksigent, Dublin, CA) and a Spark autosampler NLC 400, was used for separation of the complex peptide mixture for highly sensitive identification. Mobile phases A and B were 0.1% formic acid in 1% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Samples were loaded onto a large ID trap (300 μm ID × 5 mm, packed with Zorbax 5 μm C18 material) with 1% B at a flow rate of 10 μl/min for 3 min. A series of nanoflow gradients were used to back-flush the trapped samples onto the nano-LC column (75 μm ID × 100 cm, packed with Pepmap 3-μm C18 material). The column was heated to 52°C to improve both chromatographic resolution and reproducibility. Gradient profile was as follows: (i) a linear increase from 3 to 8% B over 5 min; (ii) an increase from 8 to 27% B over 117 min; (iii) an increase from 27 to 45% B over 10 min; (iv) an increase from 45 to 98% B over 20 min; and (v) isocratic at 98% B for 20 min.
The nano-LC was coupled to a high-resolution Orbitrap Fusion Tribrid mass spectrometer with ETD (Thermo Fisher Scientific, San Jose, CA), which was operating under data-dependent product ion mode. For the identification of the binding protein, a 3 second scan cycle was used, including an MS1 scan followed by MS2 scans by CID activation. For the arginine methylation identification, MS2 scan includes alternating CID and ETD, to fragment the most intense precursors found in the MS1 spectrum. The parameters used for MS and MS/MS data acquisition under the CID mode were: top speed mode for cycles; FTMS: scan range (m/z) = 400–1600; resolution = 120 K; AGC target = 5 × 105; maximum injection time (ms) = 50; charge states of 2–7 were fragmented; dynamic exclusion was used for sensitive identification. A duration time of 60 s with data dependent mode was employed. The isolation device was the front end quadrupole with an isolation window = 1.6 units. In-trap CID was used for CID fragmentation with collision energy of 30%, while the dual-cell ion trap was used as the detector. For ETD, reaction time of 200 ms was used with a reagent target of 2.0 × 105.
Database searching and methylation calculation
CID and ETD activation spectra were processed using Proteome Discoverer (Thermo Scientific) incorporating the SEQUEST algorithm, and merged with Scaffold. Briefly, for associated protein identification, raw files were searched against a decoyed database of TriTrypDB_7.0_TbruceiTREU927_AnnotatedProteins, with the precursor mass tolerance of 15 ppm and peptide fragment mass tolerance of 1 Da. The static carbamidomethyl modification (57.021) was assumed on cysteine, and dynamic side modifications of Oxidation (15.995) was used. The searching result was merged with scaffold and peptide FDR at 0.5% was used for cutoff, with 2-peptide per protein requirement. For identification of arginine methylation, differential modifications of MMA (14.0156 Da) and DMA (28.0313 Da) on Arg residues were examined for both CID and ETD spectra, and confirmation of the most probable assignment was obtained by manual inspection of c and z ions, for putatively identified methylated peptides. Based on the data search, the methylation types were identified via characteristic neutral losses under CID activation, combined with high-resolution product ion scan. For each methylated peptide identified by ETD, the corresponding CID spectrum was manually inspected to determine the symmetry of the methylation. The relative quantification of different methylated peptides was calculated by extracting ion currents of the precursors obtained by the Fusion Orbitrap. For each identified methylated peptide, the extracting ion currents were extracted in a narrow m/z window (0.005 units) around the monoisotopic m/z for each available charge state. The area under the curve for each precursor at each charge state was calculated using Qualbrowser (Thermo Scientific) and then the percentage of each product was calculated. The calculation of different methylation type of the same molecular weight was based on the spectra count information generated by Sequest.
Antibodies
The entire ORF of DRBD18 was cloned into pGEX4T-1 (GE Healthcare Life Sciences) to generate an N-terminal GST tagged DRBD18. GST-DRBD18 was expressed in BL21 E. coli cells and purified using glutathione resin followed by anionic exchange. The resultant eluate was dialyzed against a physiological buffer and used to immunize rabbits (Bethyl Laboratories). For affinity purification of anti-DRBD18 antibodies, GST-DRBD18 was separated by SDS-PAGE and immobilized on nitrocellulose membrane. GST-DRDB18 was stained with Ponceau stain, excised as membrane chips and bound in batch with anti-DRDB18 serum. The antibodies were washed using PBS with 0.1% Tween-20, and then eluted with 100 mM glycine (pH 2.5). Resultant eluates were neutralized with 1M Tris (pH 8.0). Anti-myc antibodies were purchased from ICL labs and used at a dilution of 1:3000. Controls for subcellular fractionation consisted of α NOG1(1:5000) and α Hsp70 (1:2000), which were generous gifts from Marilyn Parsons and Jay Bangs, respectively. α p22 (1:5000) (38) was used as a loading control.
qRT-PCR analysis
Total RNA was extracted from 1 × 108 cells using Trizol. Four μg of RNA was treated with a DNase kit (Ambion) to remove any residual DNA. RNA was reverse transcribed using random hexamers or oligo(dT) primers (Applied Biosystems). To detect endogenous DRDB18 transcripts, the following primers were used that are complementary to the 5′ UTR of DRBD18: 5′ end of DRBD18 5′ UTR (5′- AGCAAAAAGAATAAAAAGGAGAGA-3′) and to the 3′ end of DRBD18 5′ UTR (5′-GCTCTTTTCTTCTCTTCCCCTA-3′). The following primers pairs specific for each RNAseq transcript were utilized: primers complementary to the 5′ end of Tb427.07.2030 ORF (5′-GAGCCTACTCGCTTAAAACA-3′) and to the 3′ end of the Tb.427.07.2030 ORF (5′-CTTCGGAGGTAGTTCAAATG-3′); to the 5′ end of Tb427.07.2160 ORF (5′-GGTTGTACTCTCAAGGGTGA-3′) and to the 3′ end of the Tb427.07.2160 ORF (5′-CTGTATGTATCGCATTGTGG-3′); to the 5′ end of Tb427.10.1500 ORF (5′-TGAGCCTAGTGGATGACTTT-3′) and to the 3′ end of the Tb427.10.1500 ORF (5′-ATGTCTTTCCCAATAACGTG-3′); to the 5′ end of Tb427.10.5250 ORF (5′-CCTCAAGGCACC TATTACTG-3′) and to the 3′ end of the Tb427.10.5250 ORF (5′-CCACCACTGTTTCCATTACT-3′); to the 5′ end of Tb427.08.4200 ORF (5′-CCAGTGGATATGTTGGAACT-3′) and to the 3′ end of the Tb427.08.4200 ORF (5′-AGTTGAGACCACGAAAAAGA-3′); to the 5′ end of Tb427.10.5150 ORF (5′-TAACGCAACGTAATCAACAG-3′) and to the 3′ end of the Tb427.10.5150 ORF (5′-ACGTGGTGGACTTATCTGAC-3′). To assay mRNA stability, cells either uninduced or induced for DRBD18 RNAi for 24 h before being treated with 10 μg/ml Actinomycin D. Total RNA was extracted at 0, 20, 30 and 60 min, reverse transcribed using a mix of oligo(dT) and gene specific primers and analyzed by qRT-PCR. All mRNA levels were normalized to mitochondrial 9S rRNA and the percentage of mRNA remaining from time 0 was calculated.
Biochemical fractionation
PF cells harboring the MHT-DRDB18 constructs were induced with 4μg/ml of tet. Cells were harvested by centrifugation, washed with buffer A containing 20 mM Hepes pH 7.9, 20 mM KCl, 150 mM sucrose, 3 mM MgCl2 and 1 mM DTT and resuspended at 5 × 108 cells/ml. Cells were permeablized using 0.2% NP40 and passed through a 26-gauge needle. The extract was centrifuged and the resultant supernatant comprises the cytoplasmic fraction. The pellet was resuspended in buffer A and re-extracted through a 26-gauge needle. The rinsed pellet was resuspended in buffer A and is considered the nuclear fraction.
Immunofluorescence
PF cells harboring the MHT-DRDB18 constructs were either left uninduced or induced with 4μg/ml of tet. Cells were harvested by centrifugation, washed with PBS and resuspended at 5 × 107 cells/ml. Cells were fixed with 4% formaldehyde in PBS for 30 min, before permeablization using 0.2% NP40. Permeable cells were blocked using 10% normal rabbit serum and then incubated for 1 h with monoclonal mouse anti-myc antibody (c-Myc(9E10), Santa Cruz) diluted 1:50 in PBS/BSA. The secondary Alexa Fluor 488 goat anti-mouse antibody was diluted 1:200 and incubated with cells for 30 min at room temperature. Cells were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen). Images were taken with a Zeiss Axio Imager.M2 microscope using Volocity software.
RNAseq
Two biological replicates of DRBD18 RNAi cells were induced with 4 μg/ml of tet for 24 h. Cells were harvested and total RNA was isolated using Trizol. Residual DNA was removed using DNAase away (Ambion) and the RNA was purified using RNAaseEasy. RNA was quantified using the Ribogreen Assay (Invitrogen), and its quality checked using Agilent Bioanalyzer 2100 RNA nano 6000 chip (Agilent). The Illumina TruSeq RNA sample preparation kit was used to prepare cDNA libraries from RNA samples. Briefly, samples were poly(A) selected to isolate mRNA, the mRNA was cleaved into fragments, the first strand reverse transcribed to cDNA using SuperScript II reverse Transcriptase (Invitrogen) and random primers, followed by second strand cDNA synthesis using Second Strand Master Mix supplied with the kit. Following end repair, the addition of a single ‘A’ base, and ligation with adapters, the products were enriched and purified with PCR to create the final cDNA library as per manufacturer's protocol. cDNA libraries were quantified using the Picogreen Assay (Invitrogen) and Library Quantification kit (Kapa Biosystems). An Agilent Bioanalyzer 2100 DNA 7500 chip was used to confirm the sizes of the cDNA libraries. The cDNA products were then sequenced using the Illumina HiSeq2500 at the UB Genomics and Bioinformatics Core Facility (Buffalo, NY). RNAseq reads were aligned to the genome of Trypanosoma brucei TREU927 using TopHat v2.0.9 (39) with the genes annotated by TriTrypDB v6.0. Reads were mapped with at most two mismatches using Bowtie1. We used Cufflinks v2.1.1 to detect genes that were differentially expressed in uninduced and induced DRBD18 RNAi cells with the default parameters (40).
Cross-linking immunoprecipitation
Cells harboring either MHT-DRBD18(WT), MHT-DRBD18(R→K), or MHT-DRBD18(R→F) were induced with 4 μg/ml tet for 2 days. Cells were resuspended in PBS and CLIP was carried out essentially as in (41,42). Briefly, cells resuspended in PBS were UV irradiated before snap freezing in liquid nitrogen. Cells were thawed, lysed with NP40, treated with 1 U/μl of RNAase T1 (Ambion) and precleared with uncoated beads. The sample was then incubated with anti-myc conjugated resin at 4°C for 2 h. The supernatant was removed, and the beads were washed several times to remove all unbound proteins and RNAs. A second RNAase T1 incubation step was performed at a final concentration of 10 U/μl followed by dephosphorylation using calf intestine phosphatase (NEB). The sample was further washed and resuspended in one bead volume of polynucleotide kinase (PNK) buffer. γ-32P-ATP was added to a final concentration of 0.5 μCi/μl and T4 PNK (NEB) to 1 U/μl before incubation at 37°C for 30 min. The sample was washed, electroeluted in SDS-PAGE loading buffer and separated via SDS-PAGE. The protein signal was quantified using the ImageLab software. The 32P RNA signal was detected using the Typhoon Scanner Control v5.0 software and quantified using ImageJ software. For qRT-PCR analysis RNA was extracted prior to RNAase T1 treatment, reverse transcribed using gene specific primers, and analyzed by qRT-PCR. mRNA levels were normalized against 18S rRNA.
RESULTS
Identification of arginine methylation sites in DRBD18
DRBD18 was initially identified as a substrate for arginine methylation through our global mass spectrometry analysis (15). As shown in Figure 1A, four peptides containing three sites of methylation were mapped to this protein. Arg199 is methylated in three of the four peptides, in all cases decorated by MMA. Arg202 is also methylated in three out of the four recovered peptides, but the type of methylation differs in the distinct peptides. In peptides 1 and 3, Arg202 is MMA, and in peptide 4 it is DMA. Because the surrounding arginines in peptides 1 and 4 are identically modified, it suggests that other factors determine whether Arg202 becomes mono- or dimethylated. Arg204 is methylated in two peptides, and each time is observed as MMA. To confirm these sites of methylation, we generated a cell line expressing Myc-His-TAP (MHT)-DRBD18 in a tetracycline (tet)-inducible manner, immunoprecipitated MHT-DRBD18, and subjected it to mass spectrometry to detect posttranslational modifications. In this targeted experiment, we recovered the identical four peptides covering this region, containing the same methylarginines, as were detected in our methylome screen. These data suggest that Arg199, 202 and 204 are bona fide sites of arginine methylation on DRBD18 and that the identified modifications constitute the predominant types of methylation at these sites.
