

Abstract
APE1 is a multifunctional protein with a fundamental role in repairing nuclear and mitochondrial DNA lesions caused by oxidative and alkylating agents. Unfortunately, comprehensions of the mechanisms regulating APE1 intracellular trafficking are still fragmentary and contrasting. Recent data demonstrate that APE1 interacts with the mitochondrial import and assembly protein Mia40 suggesting the involvement of a redox-assisted mechanism, dependent on the disulfide transfer system, to be responsible of APE1 trafficking into the mitochondria. The MIA pathway is an import machinery that uses a redox system for cysteine enriched proteins to drive them in this compartment. It is composed by two main proteins: Mia40 is the oxidoreductase that catalyzes the formation of the disulfide bonds in the substrate, while ALR reoxidizes Mia40 after the import. In this study, we demonstrated that: (i) APE1 and Mia40 interact through disulfide bond formation; and (ii) Mia40 expression levels directly affect APE1's mitochondrial translocation and, consequently, play a role in the maintenance of mitochondrial DNA integrity. In summary, our data strongly support the hypothesis of a redox-assisted mechanism, dependent on Mia40, in controlling APE1 translocation into the mitochondrial inner membrane space and thus highlight the role of this protein transport pathway in the maintenance of mitochondrial DNA stability and cell survival.
INTRODUCTION
APE1 is the main apurinic/apyrimidinic (AP) endonuclease in the base excision repair (BER) pathway where it plays a central role in the maintenance of the genome stability being the only cellular protein that can process AP sites generated by DNA glycosylases (1). However, APE1 has at least another major cellular function, since it works as a redox signaling protein interacting with many transcriptional factors involved in cancer promotion and progression, such as NF-κB, p53, HIF-1α and more, and facilitating their DNA binding via reduction of cysteine residues (2). These two major functions of APE1, redox and repair, are completely independent: the N-terminus, containing a bipartite nuclear localization signal (NLS), is mainly devoted to redox-mediated activity, while the C-terminus exerts the enzymatic activity on the abasic sites of DNA (3). The importance of APE1 function in cell survival is demonstrated by the lethality of the APE1 gene-knockout in mice (4). APE1's localization is eminently nuclear although the protein is also present within the mitochondrial matrix, as an essential component of the mitochondrial BER (mtBER) pathway (5,6). Notably, the mechanisms regulating APE1 intracellular trafficking are still largely unknown. A description of APE1 mitochondrial localization was proposed by Chattopadhyay et al. by which proteolysis at Asn33 was responsible for the loss of NLS, therefore redirecting truncated NΔ33APE1 protein toward non-canonical subcellular compartments such as mitochondria (7). However, data from our previous work highlight the presence of the full-length APE1 protein within mitochondria of follicular thyroid cells (8). Also recent data from our lab confirm the presence of APE1 within mitochondria in its full-length form and that, during the trafficking from cytosol to the mitochondrial matrix, APE1 tends to accumulate within the inner membrane space (IMS) (6). In 2011 Li et al. demonstrated that the MTS of APE1 resides within residues 289–318 of the C-terminus, which is masked by the N-terminal domain, therefore suggesting the occurrence of specific and regulated mechanisms of protein unfolding–refolding to ensure proper APE1 localization (9). Data reported identified residues Lys299 and Arg301 as critical sites which mutation abolishes APE1 interaction with Tom20 and consequently the mitochondrial translocation of the protein (9).
Mitochondria are the main endogenous source of Reactive Oxygen Species (ROS) and, like nuclear DNA (nDNA), the integrity of mitochondrial DNA (mtDNA) is also constantly threatened by exposure to different damaging agents. Because of the association with the inner membrane, close to the ROS-generating electron transport chain, mtDNA is much more susceptible to ROS-induced damage than genomic DNA showing 16-fold higher levels of 8-oxoG formation (10). ROS-induced oxidative lesions and alkylated bases in DNA are repaired via BER, the major DNA repair mechanism acting in mitochondria (11). BER is the primary pathway known for repairing of small helix non-distorting DNA lesions and taking place both in the nucleus and in mitochondria, and is based on a cascade of multiple steps. All the components of the mtBER pathway are nuclear encoded and imported into mitochondria (12). The BER pathway includes four distinct steps: (i) recognition and removal of the modified base operated by a DNA glycosylase; (ii) processing of the generated apurinic/apyrimidinic (AP) site by APE1 protein; (iii) incorporation of the correct nucleotide(s) through the activity of the mitochondrial polymerase γ; and (iv) nick sealing catalyzed by the DNA ligase III.