Figure 1.
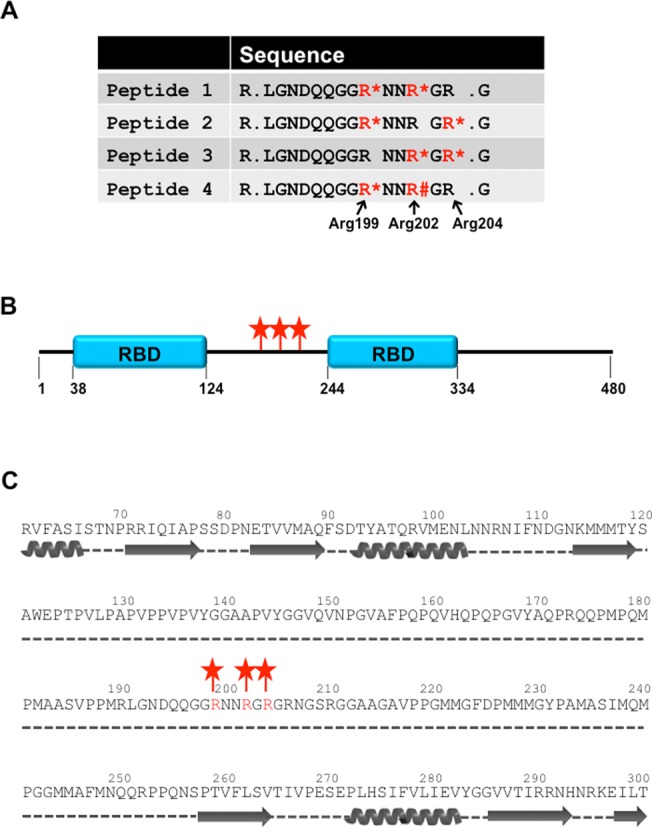
Methylated arginine residues in DRBD18. (A) Peptides recovered from mass spectrometry. Four peptides encompassing three sites of arginine methylation were mapped to DRBD18. Methylated arginines are in red, MMA is denoted with asterisks and DMA is denoted with the pound sign. (B) Schematic representation of DRBD18 domain structure with amino acid numbers indicated below. Blue boxes represent RBDs as defined by InterProScan SSF54928. The three sites of arginine methylation are indicated with red stars. (C) Secondary structure prediction of DRBD18 using the Phyre2 algorithm. Alpha helices (coils) and B sheets (arrows) are depicted under the primary sequence. Dotted lines indicate unstructured regions. The three sites of arginine methylation are indicated with red stars.
DRBD18 contains two predicted RNA binding domains (RBDs; InterProScan SSF54928), the first spanning amino acids 38–124, and the second from residues 244–334. The three sites of arginine methylation lie between the two RBDs (Figure 1B). To further address the likely structural context of these methylarginines, we performed a secondary structure prediction of DRBD18 using the Phyre2 program (43). As shown in Figure 1C, the three sites of methylation are predicted to lie in a proline-rich unstructured region of the protein. We previously reported that in T. brucei methylarginines are generally found in unstructured regions and that, in RBPs, they typically reside outside of RBDs (15). Thus, DRBD18 follows these global trends with regard to methylarginine context. Together, our data show that DRBD18 contains three methylarginines in an unstructured region in between its two predicted RBDs. Based on their location within DRBD18, it is unlikely that these three methylarginines are integral to the overall structure of DRBD18.
DRBD18 is an abundant and essential protein that is localized primarily to the cytoplasm
To begin to understand DRBD18 function, we down-regulated its expression through tet-inducible RNAi and assayed PF T. brucei growth. qRT-PCR analysis showed that upon tet induction, DRBD18 mRNA levels were reduced by approximately 90% compared to uninduced cells (Figure 2A, top). To assess DRBD18 protein expression, we generated an antibody against recombinant GST-DRBD18. Anti-DRBD18 antibodies recognize one band at the correct molecular weight, and upon induction of RNAi we observe a robust reduction in DRBD18 protein (Figure 2A, bottom). Cells in which DRBD18 expression was repressed exhibited slowed growth on day two post-induction and began to die four days post-induction. By day 6 post-induction, few live cells remained (Figure 2B). We next used our specific anti-DRBD18 antibodies to determine the abundance of DRBD18 in parental (strain 29–13) cells. Increasing amounts of recombinant GST-DRBD18 protein were separated by SDS-PAGE alongside extract from 106 PF T. brucei cells, and the anti-DRBD18 signals were quantified. A representative western blot and quantification from triplicate determinations are depicted in Figure 2C. There are approximately 400 000 molecules of DRBD18 per cell, making DRBD18 one of the more abundant RBPs characterized to date (12,44,45). Given that there are approximately 50 000 mRNA molecules per cell (46), there are roughly eight molecules of DRBD18 per mRNA, suggesting that DRBD18 may pose a significant impact on the PF T. brucei transcriptome. Finally, we used our anti-DRBD18 antibodies to probe the subcellular localization of DRBD18 by western blot of subcellular fractions (these antibodies proved unsuitable for immunofluorescence). We performed biochemical fractionation followed by western blot analysis on parental (strain 29–13) cells, and used HSP70 and NOG1 as cytoplasmic and nuclear markers, respectively. These analyses demonstrated that DRBD18 localizes primarily to the cytoplasm (Figure 2C). From these data, we conclude that DRBD18 is an abundant cytoplasmic protein that is essential for PF T. brucei growth.
Figure 2.
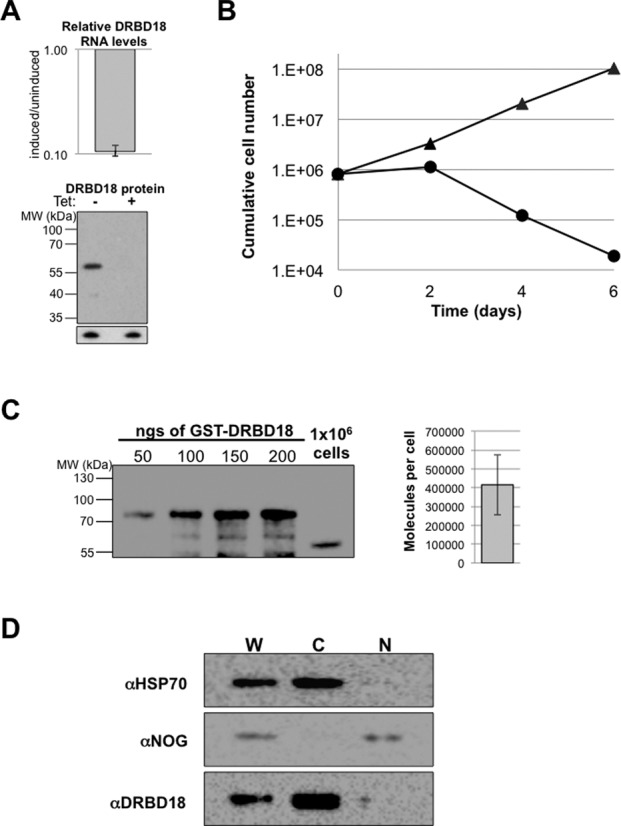
DRBD18 is an abundant, cytoplasmic protein essential for viability of procyclic form T. brucei. (A) DRBD18 RNAi leads to a reduction of DRBD18 mRNA and protein levels. Top: qRT-PCR analysis of DRBD18 mRNA levels represented as the value in cells induced for RNAi for one day relative to that in uninduced cells. RNA levels were normalized against 18S rRNA and represent the average and standard error of six determinations. Bottom: western blot analysis of uninduced (−) or tet induced (+) cells using affinity purified anti-DRBD18 antibodies. Bottom panel shows the p22 protein which serves as a load control. (B) PF cells either expressing (triangles) or repressed for (circles) DRBD18 were assessed for parasite growth over the course of six days. Cells were counted every two days and diluted back to a starting concentration of 8 × 105 cells/ml. (C) Quantification of DRBD18 abundance. GST-DRBD18 (50–200 ngs) and 1 × 106 strain 29–13 cells were separated via 10% SDS-PAGE and analyzed by western blot using anti-DRBD18 antibodies. The strain 29–13 cell signal was converted into molecules of DRBD18 per cell. A representative blot is shown on the left and quantification of triplicate determinations +/- standard error is shown on the right. (D) Western blot analysis of subcellular fractions of strain 29–13 PF cells. 2 × 106 cellular equivalents of cytoplasmic and nuclear fractions were separated via 10% SDS-PAGE. HSP70 and NOG1 represent cytoplasmic and nuclear markers, respectively. W, whole cell; C, cytoplasmic fraction; N, nuclear fraction.
Depletion of DRBD18 leads to extensive changes in the PF transcriptome
The amino acid sequence of DRBD18 implies RNA binding activity, and the protein's cytoplasmic localization suggests a possible role in modulation of RNA stability. Therefore, we next sought to determine the impact of DRBD18 on global RNA levels. To this end, we down-regulated DRBD18 expression in PF T. brucei, and subjected total RNA from uninduced and induced cells to Illumina sequencing. To ensure the validity of our RNAseq analysis, we analyzed RNA isolated from biological replicates of DRBD18 RNAi cells in which DRBD18 was depleted to similar levels. We achieved an average of 29.4 and 29.6 million uniquely mapped reads for uninduced and DRBD18 knockdown induced samples, respectively. Upon depletion of DRBD18, 985 transcripts showed a significant change in abundance in both biological replicates compared to uninduced cells at a 5% false discovery rate (Figure 3A; Supplementary Table S1). Of these, 568 RNAs were up-regulated and 417 RNAs were down-regulated. Up-regulated RNAs increased in abundance anywhere from 1.4-fold to 40-fold, with an average of a 3.3-fold increase in abundance. Down-regulated RNAs were decreased in abundance upon induction of DRBD18 RNAi to between 0.16 and 0.75 of uninduced levels, with an average value of 0.59 of uninduced levels.
Figure 3.
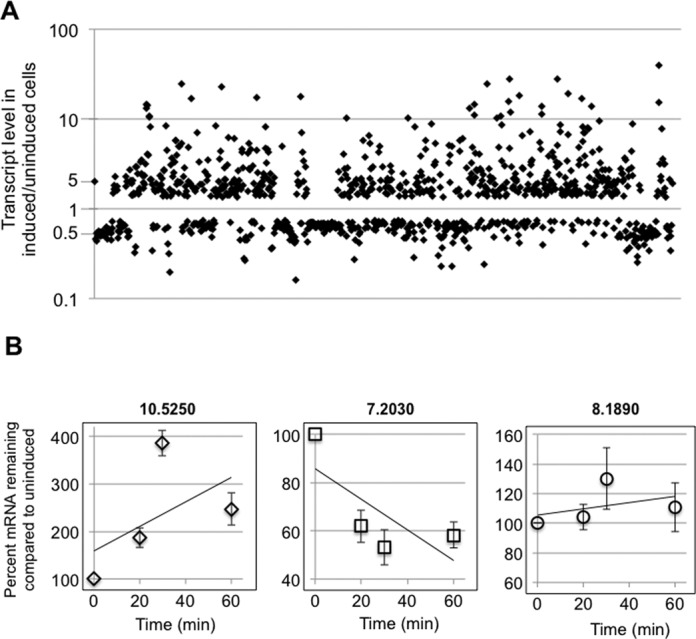
RNAseq analysis of DRBD18 depleted cells reveals extensive changes to the transcriptome. (A) Scatter plot showing transcript abundance (in induced/uninduced DRBD18 RNAi cells) for all transcripts that were significantly affected in biological replicate samples. (B) Degradation kinetics of transcripts that were either up-regulated (10.5250 (Tb927.10.5250, ZC3H32)), down-regulated (7.2030 (Tb927.7.2030, RHS7)), or unchanged (8.1890 (Tb927.8.1890, cytochrome c1)) in RNAseq analyses upon DRBD18 depletion. DRBD18 RNAi cells were either untreated or treated with tetracycline to induce RNAi for 24 h, actinomycin D was added at time zero, and total RNA was isolated at the indicated time points and quantified using qRT-PCR. The quantified results are expressed as the percent of mRNA remaining in RNAi induced cells relative to the amount remaining in uninduced levels. Transcript levels were normalized against mitochondrial 9S rRNA, and are plotted as the average and standard error of 4–6 determinations.