Little is known about the mechanisms of BER induction in mitochondria, except that in the presence of mtDNA damage, the mtBER pathway is activated (13) and that APE1 translocates into mitochondria in response to oxidative stress, increasing mtDNA repair rate and therefore promoting cell survival (14). Due to its significant molecular mass, mitochondrial localization of APE1 should require active transportation processes. The main chaperone responsible for redox-assisted folding processes within IMS of mitochondria in mammals is Mia40 (15). Mitochondrial intermembrane space import and assembly protein 40 (Mia40) and Augmenter of liver regeneration (ALR) represent the central components of the MIA import machinery that uses a redox system to drive cysteine-enriched proteins into the mitochondrial compartment. After the passage throughout the inner membrane via Translocase of the outer membrane (TOM), substrate proteins are recognized by Mia40 through a process that involves the formation of disulfide bonds between Mia40 and the precursor protein. Mia40 is composed of a highly conserved C-terminal domain, which contains the catalytic CPC (cysteine–proline–cysteine) motif and a hydrophobic cleft. Once Mia40 has oxidized its substrate, its CPC motif remains in a completely reduced form and has to be re-oxidized for a further cycle of import (16). The protein responsible for this process is ALR, a sulfhydryl oxidase coupled with one molecule of Flavin Adenin Dinucleotide (FAD) that acts as a homodimer in solution (17–19). Mia40 is ubiquitously expressed in human tissues and the highest levels of Mia40 were found in the liver, brain and kidney at both transcript and protein levels (20,21). In human Mia40, the CPC motif is formed by Cys53 and Cys55 residues and the cysteine responsible for the intermolecular disulfide bond is the Cys55 (22). Mutation of this cysteine residue results in the loss of function of Mia40 that is no more able to interact with the substrate. On the contrary, when Cys53 is mutated, Mia40 is still able to interact with the substrates but cannot efficiently complete the oxidation reaction (23–25).
The majority of Mia40-dependent proteins have been identified in yeast. Typical substrates of the MIA pathway are small proteins, around 10 kDa, whose destination is the IMS or the matrix. These proteins are cysteine enriched, belong to the families of the twin CX3C and CX9C proteins and fulfill different functions in the mitochondria (16,26,27). Recently, also the tumor suppressor protein p53 was found to localize within human mitochondria through the MIA pathway. Authors demonstrated that the import is an active process activated by oxidative stress, respiration-dependent, and that it contributes to the maintenance of mtDNA integrity (28).
The present study focuses on the identification and characterization of the import machinery that mediates APE1 translocation into the mitochondrial compartment. Our data identify APE1 as a new substrate of Mia40 and correlate its expression with the translocation of APE1 and the repair capacity of mtBER supporting the involvement of the MIA pathway in controlling the mitochondrial genome stability.
MATERIALS AND METHODS
Cell culture and treatments
HeLa and Huh7 cells were grown in DMEM (Dulbecco's modified Eagle's medium), JHH6 cells were grown in William's medium E, both supplemented with 10% fetal bovine serum (FBS), 100-U/ml penicillin and 10-μg/ml Streptomycin sulphate. One day before treatment HeLa cells were seeded 4 × 106 cells/plate. For mtDNA damage measurements HeLa and JHH6 cells were treated with 400 μM and 1.2 mM of H2O2, respectively, in medium without serum for 15 min, and then cells were grown for 1 h in the presence of 10% FBS before harvesting. For cell viability analysis 5 × 104 JHH6 and 10 × 104 Huh7 cells were seated on 96 multiwell plates and after 24 h treated with the reported amount of Methyl methanesulfonate (MMS) or H2O2 for 8 h in medium without FBS. For inducible silencing of endogenous APE1, HeLa cell clones were developed as described in Vascotto et al. (29). For inducible siRNA experiments, doxycycline (Sigma) was added to the cell culture medium at the final concentration of 1 μg/ml, and cells were grown for 10 days.
Transient transfection experiments
One day before transfection cells were seeded in 10-cm plates at the density of 1.2 × 106 cells/plate. Cells were then transfected with 100 nM of either shRNA Mia40 scramble (control) or shRNA Mia40 (encoding for siRNA against Mia40) per plate using Oligofectamine Reagent (Invitrogen) according to the manufacturer's instructions. Cells were harvested 72 h after the transfection.
For overexpression of APE1 and Mia40 proteins, 2.5 × 106 cells/plate were seeded 24 h before transfection. Cells were then transfected with 2 μg of pCMV 5.1 APE1-FLAG, C65S, C93S, C99S, C65S+C93S, C65S+C99S or Mia40-HisTag per plate using Lipofectamine Reagent (Invitrogen) according to the manufacturer's instructions. Cells were harvested 24 h after the transfection. For transient APE1 silencing HeLa cells were seeded at the density of 1.2 × 106 cells/plate. Then, cells were transfected with 6 μg of pSuper-scramble or pSuper-APE1, in order to obtain silencing of endogenous APE1, and with 6 μg per plate of pCMV 5.1 Empty, APE1 WT, C65S or C93S to re-express APE1. Cells were harvested 48 h after the transfection.
Preparation of total cell extracts and anti-Flag affinity purification
For the preparation of total cell lysates cells were harvested by trypsinization and centrifuged at 250 × g for 5 min at 4°C. Supernatant was removed, and the pellet was washed once with ice-cold phosphate-buffered saline (PBS) and then centrifuged again as described before. Cell pellet was resuspended in lysis buffer [50-mM Tris HCl (pH 7.4), 150-mM NaCl, 1-mM ethylenediaminetetraacetic acid (EDTA), 1% [wt/vol] Triton X-100, protease inhibitor cocktail (Sigma), 0.5-mM phenylmethylsulfonyl fluoride] at a cell density of 107 cells/ml and rotated for 30 min at 4°C. After centrifugation at 12 000 × g for 10 min at 4°C, the supernatant was collected as total cell lysate (WCE). The protein concentration was determined using Bio-Rad protein assay reagent (Bio-Rad). For affinity purification analysis total cell lysates were incubated with anti-Flag M2 affinity gel (Sigma) for 3 h at 4°C and then processed following the manufacturer's instructions. Proteins were eluted by incubation with 0.15 mg/ml 3xFlag peptide in Tris-buffered saline and then subjected to western blot analysis.
Preparation of subcellular fractions
Subconfluent HeLa and JHH6 cells were collected and suspended in Grinding buffer (250-mM sucrose, 1-mg/ml bovine serum albumin (BSA), 2-mM EDTA, pH 7.4) with 1:100 protease inhibitor cocktail. Cell suspensions were sonicated on ice under mild controlled conditions to disrupt selectively plasma membranes (4-s sonication one time). The homogenates were then fractionated by differential centrifugation at 800 × g and 16 000 × g at 4°C, to obtain the enriched cytoplasmic, nuclear and mitochondrial fractions. Samples were then subjected to Bradford analysis for the evaluation of protein content before separation on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis. Purity of mitochondria and nuclei was evaluated using specific marker proteins (LSD1 for nuclei and ATP 5A for mitochondria) to ascertain the absence of cross contamination between the two compartments.