To determine whether the changes in RNA abundances upon DRBD18 depletion are caused by changes in mRNA decay rates, we measured mRNA levels by qRT-PCR at 0, 20, 30 and 60 min following inhibition of RNA synthesis with actinomycin D. One representative mRNA from each class of up-regulated, down-regulated, and unaffected transcripts was chosen for analysis. As expected, we found that the up-regulated mRNA (10.5250) (Tb927.10.5250, ZC3H32) is stabilized and the down-regulated mRNA (7.2030) (Tb927.7.2030, RHS7) is destabilized in DRBD18 depleted cells compared to uninduced control cells (Figure 3B). A transcript whose abundance was not changed upon DRBD18 RNA in RNAseq analysis (8.1890) (Tb927.8.1890, cytochrome c1) decayed at a similar rate in both induced and uninduced DRBD18 RNAi cells. As a confirmation that DRBD18 regulates mRNA abundance at the level of stability rather than transcription, we analyzed the genomic context of a subset of our most significantly up- and down-regulated RNAs. Because RNA polymerase II transcribed genes in T. brucei are arranged as polycistronic transcription units (2,47,48), adjacent genes are transcribed at comparable rates. RNAs whose abundance changed dramatically upon DRBD18 down-regulation were commonly flanked by RNAs whose abundance was unchanged or even changed in the opposite direction, validating a role for DRBD18 in post-transcriptional regulation of RNA levels. Together, these data demonstrate that DRBD18 affects the abundance of 985 transcripts by destabilizing 568 mRNAs and stabilizing 417 mRNAs.
Depletion of DRBD18 affects different classes of RNAs
To determine if depletion of DRBD18 affects specific classes of RNAs, we mined the fully annotated genome (TriTrypDB) and grouped affected RNAs based on their combined molecular function/biological process GO terms and InterPro domains for their encoded proteins. Depletion of DRBD18 results in two differentially affected RNA populations, comprising transcripts that are up-regulated upon DRBD18 repression (i.e. normally destabilized by DRBD18), or transcripts that are down-regulated upon repression (i.e. normally stabilized by DRBD18) (Figure 3). Here, we refer to these populations as DRBD18-destabilized and DRBD18-stabilized, respectively, to reflect the effect of the DRBD18 protein itself (as opposed to the effect of depleting DRBD18). Both the DRBD18-destabilized and DRBD18-stabilized populations contain transcripts encoding proteins involved in a multitude of cellular processes (Figure 4A). Hypothetical proteins with predicted trans-membrane domains and no other distinguishing features constituted a large fraction of both populations. We also observed differential regulation of certain classes of RNAs. For example, kinases and phosphatases constituted 20% of DRBD18-destabilized RNAs, but only 2% of DRBD18-stabilized RNAs. Conversely, retrotransposon hot spot RNAs were enriched in the DRBD18-stabilized RNA population but were absent from the DRBD18-destabilized population. Proteins implicated in RNA binding and processing were prominent in both the stabilized and destabilized RNA populations, although the specific classes of proteins differed in the two populations. DRBD18-stabilized RNAs encode many RNA processing enzymes such as tRNA synthetases and predicted RNA methyltransferases, while the DRBD18-destabilized RNA population was enriched for canonical RNA binding proteins of the RRM, Puf and ZC3H families (Supplementary Table S1).
Figure 4.
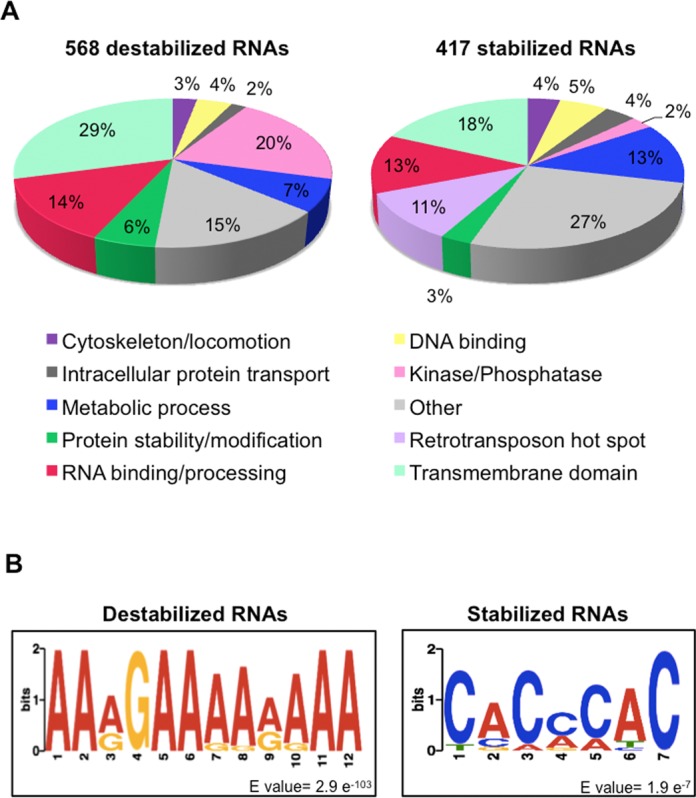
Bioinformatic analysis of mRNAs regulated by DRBD18. (A) Predicted biological processes/molecular functions of DRBD18-destablized and DRBD18-stabilized RNAs according to their GO terms and InterProScan domains listed on TriTrypDB. (B) Predicted consensus motifs for highly destabilized and stabilized transcripts. The published 3′ UTR sequences (49) from transcripts destabilized at least 6-fold or stabilized at least 2.5-fold were searched for consensus motifs using MEME (52) with the following parameters; motif width between 6 and 12 nucleotides, search on given strand only, and the motif must occur in each 3′ UTR at least once. Motif discovery was repeated with shuffled sequences to determine a reasonable E-value cutoff.
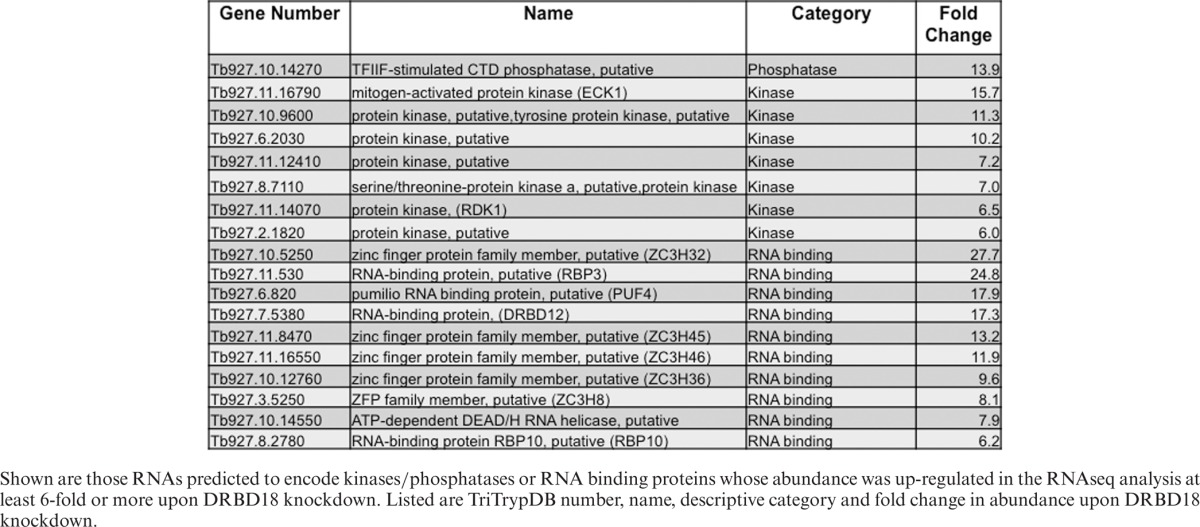
We also observed that the magnitude of the changes upon DRBD18 depletion differed for stabilized and destabilized RNAs. Most DRBD18-stabilized RNAs were decreased approximately 2-fold upon DRBD18 RNAi, and the most significantly affected RNA was decreased approximately 6-fold (Figure 3A and Supplementary Table S1). In contrast, within the DRBD18-destablized population, we detected 56 RNAs whose abundance increased between 6- and 40-fold upon DRBD18 RNAi (Figure 3 and Supplementary Table S1). Interestingly, several of the most highly DRBD18-destablized RNAs were in the categories of kinases/phosphatases and RNA binding proteins (Supplementary Table S1 and Table 1). We detected 56 DRBD18-destabilized transcripts whose abundance changed 6-fold or greater in our RNAseq analysis. Among these are a putative TFIIF-stimulated CTD phosphatase, the ECK1 and RDK1 kinases, a putative protein kinase A, and a putative RNA helicase (Table 1). Also in this category were nine RNAs encoding canonical RBPs, including those encoding ZC3H32 and RBP3, which increased 28- and 25-fold, respectively, upon DRBD18 down-regulation (Table 1). Together, these data demonstrate that DRBD18 specifically stabilizes one set of RNAs while preferentially and strongly destabilizing a different set of RNAs. DRBD18's dramatic effects on proteins involved in signal transduction and gene regulation indicate a capacity for profoundly affecting cell function.
Table 1. Highly DRBD18-destabilized mRNAs encoding kinases/phosphatases and RNA binding proteins.
Many RBPs act through binding mRNA 3′ UTRs, and 3′ UTRs are important elements in trypanosome gene regulation (3). To gain insight into potential DRBD18 target sequences, we analyzed the 3′ UTRs of the DRBD18-destabilized and DRBD16-stabilized RNAs separately. Using the published set of T. brucei UTR sequences (49), we were able to analyze 320 of the 568 destabilized RNAs and 247 UTRs of the 417 stabilized RNAs. One striking feature we observed was 3′ UTR length. Genome-wide studies in T. brucei identified average 3′ UTR lengths of 400–600 nucleotides (49–51). Considering the most abundant 3′ UTR population, we determined an average 3′ UTR length of the DRBD18-stabilized RNAs of 756 nucleotides. Remarkably, the average 3′ UTR length in DRBD18-destabilized RNAs is 1830 nucleotides, three to four times as long as the genome average, suggesting that DRBD18 acts combinatorially with other RBPs in the regulation of this mRNA population. The presence of long 3′ UTRs was even more striking when we focused on those RNAs that were highly DRBD18-destabilized (those with a >6-fold increase in abundance upon DRBD18 knockdown). Analysis of these RNAs revealed an average 3′ UTR length of 3222 nucleotides. Further probing also revealed that this RNA population had an average of 24 polyadenylation sites (PASs), more than double the genome-wide average (51). In addition, highly DRBD18-destabilized transcripts exhibited remarkable variability in 3′ UTR length, with the average difference in length between the shortest and longest identified 3′ UTR being 4857 nucleotides. Using this stringent set of highly affected RNAs, we next searched for common sequence motifs using MEME (52) As seen in Figure 4B, highly DRBD18-destabilized RNAs share a 12-nucleotide motif comprised of primarily adenosines with an invariant guanosine in the fourth position. This motif occurs in all analyzed 3′ UTRs at least once, with an average occurrence of four times per 3′ UTR. Because PASs are more likely to occur adjacent to adenosines (51), and these 3′ UTRs have an increased number of PASs, it is possible that DRBD18 plays a role in polyadenylation of a certain set of RNAs. We also analyzed 3′ UTRs of the most highly DRBD18-stabilized RNAs (those with a >2.5-fold change in abundance upon DRBD18 knockdown) using the same parameters. In 3′ UTRs of stabilized RNAs, we identified a different, 7-nucleotide motif, comprised primarily of cytosines and adenosines. Thus, mRNAs that are destabilized by DRBD18 have long 3′ UTRs, and DRBD18-destabilized and DRBD18-stablized mRNAs harbor unique 3′ UTR sequence motifs.