Western blot analysis
The indicated amounts of total, cytoplasmic, nuclear or mitochondrial extracts were electrophoresed onto a 12% SDS-PAGE. Then, proteins were transferred to nitrocellulose membranes (Schleicher & Schuell). Membranes were saturated by incubation with 5% non-fat dry milk in PBS/0.1% Tween 20 for 1 h at room temperature, and incubated with the specific primary antibody [anti-APE1 monoclonal (Novartis): overnight 4°C, dilution 1:2000; anti-FLAG monoclonal (Sigma): 3 h at 25°C, dilution 1:3000; anti-Actin polyclonal (Sigma): overnight at 4°C, dilution 1:2000; anti-ATP 5A monoclonal (Abcam): overnight at 4°C, dilution 1:10000; anti-LSD1 polyclonal (Abcam): overnight at 4°C, dilution 1:10000; anti-Mia40 polyclonal (gift from the laboratory of Prof. Matthias Bauer) overnight at 4°C, dilution 1:1000; anti-HisTag polyclonal (Sigma): overnight at 4°C, dilution 1:2000]. Membranes were washed three times with PBS/0,1% Tween 20 and incubated for 2 h with the secondary antibody. After three washes with PBS/0.1% Tween 20 signals were detected with the enhanced chemiluminescence procedure (Bio-Rad) (Figures 1A, 2A, 3B and 4A) or Odyssey CLx scanner (Li-Cor Biosciences) (Figures 1B and C, 5B and C, 6A and 3A and C). Normalization was performed with polyclonal antibodies anti-Actin for total extracts and with ATP 5A for mitochondrial extracts. Blots were then quantified by using a Gel Doc video-densitometer (Bio-Rad) or ImageStudio software (Li-Cor Biosciences).
Figure 1.
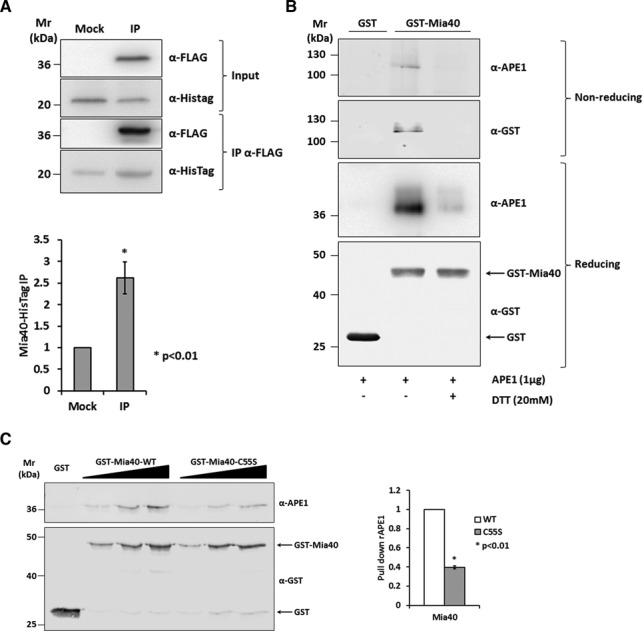
Mia40 Cys55 is responsible for the binding to APE1. (A) Western blot (top) and relative quantification (bottom) of the affinity purification analysis of HeLa cells expressing APE1-FLAG and Mia40-HisTag proteins. Control sample (Mock) and APE1-FLAG (IP) were immunoprecipitated under native conditions from total cell extracts, separated by 12% SDS-PAGE, and analyzed by western blot to evaluate the levels of each interacting partner by using specific antibodies (anti-APE1, anti-HisTag). Mia40-HisTag protein resulted enriched in IP fraction. (B) GST pull-down assay under reducing and non-reducing conditions. Fifteen picomoles of recombinant GST-Mia40 protein were used as bait, GST alone as control and APE1 protein as prey. Interaction between the two proteins was disrupted after treatment with reducing agent (DTT 20 mM). Under non-reducing conditions the complex formed by APE1 and Mia40 is visible and is disrupted when proteins were incubated with DTT. (C) Western blot (left) and relative histogram (right) of GST pull-down analysis of recombinant APE1 WT protein. GST-Mia40 WT and C55S mutant (5, 10 and 15 pmoles) were used as prey and GST (15 pmoles) alone as control. The binding of APE1 (5, 10 and 15 pmoles) was normalized to the western blotting signal of GST-Mia40 and resulted strongly decrease by the mutation of Mia40 Cys55 residue.
Figure 2.
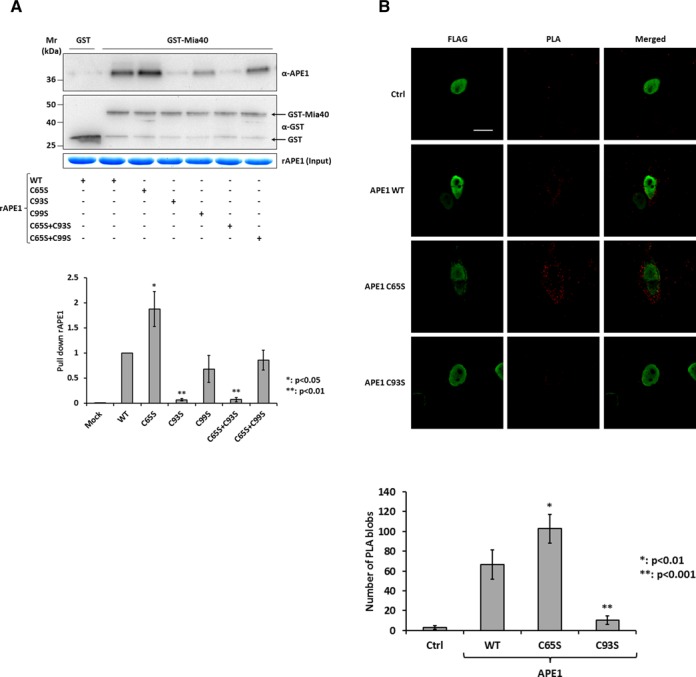
APE1 interacts with Mia40 through the redox Cys93 residue. (A) Western blot (top) and relative histogram (bottom) of GST pull-down analysis of recombinant APE1 (15 pmoles) WT and redox Cys65, Cys93, Cys99, Cys65+Cys93 and Cys65+Cys99 to Ser mutants. GST-Mia40 (15 pmoles) was used as prey and GST (15 pmoles) alone as control. The binding of APE1 WT and mutants was normalized to the western blotting signal of GST-Mia40. (B) Representative immunofluorescence images (top) and histogram (bottom) of PLA analysis between ectopic flagged APE1 and endogenous Mia40. HeLa cells were transiently transfected with pCMV5.1-FLAG vector expressing APE1 WT, Cys65 and Cys93 to Ser mutants. APE1 expression was detected by using an anti-FLAG antibody (green), while PLA signal are visible as red dots. Control reaction was carried out omitting anti-Mia40 antibody and shows no or little PLA signal. White bar corresponded to 10 μm. Data reported in the histogram accounted for the average number of PLA signals of at least 15 randomly selected cells per condition.
Figure 3.
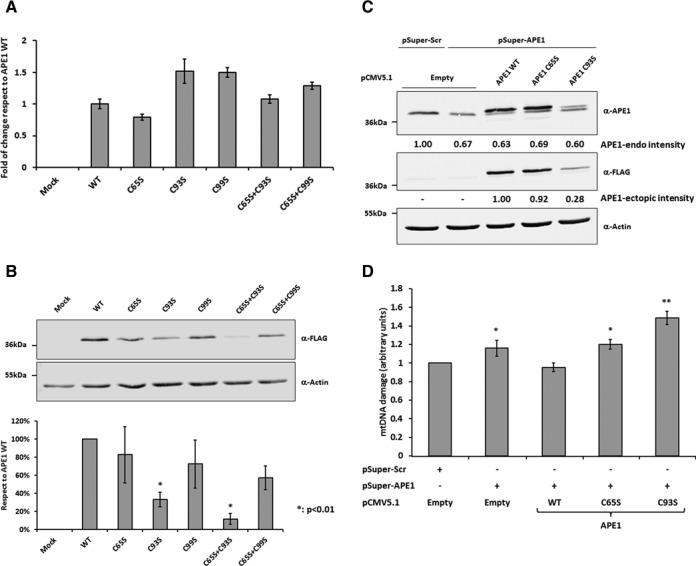
Cys93 mutation alters APE1's stability increasing mtDNA damage. (A) mRNA and (B) protein expression analysis of APE1 redox mutants in HeLa cells. Ten micrograms of total cell extracts from HeLa cells expressing APE1 WT or redox mutant were analyzed by western blot to evaluate the effect of mutation on APE1 stability. Mutation of Cys93 significantly reduces the accumulation of the protein within the cell with respect to the WT form. Actin was used as loading control to normalize APE1's signal intensity. (C) Western blot analysis of HeLa cells after endogenous APE1 silencing and re-expression of APE1 WT, Cys65 or Cys93 to Ser mutant proteins. Endogenous APE1 protein is significantly reduced after transfection while exogenous APE1 WT, C65S and C93S proteins are expressed. Actin was used as loading control. (D) mtDNA damage analysis of HeLa cells after endogenous APE1 loss of expression and re-expression of ectopic APE1 WT, Cys65 or Cys93 to Ser mutant proteins. Silencing of APE1 leads to increased levels of mtDNA damage rescued by the re-expression of APE1 WT. APE1 C65S and C93S proteins are not able to rescue the damage after APE1 silencing.