Arginine methylation of DRBD18 does not impact its subcellular localization
Having shown that DRBD18 affects the abundance of hundreds of mRNAs, we next wanted to determine how arginine methylation impacts the protein's function. To address this question, we created two methylmutants. In the first, DRBD18(R→K), we mutated the three identified methylarginines to lysines, thereby creating a hypomethylated DRBD18 while preserving the residues’ positive charge. In the second methylmutant, DRBD18(R→F), we replaced the methylarginines with phenylalanine, which studies have shown can act as a methylarginine mimic (53–55). Because the methylarginines in DRBD18 are located in a long unstructured region of the protein (Figure 1), it is unlikely that mutations in this region will dramatically alter DRBD18 structure. We generated three cell lines in which either wild type DRBD18(WT) or one of the two methylmutants were expressed as RNAi-resistant Myc-His-TAP (MHT) fusions, in the DRBD18 RNAi background (Figure 5A). Western blot analysis showed that endogenous DRBD18 was similarly depleted in all cell lines and that the larger MHT-DRBD18 variants were expressed to approximately equal levels (Figure 5B). Addition of the MHT tag to DRBD18 variants allowed us to specifically compare properties of WT and methylmutant proteins without interference from residual endogenous protein. To begin to understand the effects of methylation on DRBD18, we asked whether its nucleocytoplasmic localization was altered by methylation status, as has been reported for several other methylated RBPs (56–58). Cells harboring MHT-DRBD18(WT) or its mutant variants were subjected to subcellular fractionation and anti-myc western blot (Figure 6A). These analyses demonstrated that MHT-tagged WT, hypomethylated and methylmimic DRBD18 are all primarily cytoplasmic, like endogenous DRBD18 (Figure 2C). To validate the biochemical fractionation studies, we performed indirect immunoflouresence on cells harboring MHT-DRBD18(WT) or its mutant variants using anti-myc antibodies. As seen in Figure 6B, all MHT-DRBD18 variants are both dispersed throughout the cytoplasm and exhibit a prominent enrichment at the nuclear periphery. These data demonstrate that neither the overexpression of the MHT-tagged protein, nor methylation status of arginines 199, 202, or 204, alters the cytoplasmic localization of DRBD18.
Figure 5.
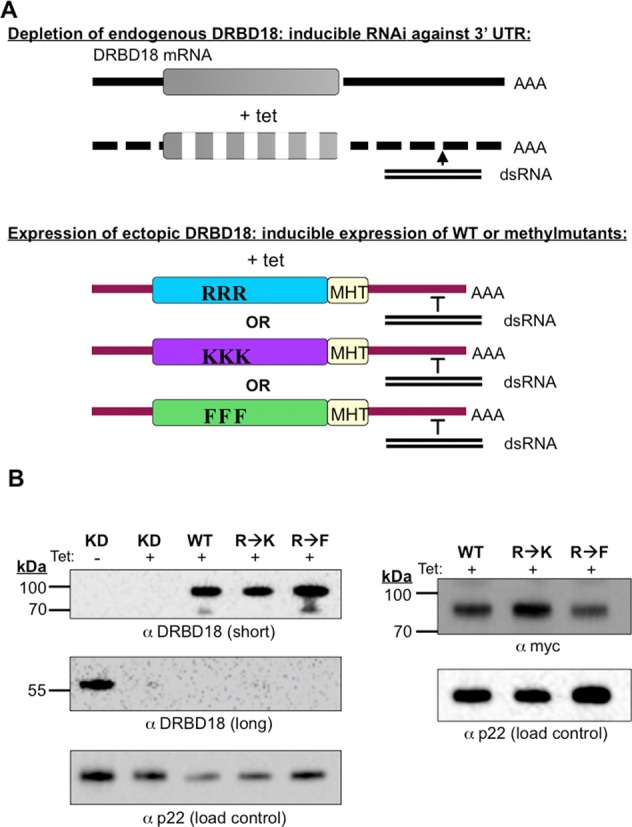
Generation of DRBD18 RNAi cells complemented with MHT-tagged DRBD18(WT), DRBD18(R→K), or DRBD18(R→F). (A) Schematic of the method used to generate complemented cell lines. Addition of tet produces dsRNA targeting the DRBD18 3′ UTR and leads to depletion of the endogenous DRBD18 transcript. Simultaneous expression of the ectopic DRBD18 constructs is induced by tet, and these bear 3′ UTRs that are resistant to RNAi. RRR indicates MHT-DRBD18(WT), KKK indicates MHT-DRBD18(R→K) and FFF indicates MHT-DRBD18(R→F) mutants. (B) Validation of cell lines. Complemented cells were harvested two days post-induction, lysed in SDS-PAGE sample buffer and resolved via 10% SDS-PAGE. A western blot with anti-DRBD18 antibodies (left) shows that tagged DRBD18 protein (expected size ∼79 kDa) is expressed at similar levels in all cell lines (short exposure), and endogenous DRBD18 (expected size ∼52 kDa) (long exposure) is similarly decreased in all cell lines. A western blot targeting the myc-tag on the exogenous proteins (right) confirms similar expression levels. The p22 protein serves as a loading control.
Figure 6.
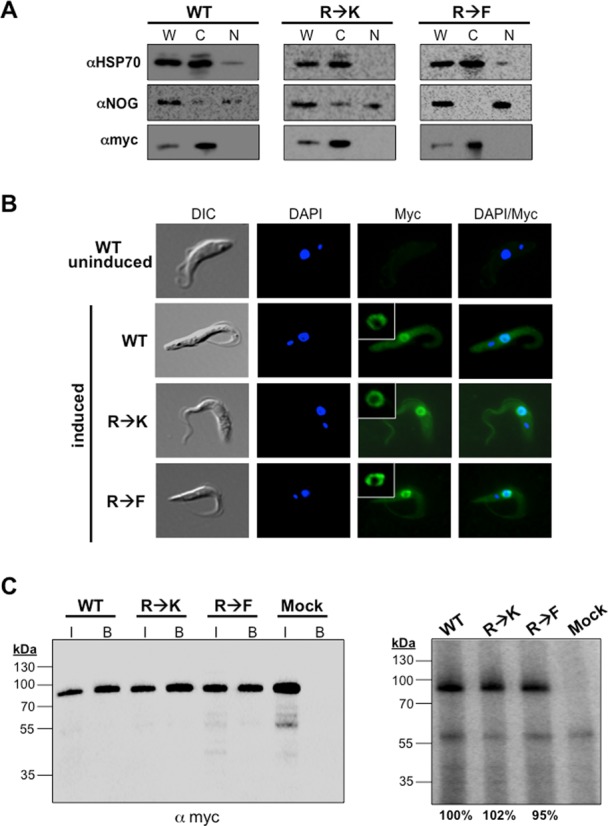
Effects of hypomethylation and methylmimic on subcellular localization and RNA binding capacity of DRBD18. (A) Biochemical fractionation and western blot analysis of MHT-DRBD18(WT), MHT-DRBD18(R→K) and MHT-DRBD18(R→F) expressing cell lines. Cells were harvested two days post-induction and 1 × 106 cellular equivalents of cytoplasmic and nuclear fractions were separated via 10% SDS-PAGE. HSP70 and NOG are cytoplasmic and nuclear markers, respectively. MHT-DRBD18 constructs were visualized using anti-myc antibody. W, whole cell; C, cytoplasmic fraction; N, nuclear fraction. (B) Indirect immunofluorescence of uninduced MHT-DRBD18(WT) expressing cells and MHT-DRBD18(WT), MHT-DRBD18(R→K), and MHT-DRBD18(R→F) expressing cell lines 2 days post-induction with tet using anti-myc antibody. Nuclei and kinetoplasts were stained with DAPI, and merged signals (myc/DAPI) are shown on the right. DIC, differential interference contrast. Inset shows higher magnification image of perinuclear labeling. (C) CLIP analysis of cells expressing MHT-DRBD18 variants. Cells expressing MHT-DRBD18 variants were UV crosslinked at 254 nm and proteins were precipitated using anti-myc conjugated resin. Cell lysate pooled from all three samples and incubated with uncoated beads served as a negative control sample (mock). Left panel shows anti-myc western blot analysis of input samples (7.5 × 105 cell equivalents). Also shown are immunoprecipitated (bound) MHT-DRDB18 variant proteins following in vivo crosslinking and immunoprecipitation and just prior to labeling with γ32P-ATP by polynucleotide kinase. These bound samples were used for the analyses shown in the right panel. I, input, B, bound samples. Right panel shows phosphorimage analysis of bound MHT-DRBD18 variant RNPs. Values below the image represent the average signals from three replicate experiments.
Arginine methylation of DRBD18 does not significantly reduce its RNA binding capacity
The presence of two RBDs implicates DRBD18 in RNA binding. Thus, we next asked if DRBD18 binds RNA in vivo, and if so, whether this parameter is modulated by arginine methylation. To this end, we performed in vivo cross-linking immunoprecipitation (CLIP) (41) on cells expressing MHT-DRBD18(WT), MHT-DRBD18(R→K), or MHT-DRBD18(R→F) to assess total in vivo RNA binding capacity. Cells were UV irradiated and DRBD18-containing ribonucleoprotein (RNP) complexes were immunoprecipitated with anti-myc antibody. The immunoprecipitated sample was subjected to RNase digestion, followed by dephosphorylation and 5′-end-labeling of the bound RNAs using 32P. We next normalized samples from MHT-DRBD18(WT), MHT-DRBD18(R→K) and MHT-DRBD18(R→F) immunoprecipitates based on the amount of DRBD18 protein in each sample as indicated by anti-myc western blot. Proteins were then resolved via SDS-PAGE and visualized by phosphorimaging to determine the overall capacity of each DRBD18 variant to bind RNA in vivo. As seen in Figure 6C (right), in the MHT-DRBD18(WT) sample we observe a band of radiolabeled protein at ∼80 kDa that corresponds to the molecular weight of MHT-DRBD18(WT) seen in the anti-myc western blot (Figure 6C, left). These data indicate that DRBD18 binds RNA in vivo. Both hypomethylated MHT-DRBD18(R→K) and methylmimic MHT-DRBD18(R→F) also bind RNA, as indicated by the radiolabeled bands similar to that observed with WT protein. Quantification from three replicates indicates that there is no appreciable difference in RNA binding affinity between MHT-DRBD18(WT) and its methylmutants. Together, these data demonstrate that DRBD18 binds RNA in vivo, as predicted from its sequence, irrespective of its methylation status.
Arginine methylation of DRBD18 impacts its protein–protein interactions
Many RBPs do not function alone but rather act in the context of larger RNP complexes. Therefore, to better understand how DRBD18 functions in T. brucei and how arginine methylation modulates its functions, we purified DRBD18-containing RNPs by tandem affinity chromatography and identified associated proteins by mass spectrometry analysis. To obtain the largest set of DRBD18-associated proteins, we began by identifying proteins that co-purify with MHT-DRBD18(WT) from cell extracts in which RNA was left intact. Consistent with the RNAseq data in which depletion of DRBD18 causes both up- and down-regulation of specific sets of mRNAs, MHT-DRBD18(WT) associated with proteins implicated in both RNA stabilization and decay (Supplementary Table S2). For example, MHT-DRBD18(WT) co-purified with ZC3H39 and DRBD3, both of which have been implicated in RNA regulation during the trypanosome stress response (59,60). MHT-DRBD18(WT) also associated with four components of the CAF1-NOT complex, which comprises the primary mRNA degradation complex in trypanosomes (61). From these data, we conclude that DRBD18 associates with multiple RNP complexes that presumably contribute to the effects of DRBD18 on RNA metabolism.