Figure 4.
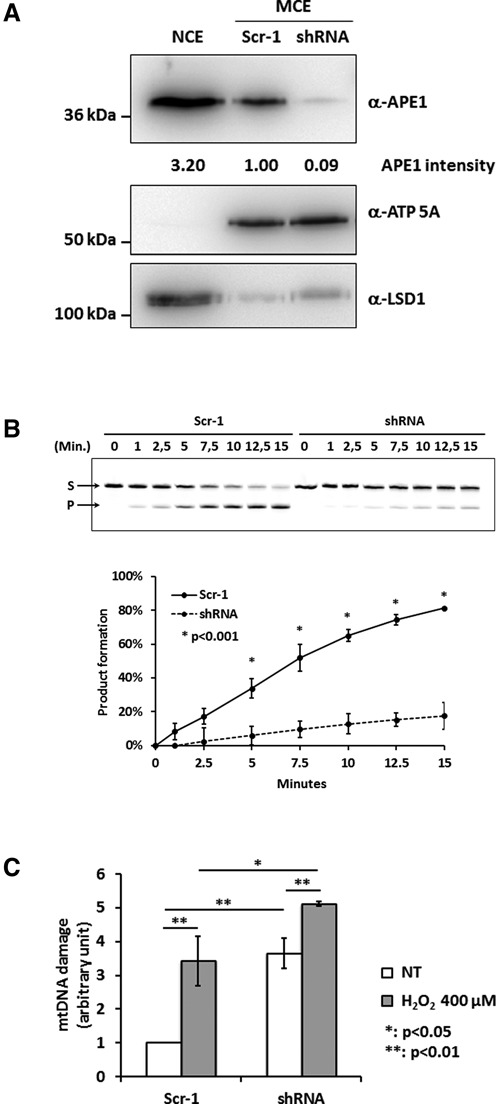
Loss of APE1 expression determines increased levels of mtDNA damage. (A) Western blot analysis of mitochondrial fractions (MCE) from control (Scr-1, lane 2) and APE1-shRNA stable clones (lane 3). APE1-shRNA cells show APE1 reduction up to 90% with respect to the Scr-1 cells. ATP 5A and LSD1 were used as mitochondrial and nuclear markers, respectively. A nuclear extract (NCE, lane 1) from HeLa cells was used as control to exclude nuclear/mitochondrial cross contamination. (B) Endonuclease activity analysis of mitochondrial extracts from Scr-1 and shRNA cells. The conversion of the fluorescent tetrahydrofuran-containing oligonucleotide substrate (S) to the shorter incised product (P) was evaluated for the reported times on a denaturing 20% (wt/vol) polyacrylamide gel. A representative image (top) and average values of incision percentage ±SD of three independent experiments (bottom) are shown. Endonuclease activity is almost abolished upon loss of APE1 expression. (C) mtDNA damage analysis of Scr-1 and shRNA clones under basal conditions and after H2O2 treatment (400 μM). Levels of mtDNA damage are increased in cells lacking APE1 both under basal and oxidative stress conditions.
Figure 5.
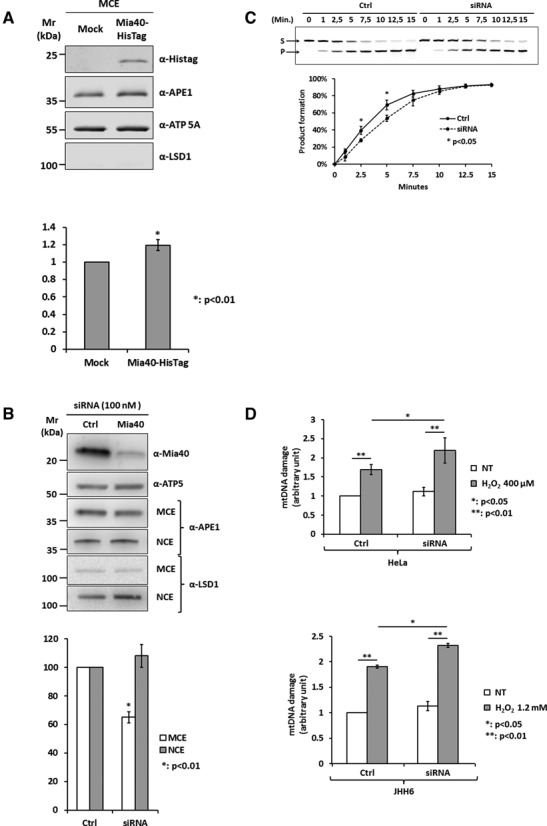
Loss of Mia40 expression reduces mitochondrial APE1 content affecting mtDNA stability. (A) Western blot analysis (top) of mitochondrial extracts from HeLa cells overexpressing Mia40-HisTag. HeLa cells were transiently transfected with pCMV 5.1 empty vector (Mock) or codifying for Mia40-HisTag protein. Densitometric analysis of mitochondrial APE1 levels (bottom) highlights a statistically significant increase of APE1 content into the mitochondrial fraction as a consequence of Mia40 overexpression. Data are the mean ± SD of three independent experiments. (B) Western blot analysis of nuclear (NCE) and mitochondrial (MCE) HeLa cell extracts of Mia40 siRNA cells (top). Densitometric analysis of mitochondrial and nuclear APE1 levels (bottom) highlights a statistically significant reduction of APE1 content into the mitochondrial fraction as a consequence of Mia40 loss of expression. Data are the mean ± SD of five independent experiments. (C) Endonuclease activity analysis of mitochondrial fractions from scramble (Ctrl) and Mia40-siRNA-treated cells (siRNA). Data analysis reported in the diagram (bottom) shows that endonuclease activity is reduced after Mia40 silencing. (D) mtDNA damage analysis from HeLa cells (top) and JHH6 hepatic cell line (bottom) after Mia40 loss of expression under basal conditions and after H2O2 treatment (HeLa: 400 μM; JHH6: 1.2 mM). Silencing of Mia40 leads to an increase of mtDNA damage under oxidative stress conditions.
Figure 6.
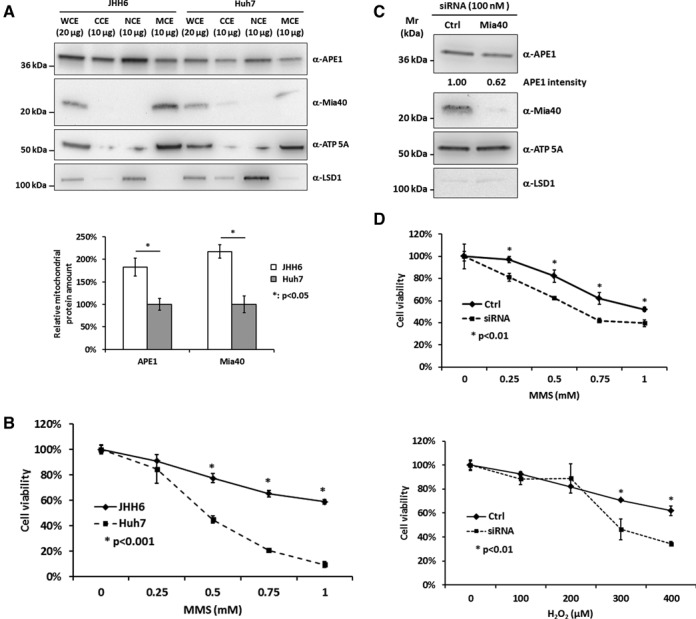
Loss of Mia40 expression sensitizes JHH6 cells to MMS and H2O2 treatment. (A) Western blot analysis of total (WCE), cytoplasmic (CCE), nuclear (NCE) and mitochondrial (MCE) cell extracts from JHH6 (lanes 1–4) and Huh7 (lanes 5–8) cell lanes (top). Densitometric analysis shows a direct correlation between expression levels of Mia40 and APE1 (bottom). ATP 5A and LSD1 were used as mitochondrial and nuclear markers, respectively. (B) MTS cell viability assay on JHH6 and Huh7 cell lines after 8 h of treatment with increased amounts of MMS shows an increased sensitivity of Huh7 to the treatment. (C) Western blot analysis of mitochondrial extracts of Mia40 siRNA and relative control from JHH6 cells. Loss of Mia40 expression negatively affects the mitochondrial levels of APE1 protein. (D) MTS cell viability assay on control (Ctrl) and Mia40 siRNA JHH6 cells after 8 h of treatment with reported amounts of MMS (top) and H2O2 (bottom). Reduction of mitochondrial APE1 content as a consequence of Mia40 silencing sensitizes cells to oxidative and alkylating treatments.