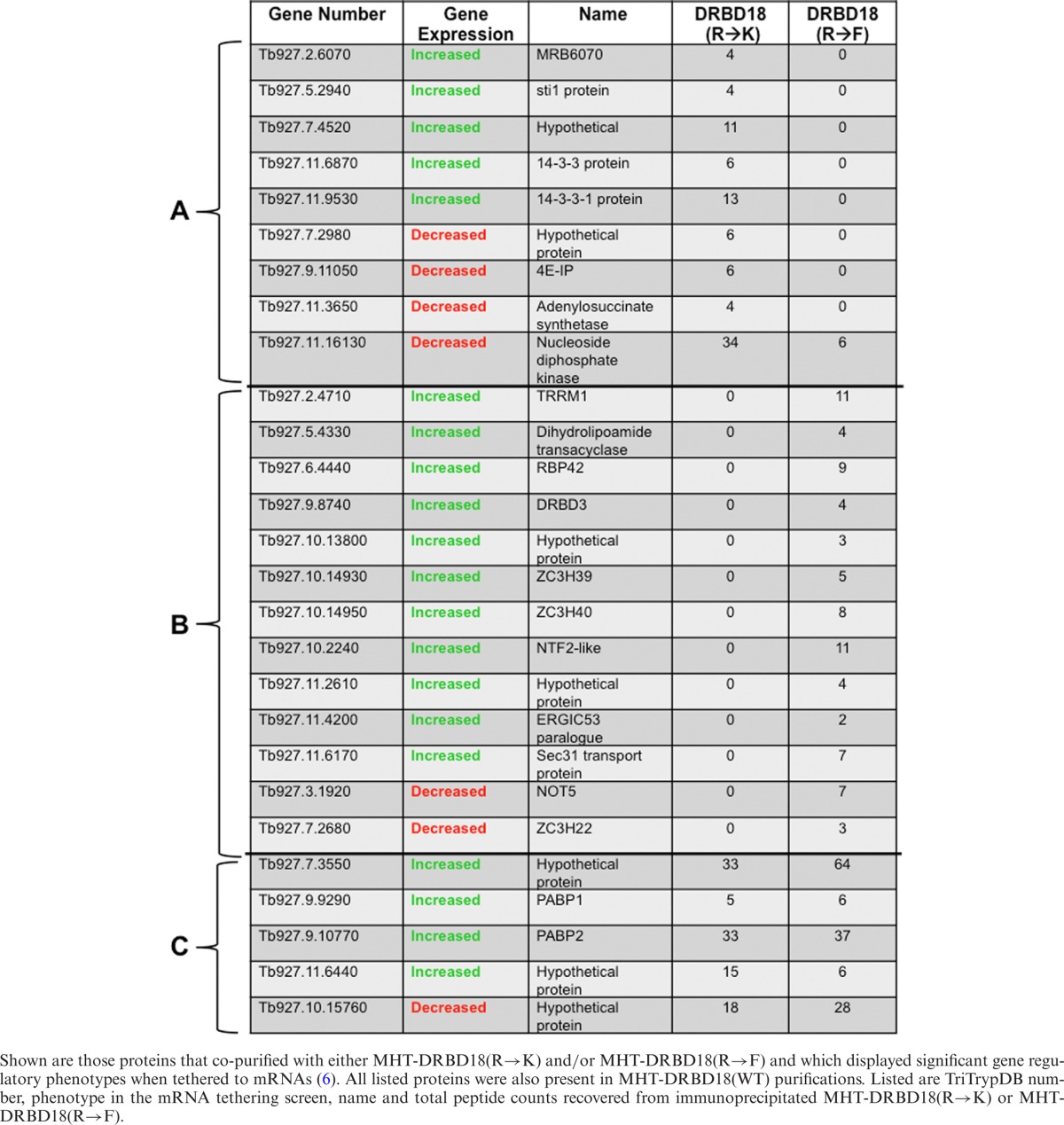
Arginine methylation can influence protein–protein interactions with examples of both positive and negative impacts (55,62,63). Having identified numerous proteins with which DRBD18 associates, we next sought to determine how methylation of DRBD18 affects these interactions. We treated cell extracts with RNases prior to tandem affinity purification allowing us to focus on direct protein interactions of hypomethylated and methylmimic DRBD18. In all, we identified 115 proteins that associated with hypomethylated MHT-DRBD18(R→K), but not with the methylmimic MHT-DRBD18(R→F). Conversely, a different set of 131 proteins were associated with MHT-DRBD18(R→F) but were absent in the pool of MHT-DRBD18(R→K) interacting proteins. To begin to decipher how these DRBD18-associated proteins might contribute to RNA fate, we cross-referenced proteins that were exclusively associated with either MHT-DRBD18(R→K) or MHT-DRBD18(R→F) with a set of proteins that was shown in a recent T. brucei global mRNA tethering study to increase or decrease expression of an mRNA to which they are artificially bound (6). Table 2 shows those proteins that were recovered in purifications of MHT-DRBD18(WT) and at least one of the methylmutant variants, and which presented an increased or decreased gene expression phenotype in the global tethering study. Of the 27 proteins fitting these criteria, only five were associated with both MHT-DRBD18(R→F) and MHT-DRBD18(R→K) to similar levels based on peptide counts, including the poly(A) binding proteins PABP1 and PABP2 (Table 2, section C). Thirteen proteins were exclusively complexed with MHT-DRBD18(R→F) (Table 2, section B), and the majority of these (11 of 13) facilitated increased gene expression when tethered to reporter RNA (6). The remaining nine proteins preferentially associated with MHT-DRBD18(R→K). These included approximately equal numbers of proteins reported to increase and decrease gene expression. Interestingly, we note that 4E-IP (Tb927.9.11050) is among the proteins that complexes exclusively with MHT-DRBD18(R→K), and this protein was identified as one of the most significant suppressors of gene expression by mRNA tethering (6). Collectively, these data demonstrate that arginine methylation significantly impacts the protein–protein interactions of DRBD18, shaping the composition of DRBD18-containing RNPs and likely its impact on mRNA fate.
Table 2. Subset of proteins associated with MHT-DRBD18(R→K) and/or MHT-DRBD18(R→F).
Hypomethylated and methylmimic DRBD18 mediate opposite effects on RNA levels
Having shown that arginine methylation impacts protein–protein interactions of DRBD18, we next wanted to ask how these biochemical differences translate into changes in the ability of DRBD18 to modulate mRNA levels. To this end, we used a similar strategy to that described above, depleting DRBD18 using RNAi against its 3′ UTR, and complementing with WT or methylmutant proteins bearing a different, RNAi resistant 3′ UTR (Figure 5A). Growth assays with cells complemented with the MHT-DRBD18(WT) fusion showed only a modest rescue of cell growth (data not shown), suggesting that the MHT tag partially inhibits DRBD18 function. For analysis of cellular mRNA levels we, therefore, generated complemented cell lines using untagged DRDB18. As shown in Figure 7A, untagged DRBD18(WT) fully rescued the knockdown growth phenotype (compare growth of complemented cells to knockdowns in Figure 2B). These data confirm that the drastic growth defect exhibited in the DRBD18 knockdown cell line (Figure 2B) is due solely to the loss of DRBD18 rather than to off-target affects of the RNAi. We next created complemented cell lines expressing untagged versions of either hypomethylated DRBD18(R→K) or methylmimic DRBD(R→F) mutants. WT and methylmutant DRBD18 were expressed to similar levels (Figure 7B). However, the endogenous DRBD18 mRNA levels in these cell lines were not depleted to exactly the same degree as in the DRBD18(WT) complemented cells. Endogenous DRBD18 mRNA levels were depleted to 18.5% of wild type levels in the DRBD18(WT) complemented cells, whereas endogenous levels in DRBD18(R→K) and DRBD(R→F) complemented cells were depleted only to 37.2% and 36.9%, respectively. Nonetheless, cell lines complemented with untagged DRBD18 methylmutants displayed small, but reproducible growth phenotypes (data not shown).
Figure 7.
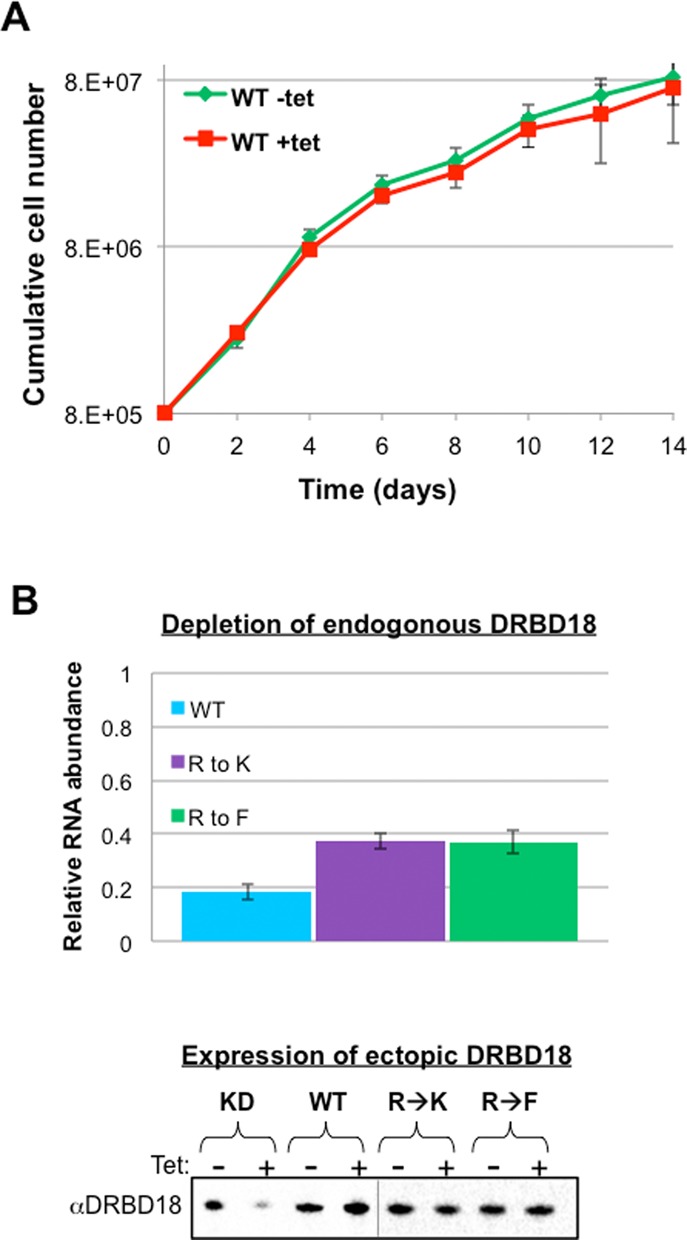
Generation of DRBD18 knockdown cell lines complemented with untagged DRBD18 variants. (A) Expression of untagged DRBD18 restores cell growth. Green line indicates cells in which neither DRBD18 RNA nor expression of exogenous DRBD18 has been induced. Red line indicates cells in which endogenous DRBD18 is repressed and untagged wild type protein is expressed. (B) DRBD18 RNA and protein levels in cell lines complemented with untagged DRBD18. Top panel shows qRT-PCR analysis of endogenous DRBD18 transcript levels normalized against 18S rRNA. Error bars depict standard deviation from 3–6 determinations. Bottom panel shows anti-DRBD18 western blot analysis of cells expressing ectopic DRBD18 constructs. Complemented cells were harvested 2 days post-induction, lysed in SDS-PAGE sample buffer, and resolved via 10% SDS-PAGE.
To begin to understand if methylation differentially impacts the RNA populations that are stabilized or destabilized by DRBD18, we first validated a subset of our RNAseq data using qRT-PCR against specific transcripts whose abundances were increased or decreased in DRBD18 knockdown cells. To ensure a representative sampling, we selected three DRBD18-destabilized and three DRBD18-stabilized RNAs, each one representing a highly enriched GO term. We also confirmed that transcripts whose abundances were unchanged in the RNAseq analysis were unaffected in our qRT-PCR measurements (data not shown). qRT-PCR analysis of three DRBD18-stabilized RNAs showed that the abundance of these transcripts was reduced to between 0.61 and 0.83 of uninduced levels when DRBD18 was depleted (Figure 8A, red). Likewise, analysis of three DRBD18-destabilized RNAs showed that each of these RNAs increase in abundance between 2.1 and 5.4-fold when DRBD18 is depleted (Figure 8B, red). Therefore, qRT-PCR analysis confirmed the trends observed in the RNAseq study for the subset of RNAs tested. We next interrogated the ability of DRBD18(WT) to restore these transcripts to levels observed in uninduced RNAi cells. Consistent with its ability to complement growth, complementation of the knockdown with DRDB18(WT) restored all tested RNAs to levels comparable to those seen in uninduced cells (Figure 8A and B, blue). These data confirm that DRBD18 mediates both positive and negative impacts on RNA abundance.
Figure 8.
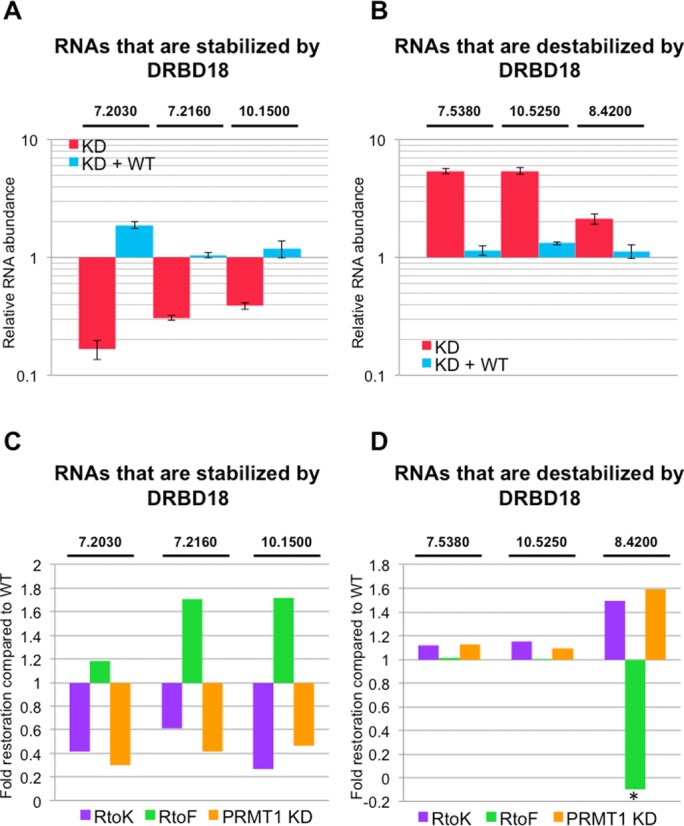
Effects of DRBD18 methylation on target RNA abundances. (A) qRT-PCR analysis of three selected RNAs in DRBD18 repressed cells (KD; red bars) and DRBD18(WT) complemented cells (KD + WT; blue bars). These RNAs were among those whose abundance decreased upon DRBD18 depletion in RNAseq, and are thus normally stabilized by DRBD18. The specific transcripts that were tested are indicated by the last five or six digits of their TriTrypDB number above the graph. RNA abundances were normalized against 18S rRNA and are plotted relative to abundances in uninduced cells. Average and standard deviation from at least three replicates is shown. (B) qRT-PCR analysis as in A, except the three RNAs were among those whose abundance increased upon DRBD18 depletion in RNAseq, and are thus normally destabilized by DRBD18. (C) qRT-PCR analysis of the same transcripts as in panel A in DRBD18(R→K) (purple) or DRBD18(R→F) (green) complemented cells, and in TbPRMT1 knockdown cells (orange). Relative RNA abundances (induced/uninduced) are plotted to show the fold restoration of RNA levels compared that in DRBD(WT) complemented cells. (D) qRT-PCR analysis as in C, except measuring the same transcripts as in panel B. Asterisk, the value is less than one because the transcript level was depleted to a level lower than in the DRBD18 knockdown alone.
We next probed the effect of arginine methylation on the ability of DRBD18 to modulate these different RNA populations. To begin, we assessed the ability of methylmimic DRBD18(R→F) and hypomethylated DRBD18(R→K) to stabilize RNAs by repeating the targeted qRT-PCR analyses in Figure 8A in each complemented cell line. We analyzed the ability of each DRBD18 methylmutant to restore transcript levels relative to that of DRBD18(WT) complemented cells, and the results are presented as ‘Fold restoration compared to wild type’ (Figure 8C). A value of one indicates the methylmutant stabilizes the transcript as well as DRBD18(WT), a value less than one indicates that the mutant stabilizes RNA less efficiently that DRBD18(WT), while a value greater than one demonstrates that the methylmutant protein exacerbates the stabilization of a given mRNA. As seen in Figure 8C (green bars), for those transcripts that are stabilized by DRBD18 (Tb927.7.2030, Tb927.7.2160, Tb927.10.1500), methylmimic DRBD(R→F) stabilizes these RNAs to the same extent or better than DRBD18(WT). Conversely, hypomethylated DRBD18(R→K) is unable to fully stabilize these RNAs (Figure 8C, purple bars). These data suggest that DRBD18 arginine methylation is required for the protein's full capacity for mRNA stabilization.
To this point, we have used DRBD18 proteins mutated at the three arginine residues to approximate methylated or hypomethylated protein. To ensure that the observed phenotype was due to the change in DRBD18 methylation status rather than mutation of its amino acid sequence, we repeated the qRT-PCR analysis in cells that were wild type with respect to DRBD18, but that were depleted of TbPRMT1. TbPRMT1 is the major T. brucei PRMT in vivo (27), and several TbPRMT1 peptides were identified by mass spectrometry of tandem affinity purified MHT-DRBD18 (Supplementary Table S2). Thus, we hypothesized that knockdown of TbPRMT1 would render the wild type pool of DRBD18 hypomethylated and recapitulate the phenotype of the DRBD18(R→K) mutant. We isolated RNA from previously described uninduced and induced TbPRMT1 knockdown cells expressing 26% of wild type levels of TbPRMT1 (31) and analyzed the levels of DRBD18 stabilized mRNAs by qRT-PCR. As shown in Figure 8C (orange bars), the transcript levels in cells in which TbPRMT1 was depleted were very similar to those in DRBD18 knockdown cells complemented with DRBD18(R→K) (Figure 8C, purple bars). Thus, DRBD18 that is rendered hypomethylated by either (R→K) mutation or by TbPRMT1 knockdown is unable to efficiently stabilize these mRNAs, whereas methylmimic DRBD18 stabilizes as well or better than the wild type protein. Based on these data, we conclude that arginine methylation of DRBD18 is required for efficient stabilization of a subset of mRNAs.
Having shown that DRBD18 methylation positively impacts the protein's ability to stabilize a subset of mRNAs, we next asked whether arginine methylation impacts the ability of DRBD18 to destabilize mRNAs. We isolated RNA from DRBD18 knockdown cells complemented with WT or methylmutant proteins, and performed qRT-PCR analysis of a subset of transcripts that are destabilized by DRBD18. Data are again presented as ‘Fold restoration compared to wild type’ such that a value less than one indicates an impaired ability to destabilize a given RNA and a value greater than one indicates enhanced destabilization compared to wild type. For two of the three transcripts tested (Tb927.7.5380 and Tb927.10.5250), methylation status had little effect of the ability of DRBD18 to rescue wild type RNA levels (Figure 8D, purple and green bars). However, for a third mRNA, we observed a phenotype opposite to that seen with the stabilized pool of transcripts. When we analyzed levels of the Tb927.8.4200 transcript, we found that methylmimic DRBD18(R→F) was significantly impaired in its ability to destabilize the RNA (Figure 8D, green bars), whereas DRBD18 hypomethylated by either (R→K) mutation or TbPRMT1 knockdown exhibited destabilization efficiency greater than that of DRBD18(WT) (Figure 8D, purple and orange bars). These data demonstrate that arginine methylation impairs the ability of DRBD18 to destabilize a subset of mRNAs. Collectively, our results show that arginine methylation differentially impacts the distinct functions of DRBD18, facilitating its mRNA stabilizing function while, at least for a subset of transcripts, inhibiting its ability to destabilize mRNAs.
Thus far, we have shown that DRBD18 functions in both stabilization and destabilization of mRNA, with methylation increasing its stabilization affects. We hypothesize that DRBD18 acts directly on both populations of RNAs. To confirm that the phenotypes we observe are not due to secondary affects from modulating the abundances of known RBPs, we immunoprecipitated in vivo cross-linked MHT-DRBD18(WT) and methylmutant-containing RNPs and analyzed specific associated RNAs using qRT-PCR. As shown in Figure 9, both a DRBD18-stabilized transcript (7.2030) and a DRBD18-destabilized transcript (8.4200) are enriched 3- and 4-fold, respectively, in MHT-DRBD18(WT) immunoprecipitates compared to input levels. From these data, we conclude that MHT-DRBD18(WT) associates with both stabilized and destabilized RNAs, and likely functions on both populations directly. In addition, we observed a preference for certain RNAs based on DRBD18 methylation status. While MHT-DRBD18(R→K) exhibited a similar level of enrichment of the 7.2030 transcript compared to MHT-DRBD18(WT)WT, MHT-DRBD18(R→F) displayed a fold enrichment of ∼7.4-fold, more than double the enrichment seen with MHT-DRBD18(R→K). Strikingly, analysis of the destabilized transcript, 8.4200, displayed an opposite phenotype. The 8.4200 transcript is equally enriched in MHT-DRDB18(WT) and MHT-DRBD18(R→F) immunoprecipitates, but the enrichment is elevated in MHT-DRBD18(R→K) immunoprecipitates. Therefore, these data suggest that methylation partially modulates DRBD18 RNA binding preference in a manner consistent with the effects of methylation on mRNA stability. Specifically, methylmimic DRBD18 is enriched for transcripts that are stabilized by DRBD18 binding, and hypomethylated DRBD18 preferentially associates with transcripts that are destabilized by DRBD18 binding.
Figure 9.
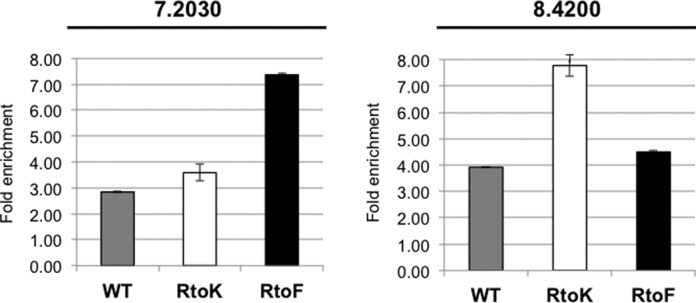
Enrichment of destabilized and stabilized mRNAs in different DRBD18-containing RNPs. qRT-PCR analysis of two selected RNAs in MHT-DRBD18(WT) (grey bars), MHT-DRBD18(R→K) (white bars) and MHT-DRBD18(R→F) (black bars) immunoprecipitations. The specific transcripts that were tested are indicated by the last five digits of their TriTrypDB numbers above each graph. RNA abundances were normalized against 18S rRNA and are plotted relative to input levels. Average and standard deviation from three replicates is shown.
DISCUSSION
RBPs are key regulators of gene expression and development in kinetoplastid parasites (2,3). However, little is known regarding how the functions of RBPs are expanded or regulated by posttranslational modifications. Here, we report the characterization of a previously unstudied T. brucei RNA binding protein, DRBD18, and demonstrate that its functions are dramatically modulated by arginine methylation. In agreement with a global screen identifying proteins whose depletion results in a change in parasite fitness (64), knockdown of DRBD18 leads to PF cell death. Quantification of DRBD18 levels indicates it is a relatively abundant RBP. In accordance with its abundant and essential nature, depletion of DRBD18 caused extensive changes in the T. brucei transcriptome. RNAseq analysis revealed that approximately one tenth of mRNAs were significantly stabilized or destabilized 24 h following knockdown of DRBD18. Complementation studies demonstrated that DRBD18(WT) is fully functional in restoring mRNAs to uninduced levels. In contrast, complementation of DRBD18 knockdown cell lines with DRBD18 methylmutants differentially affected distinct pools of mRNAs (Figures 7 and 8). Hypomethylated DRBD18 was competent for RNA destabilization but unable to efficiently stabilize target RNAs. Conversely, methylmimic DRBD18 very efficiently stabilized target RNAs, but was partially crippled for mRNA destabilization. Hypomethylated and methylmimic DRBD18 co-purified with a distinct set of proteins that likely contribute to their differential functions. Moreover, differentially methylated DRBD18 populations exhibited distinct RNA binding preferences consistent with their effects on mRNA fate. Finally, the protein arginine methyltransferase, TbPRMT1, was implicated in DRBD18 methylation and function since it co-purified with DRBD18, and TbPRMT1 depletion recapitulated the effects of hypomethylated DRBD18 on target mRNA abundance. Together, our data provide a compelling example of functional diversification of an RBP due to posttranslational arginine methylation.
Bioinformatic analysis of transcripts whose abundances were affected by knockdown of DRBD18 revealed a wide array of biological processes. While most functional GO terms were equally represented in both up- and down-regulated transcripts, and both included many hypothetical proteins, there were some classes of mRNAs that were differentially impacted by DRBD18 depletion. For example, of the 82 mRNAs encoding kinases/phosphates whose abundance was significantly altered upon DRBD18 depletion, 75 were up-regulated while only six were down-regulated. Interestingly, six of the mRNAs that were up-regulated by DRBD18 RNAi represent nearly 50% of the kinases/phosphatases shown to drive stumpy form formation in a genome-wide analysis of T. brucei quorum sensing (8). For example, mRNA encoding the YAK protein kinase (Tb927.10.15020) is up-regulated in PF T. brucei almost 4-fold upon DRBD18 depletion, while the protein kinase A regulatory subunit transcript (Tb927.11.4610) is up-regulated 2.2-fold. This may indicate that one role for DRBD18 in PF cells is to down-regulate these kinase genes upon differentiation to PF. Additionally, the mRNA encoding RDK1 kinase (Tb927.11.14070) is upregulated 6.5-fold upon DRBD18 depletion. RDK1 was identified in a kinome-wide RNAi screen as a repressor of BF to PF differentiation (65). The ability of DRBD18 to keep RDK1 mRNA levels low also supports the notion that one function of DRBD18 is maintenance of the PF state.
DRBD18 knockdown also caused up-regulation of numerous classes of RBPs (Table 1). For example, four RBPs were among the top ten most highly up-regulated mRNAs upon DRBD18 depletion: ZC3H32 (Tb927.10.5250), RBP3 (Tb927.11.530), Puf4 (Tb927.6.820) and DRBD12 (Tb927.5380). DRBD12, the only one of these that has been characterized, destabilizes transcripts with AU-rich elements (66). These findings suggest that the wide-ranging effects of DRBD18 on the PF transcriptome may be amplified by its modulation of other RBPs. We performed our RNAseq analysis 24 h post-induction to reduce the likelihood of secondary effects. Nevertheless, we cannot rule out that some of the alterations in transcript abundance are secondary effects caused by RBPs that are themselves modulated by DRBD18. However, the fact that DRBD18-meditated mRNA destabilization and DRBD18-meditated mRNA stabilization respond differently to the methylation status of the protein (Figure 8) strongly suggests that one is not a secondary effect of the other. In addition, we demonstrated that MHT-DRBD18(WT) can be cross-linked in vivo to both DRBD18-stabilized and DRBD18-destabilized mRNAs (Figure 9), consistent with direct effects on both mRNA populations.
DRBD18 displayed a cytoplasmic localization with a pronounced perinuclear staining pattern, irrespective of its methylation status. Mass spectrometry studies showed that MHT-DRBD18(WT) associates with five different members of the nuclear pore complex (nucleoporins) (67,68), some of which presumably extend into the cytoplasm as observed with nucleoporins in other eukaryotes (69). Three of these DRBD18-associated nucleoporins are hypothesized to participate in direct transport of cargoes into and out of the nucleus. Interestingly, the murine homologue of one of these proteins, TbNup158, has been shown to mediate mRNA transport (70). Furthermore, MHT-DRBD18(WT) also associates with the proposed mRNA transport protein Tb927.10.2240. Thus, the distinct accumulation of DRBD18 around the nucleus could be caused by key DRBD18 protein–protein interactions that position it in an ideal location to target mRNAs as they exit the nucleus.
Complementation of DRBD18 knockdown cells with DRBD18(WT), hypomethylated DRBD18(R→K), or methylmimic DRBD18(R→F) allowed us to develop a model for DRBD18 protein function and the role of arginine methylation in regulating its functions. As seen in Figure 10, DRBD18(WT) acts in two distinct ways on two separate mRNA populations, defined by distinct cis-acting sequences (indicated by red or green patches). DRBD18(WT) promotes decay of one set of mRNAs, while stabilizing another set. These opposing effects are evident in the RNAseq analysis of DRBD18 depleted cells, the qRT-PCR analysis of DRBD18(WT) complemented cells, as well as in the similar enrichment levels of stabilized and destabilized transcripts in MHT-DRDB18(WT) immunoprecipitations. Mass spectrometry identified at least three sites of arginine methylation in DRBD18(WT); however, the extent to which DRBD18(WT) is methylated in the cell remains unknown. That is to say, DRBD18(WT) must be thought of as a mixed population containing both methylated and unmethylated protein. When the population of DRBD18 is forced to resemble a more methylated state, as seen with the DRBD18(R→F) mutant, DRBD18 can stabilize RNA more efficiently than the WT mixed population (Figure 10, right). Conversely, when the protein is forced into a hypomethylated state, as seen with the DRBD18(R→K) mutant or TbPRMT1 knockdown, the protein loses its ability to efficiently stabilize RNAs, but can efficiently destabilize mRNAs (Figure 10, left). mRNA stabilization appears to be very sensitive to DRBD18 methylation status (Figure 8C), while only a subset of DRBD18-destabilized mRNAs tested were responsive to methylation status (Figure 8D). In this case, destabilization required hypomethylated DRBD18, and methylmimic protein was ineffective at mRNA degradation. Collectively, our data strongly support a model in which methylation acts as a switch for DRBD18, directing it to act on one subset of RNAs in a destabilizing manner, and on another set of RNAs as a stabilizing factor.
Figure 10.
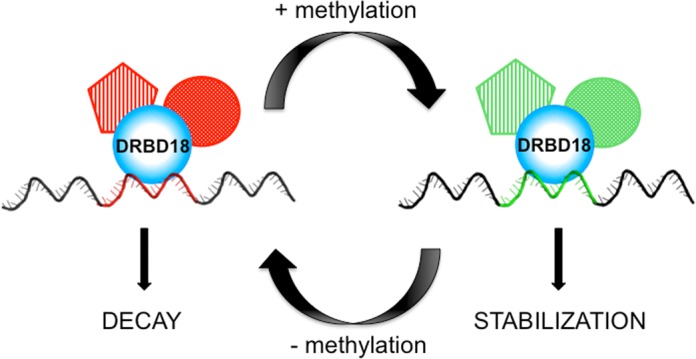
Model of DRBD18 function and the affects of arginine methylation. See text for details.
What are the biochemical mechanisms by which arginine methylation impacts DRBD18 function? Arginine methylation likely affects DRBD18 function by modulating its interactions with effector proteins (Figure 10). Indeed, we identified dozens of proteins that associate preferentially with either hypomethylated or methylmimic DRBD18. Of the thirteen proteins that preferentially interact with DRBD18(R→F) and which displayed a gene regulatory function in mRNA tethering studies, eleven lead to increased gene expression. Interestingly, methylmimic DRBD18 bound NOT5 (Tb927.3.1920), whereas hypomethylated DRBD18 did not (Table 2). NOT5 is a component of the CAF1/NOT complex, which is the major deadenylation complex in trypanosomes that promotes RNA decay (61). Thus, it is curious that a component of the primary RNA decay machinery is enriched in purifications of the DRBD18 mutant form that is most competent for mRNA stabilization. One possible scenario is that methylation of DRBD18 sequesters NOT5 from the CAF1/NOT complex in a manner that renders the CAF1/NOT complex decay incompetent. A different set of proteins preferentially associated with hypomethylated DRBD18. Of particular interest in this regard is 4E-IP, which was one of the most significant gene expression repressors in mRNA tethering studies (6). The 4E-IP protein is believed to interact with the translation initiation factor, eIF41, to block translation (71). Thus, it will be of interest to determine if some of the mRNA destabilizing affects we see when DRBD18 is hypomethylated are due to the protein's association with 4E-IP, and whether DRBD18 can directly affect mRNA translation.
Arginine methylation also appears to play a role in specifying the RNA binding preferences of DRBD18. In agreement with our model proposing that DRBD18 methylation leads to stabilization of specific RNAs, qRT-PCR analysis of in vivo CLIP samples demonstrates that DRBD18(R→F) preferentially associates with stabilized RNA, while hypomethylated DRBD18(R→K) has a stronger enrichment of a destabilized RNA (Figure 10). Methylation of specific arginine residues has been shown to modulate RNA binding for certain RBPs. For example, arginine methylation of the neuronal translation factor, FMRP, inhibits FMRP binding to one RNA while having no effect on its binding a different target RNA (26). Also, TbPRMT1-mediated methylation of the T. brucei mitochondrial protein RBP16 decreases its association with guide RNA while simultaneously augmenting its association with mRNA (32,33). In general, the mechanisms by which arginine methylation modulates RNA binding specificity are poorly understood. With regard to DRBD18, the presence of extraordinarily long 3′ UTRs on target mRNAs suggests that the protein acts combinatorially with other RBPs in regulating mRNA fate. These combinatorial interactions may be influenced by the methylation status of the protein, thereby contributing to differing RNA binding specificities of methylation and unmethylated DRBD18. Future studies will be aimed at characterizing DRBD18 RNPs, the contributions of their component proteins to control mRNA stability, and the role of arginine methylation in modulating specific protein–protein interactions.
In summary, we have demonstrated that the T. brucei RBP, DRBD18, has distinct effects on the stabilities of different sets of mRNAs. Arginine methylation acts as a switch, converting DRBD18 from a more destabilization-competent to a more stabilization-competent form. Our global arginine methylome studies (15,16), as well as similar studies in mammalian cells (20), identified RBPs as a major category of arginine methylated proteins. Thus, future studies aimed at deciphering the molecular mechanism of the methylation switch on DRBD18 may reveal a more general model for posttranscriptional gene regulation in both kinetoplastid parasites and higher organisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Marilyn Parsons for anti-NOG1 antibodies and Jay Bangs for anti-HSP70 antibodies. We are also grateful to Dr. Natalie McAdams for critical reading of the manuscript.
FUNDING
National Institutes of Health [RO1 AI060260 to L.K.R., F32AI100350 to K.L., R01 DE024523 to Y.S.]; National Science Foundation [DBI1322212 to Y.S.]; Center of Protein Therapeutics Industrial Award (NIH) [U54HD071594, AG048388, HL103411 and AI060260 to J.Q.]; American Heart Association (AHA) [12SDG9450036 to J.Q]. Funding for open access charge: National Institutes of Health [RO1 AI060260].
Conflict of interest statement. None declared.
REFERENCES
- 1.Matthews K.R. The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 2005;118:283–290. doi: 10.1242/jcs.01649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kramer S. Developmental regulation of gene expression in the absence of transcriptional control: the case of kinetoplastids. Mol. Biochem. Parasitol. 2012;181:61–72. doi: 10.1016/j.molbiopara.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Kolev N.G., Ullu E., Tschudi C. The emerging role of RNA-binding proteins in the life cycle of Trypanosoma brucei. Cell. Microbiol. 2014;16:482–489. doi: 10.1111/cmi.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunzl A. The pre-mRNA splicing machinery of trypanosomes: complex or simplified. Eukaryot. Cell. 2010;9:1159–1170. doi: 10.1128/EC.00113-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Calvillo S., Vizuet-de-Rueda J.C., Florencio-Martinez L.E., Manning-Cela R.G., Figueroa-Angulo E.E. Gene expression in trypanosomatid parasites. J. Biomed. Biotechnol. 2010;2010:525241. doi: 10.1155/2010/525241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erben E.D., Fadda A., Lueong S., Hoheisel J.D., Clayton C. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 2014;10:e1004178. doi: 10.1371/journal.ppat.1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolev N.G., Ramey-Butler K., Cross G.A., Ullu E., Tschudi C. Developmental progression to infectivity in Trypanosoma brucei triggered by an RNA-binding protein. Science. 2012;338:1352–1353. doi: 10.1126/science.1229641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mony B.M., MacGregor P., Ivens A., Rojas F., Cowton A., Young J., Horn D., Matthews K. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature. 2014;505:681–685. doi: 10.1038/nature12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Droll D., Minia I., Fadda A., Singh A., Stewart M., Queiroz R., Clayton C. Post-transcriptional regulation of the trypanosome heat shock response by a zinc finger protein. PLoS Pathog. 2013;9:e1003286. doi: 10.1371/journal.ppat.1003286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling A.S., Trotter J.R., Hendriks E.F. A zinc finger protein, TbZC3H20, stabilizes two developmentally regulated mRNAs in trypanosomes. J. Biol. Chem. 2011;286:20152–20162. doi: 10.1074/jbc.M110.139261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Archer S.K., Luu V.D., de Queiroz R.A., Brems S., Clayton C. Trypanosoma brucei PUF9 regulates mRNAs for proteins involved in replicative processes over the cell cycle. PLoS Pathog. 2009;5:e1000565. doi: 10.1371/journal.ppat.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mani J., Guttinger A., Schimanski B., Heller M., Acosta-Serrano A., Pescher P., Spath G., Roditi I. Alba-domain proteins of Trypanosoma brucei are cytoplasmic RNA-binding proteins that interact with the translation machinery. PloS One. 2011;6:e22463. doi: 10.1371/journal.pone.0022463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bedford M.T. Arginine methylation at a glance. J. Cell Sci. 2007;120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 14.Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lott K., Li J., Fisk J.C., Wang H., Aletta J.M., Qu J., Read L.K. Global proteomic analysis in trypanosomes reveals unique proteins and conserved cellular processes impacted by arginine methylation. J. Proteomics. 2013;91:210–225. doi: 10.1016/j.jprot.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisk J.C., Li J., Wang H., Aletta J.M., Qu J., Read L.K. Proteomic analysis reveals diverse classes of arginine methylproteins in mitochondria of trypanosomes. Mol. Cell. Proteomics. 2013;12:302–311. doi: 10.1074/mcp.M112.022533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Y., Maity R., Whitelegge J.P., Hadjikyriacou A., Li Z., Zurita-Lopez C., Al-Hadid Q., Clark A.T., Bedford M.T., Masson J.Y., et al. Mammalian protein arginine methyltransferase 7 (PRMT7) specifically targets RXR sites in lysine- and arginine-rich regions. J. Biol. Chem. 2013;288:37010–37025. doi: 10.1074/jbc.M113.525345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horowitz S., Trievel R.C. Carbon-oxygen hydrogen bonding in biological structure and function. J. Biol. Chem. 2012;287:41576–41582. doi: 10.1074/jbc.R112.418574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biggar K.K., Li S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015;16:5–17. doi: 10.1038/nrm3915. [DOI] [PubMed] [Google Scholar]
- 20.Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K.A., Yang V., Aguiar M., Kornhauser J., Jia X., et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics. 2014;13:372–387. doi: 10.1074/mcp.O113.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahms H., Meheus L., de Brabandere V., Fischer U., Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA. 2001;7:1531–1542. doi: 10.1017/s135583820101442x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng D., Cote J., Shaaban S., Bedford M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Hung M.L., Hautbergue G.M., Snijders A.P., Dickman M.J., Wilson S.A. Arginine methylation of REF/ALY promotes efficient handover of mRNA to TAP/NXF1. Nucleic Acids Res. 2010;38:3351–3361. doi: 10.1093/nar/gkq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Park S., Kilburn B., Jelinek M.A., Henschen-Edman A., Aswad D.W., Stallcup M.R., Laird-Offringa I.A. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J. Biol. Chem. 2002;277:44623–44630. doi: 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- 25.Bachand F., Silver P.A. PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J. 2004;23:2641–2650. doi: 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackwell E., Zhang X., Ceman S. Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum. Mol. Genet. 2010;19:1314–1323. doi: 10.1093/hmg/ddq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelletier M., Pasternack D.A., Read L.K. In vitro and in vivo analysis of the major type I protein arginine methyltransferase from Trypanosoma brucei. Mol. Biochem. Parasitol. 2005;144:206–217. doi: 10.1016/j.molbiopara.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Pasternack D.A., Sayegh J., Clarke S., Read L.K. Evolutionarily divergent type II protein arginine methyltransferase in Trypanosoma brucei. Eukaryot. Cell. 2007;6:1665–1681. doi: 10.1128/EC.00133-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisk J.C., Sayegh J., Zurita-Lopez C., Menon S., Presnyak V., Clarke S.G., Read L.K. A type III protein arginine methyltransferase from the protozoan parasite Trypanosoma brucei. J. Biol. Chem. 2009;284:11590–11600. doi: 10.1074/jbc.M807279200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisk J.C., Zurita-Lopez C., Sayegh J., Tomasello D.L., Clarke S.G., Read L.K. TbPRMT6 is a type I protein arginine methyltransferase that contributes to cytokinesis in Trypanosoma brucei. Eukaryot. Cell. 2010;9:866–877. doi: 10.1128/EC.00018-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lott K., Zhu L., Fisk J.C., Tomasello D.L., Read L.K. Functional interplay between protein arginine methyltransferases in Trypanosoma brucei. MicrobiologyOpen. 2014;3:595–609. doi: 10.1002/mbo3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goulah C.C., Pelletier M., Read L.K. Arginine methylation regulates mitochondrial gene expression in Trypanosoma brucei through multiple effector proteins. RNA. 2006;12:1545–1555. doi: 10.1261/rna.90106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulah C.C., Read L.K. Differential effects of arginine methylation on RBP16 mRNA binding, guide RNA (gRNA) binding, and gRNA-containing ribonucleoprotein complex (gRNP) formation. J. Biol. Chem. 2007;282:7181–7190. doi: 10.1074/jbc.M609485200. [DOI] [PubMed] [Google Scholar]
- 34.De Gaudenzi J., Frasch A.C., Clayton C. RNA-binding domain proteins in Kinetoplastids: a comparative analysis. Eukaryot. Cell. 2005;4:2106–2114. doi: 10.1128/EC.4.12.2106-2114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wirtz E., Leal S., Ochatt C., Cross G.A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 36.Nouri-Nigjeh E., Sukumaran S., Tu C., Li J., Shen X., Duan X., DuBois D.C., Almon R.R., Jusko W.J., Qu J. Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Analyt. Chem. 2014;86:8149–8157. doi: 10.1021/ac501380s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu C., Li J., Jiang X., Sheflin L.G., Pfeffer B.A., Behringer M., Fliesler S.J., Qu J. Ion-current-based proteomic profiling of the retina in a rat model of Smith-Lemli-Opitz syndrome. Mol. Cell. Proteomics. 2013;12:3583–3598. doi: 10.1074/mcp.M113.027847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayman M.L., Miller M.M., Chandler D.M., Goulah C.C., Read L.K. The trypanosome homolog of human p32 interacts with RBP16 and stimulates its gRNA binding activity. Nucleic Acids Res. 2001;29:5216–5225. doi: 10.1093/nar/29.24.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wurst M., Seliger B., Jha B.A., Klein C., Queiroz R., Clayton C. Expression of the RNA recognition motif protein RBP10 promotes a bloodstream-form transcript pattern in Trypanosoma brucei. Mol. Microbiol. 2012;83:1048–1063. doi: 10.1111/j.1365-2958.2012.07988.x. [DOI] [PubMed] [Google Scholar]
- 42.Das A., Morales R., Banday M., Garcia S., Hao L., Cross G.A., Estevez A.M., Bellofatto V. The essential polysome-associated RNA-binding protein RBP42 targets mRNAs involved in Trypanosoma brucei energy metabolism. RNA. 2012;18:1968–1983. doi: 10.1261/rna.033829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 44.Jha B.A., Archer S.K., Clayton C.E. The trypanosome Pumilio domain protein PUF5. PloS One. 2013;8:e77371. doi: 10.1371/journal.pone.0077371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramer S., Queiroz R., Ellis L., Hoheisel J.D., Clayton C., Carrington M. The RNA helicase DHH1 is central to the correct expression of many developmentally regulated mRNAs in trypanosomes. J. Cell Sci. 2010;123:699–711. doi: 10.1242/jcs.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haanstra J.R., Stewart M., Luu V.D., van Tuijl A., Westerhoff H.V., Clayton C., Bakker B.M. Control and regulation of gene expression: quantitative analysis of the expression of phosphoglycerate kinase in bloodstream form Trypanosoma brucei. J. Biol. Chem. 2008;283:2495–2507. doi: 10.1074/jbc.M705782200. [DOI] [PubMed] [Google Scholar]
- 47.Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D.C., Lennard N.J., Caler E., Hamlin N.E., Haas B., et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 48.Hall N., Berriman M., Lennard N.J., Harris B.R., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., et al. The DNA sequence of chromosome I of an African trypanosome: gene content, chromosome organisation, recombination and polymorphism. Nucleic Acids Res. 2003;31:4864–4873. doi: 10.1093/nar/gkg674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siegel T.N., Hekstra D.R., Wang X., Dewell S., Cross G.A. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010;38:4946–4957. doi: 10.1093/nar/gkq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manful T., Fadda A., Clayton C. The role of the 5′-3′ exoribonuclease XRNA in transcriptome-wide mRNA degradation. RNA. 2011;17:2039–2047. doi: 10.1261/rna.2837311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolev N.G., Franklin J.B., Carmi S., Shi H., Michaeli S., Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6:e1001090. doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 53.Campbell M., Chang P.C., Huerta S., Izumiya C., Davis R., Tepper C.G., Kim K.Y., Shevchenko B., Wang D.H., Jung J.U., et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 2012;287:5806–5818. doi: 10.1074/jbc.M111.289496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dillon M.B., Rust H.L., Thompson P.R., Mowen K.A. Automethylation of protein arginine methyltransferase 8 (PRMT8) regulates activity by impeding S-adenosylmethionine sensitivity. J. Biol. Chem. 2013;288:27872–27880. doi: 10.1074/jbc.M113.491092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim K.Y., Wang D.H., Campbell M., Huerta S.B., Shevchenko B., Izumiya C., Izumiya Y. PRMT4-mediated arginine methylation negatively regulates retinoblastoma tumor suppressor protein and promotes E2F-1 dissociation. Mol. Cell. Biol. 2014;35:238–248. doi: 10.1128/MCB.00945-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinha R., Allemand E., Zhang Z., Karni R., Myers M.P., Krainer A.R. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol. Cell. Biol. 2010;30:2762–2774. doi: 10.1128/MCB.01270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fronz K., Guttinger S., Burkert K., Kuhn U., Stohr N., Schierhorn A., Wahle E. Arginine methylation of the nuclear poly(a) binding protein weakens the interaction with its nuclear import receptor, transportin. J. Biol. Chem. 2011;286:32986–32994. doi: 10.1074/jbc.M111.273912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tradewell M.L., Yu Z., Tibshirani M., Boulanger M.C., Durham H.D., Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012;21:136–149. doi: 10.1093/hmg/ddr448. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Moya S.M., Garcia-Perez A., Kramer S., Carrington M., Estevez A.M. Alterations in DRBD3 ribonucleoprotein complexes in response to stress in Trypanosoma brucei. PloS One. 2012;7:e48870. doi: 10.1371/journal.pone.0048870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alves L.R., Oliveira C., Morking P.A., Kessler R.L., Martins S.T., Romagnoli B.A., Marchini F.K., Goldenberg S. The mRNAs associated to a zinc finger protein from Trypanosoma cruzi shift during stress conditions. RNA Biol. 2014;11:921–933. doi: 10.4161/rna.29622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erben E., Chakraborty C., Clayton C. The CAF1-NOT complex of trypanosomes. Front. Genet. 2014;4:299–303. doi: 10.3389/fgene.2013.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dormann D., Madl T., Valori C.F., Bentmann E., Tahirovic S., Abou-Ajram C., Kremmer E., Ansorge O., Mackenzie I.R., Neumann M., et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–4275. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boisvert F.M., Chenard C.A., Richard S. Protein interfaces in signaling regulated by arginine methylation. Sci. STKE. 2005;2007:re2. doi: 10.1126/stke.2712005re2. [DOI] [PubMed] [Google Scholar]
- 64.Alsford S., Turner D.J., Obado S.O., Sanchez-Flores A., Glover L., Berriman M., Hertz-Fowler C., Horn D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–924. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones N.G., Thomas E.B., Brown E., Dickens N.J., Hammarton T.C., Mottram J.C. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi screen. PLoS Pathog. 2014;10:e1003886. doi: 10.1371/journal.ppat.1003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najafabadi H.S., Lu Z., MacPherson C., Mehta V., Adoue V., Pastinen T., Salavati R. Global identification of conserved post-transcriptional regulatory programs in trypanosomatids. Nucleic Acids Res. 2013;41:8591–8600. doi: 10.1093/nar/gkt647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeGrasse J.A., DuBois K.N., Devos D., Siegel T.N., Sali A., Field M.C., Rout M.P., Chait B.T. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol. Cell. Proteomics. 2009;8:2119–2130. doi: 10.1074/mcp.M900038-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DuBois K.N., Alsford S., Holden J.M., Buisson J., Swiderski M., Bart J.M., Ratushny A.V., Wan Y., Bastin P., Barry J.D., et al. NUP-1 Is a large coiled-coil nucleoskeletal protein in trypanosomes with lamin-like functions. PLoS Biol. 2012;10:e1001287. doi: 10.1371/journal.pbio.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tamura K., Hara-Nishimura I. The molecular architecture of the plant nuclear pore complex. J. Exp. Bot. 2013;64:823–832. doi: 10.1093/jxb/ers258. [DOI] [PubMed] [Google Scholar]
- 70.Chakraborty P., Wang Y., Wei J.H., van Deursen J., Yu H., Malureanu L., Dasso M., Forbes D.J., Levy D.E., Seemann J., et al. Nucleoporin levels regulate cell cycle progression and phase-specific gene expression. Dev. Cell. 2008;15:657–667. doi: 10.1016/j.devcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zinoviev A., Leger M., Wagner G., Shapira M. A novel 4E-interacting protein in Leishmania is involved in stage-specific translation pathways. Nucleic Acids Res. 2011;39:8404–8415. doi: 10.1093/nar/gkr555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.