DNA extraction and mtDNA damage measurement by quantitative PCR
After treatments cells were harvested by trypsin and high-molecular weight DNA was isolated with the QUIAGEN Genomic-tip 20/G and G2, QBT, QC, QF buffers (all reagents are distributed by Qiagen) as described by the manufacturer. Briefly cells were resuspended in G2 buffer supplemented with 400 μg of RNase A and 6 mAU of proteinase K and incubated at 50°C for 2 h in a water bath. Samples were then applied to equilibrated columns, washed three times with QC buffer and then eluted with 1 ml of QF buffer twice. DNA was precipitated overnight at −80°C with the addition of 700 μl of isopropanol and then centrifuged at 4°C, 15 000 × g for 1 h. DNA was washed with EtOH 75%, centrifuged again and resuspended in 100 μl of Tris-EDTA buffer pH 8. Quantification of DNA was performed with Quant.iT-Picogreen dsDNA reagent (Invitrogen) according to the manufacturer's instructions and DNA concentration was adjusted to 10 ng/μl.
The number of mtDNA lesions was determined by Q-PCR. The following primers were used: Mitolong_for 5′-TCT AAG CCT CCT TAT TCG AGC CGA-3′ and Mito_rev 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′ which amplified an 8.9-kb mitochondrial fragment; and Mitoshort_for 5′-CCC CAC AAA CCC CAT TAC TAA ACC CA-3′ and Mito_rev, which amplified a 221-bp mitochondrial fragment. DNA was amplified using Elongase enzyme mix (Invitrogen). The polymerase chain reaction (PCR) was initiated at 94°C with hot-start for the complete denaturation of DNA and allowed to undergo the following thermocycler profile: an initial denaturation for 1 min at 94°C followed by 19 cycles of 94°C denaturation for 1 min and 64°C annealing/extension for 11 min for the 8900 bp fragment and 60°C annealing for 45 s and 72°C extension for 45 s for the 221 bp. A final extension at 72°C was performed for 10 min for both fragments. To ensure quantitative conditions a sample with the 50% of template amount was included in each amplification and, as negative control, a sample without the template was used. PCR products were quantified in triplicate by using Quant.iT-Picogreen dsDNA reagent. The mitoshort fragment was needed to calculate the relative amount of mtDNA copies and to normalize the lesion frequencies calculated with the mitolong fragment.
RNA extraction and Real time-PCR
Total RNA from HeLa cells transfected with pCMV5.1 APE1 constructs was extracted with the SV Total RNA isolation System kit (Promega). 1 µg of total RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad), according to the manufacturer's instructions. Quantitative reverse-transcription PCR was performed with a CFX96 Real-Time System (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). Primer sequences for ectopic APE1 are: sense, 5- CAG CAA GAT CCG TTC CAA -3′; antisense, 5’- TTG TCA TCG TCG TCC TTG TAA -3’. Human glyceraldehyde-3-phosphate dehydrogenase was used as internal control.
Expression and purification of recombinant proteins
pGEX-3X expression plasmids (Sigma) containing either wild-type APE1 or APE1 C65S, C93S, C99S, C65/93S, C65/99S mutants or pGEX-4T expression vectors (Sigma) containing wild-type Mia40, Mia40 C55S mutant or the empty vector were transformed into Escherichia coli BL21 (DE3) cells (Stratagene). Bacterial cells were grown at 37°C to an absorbance of 0.8 OD measured at 600 nm, and then protein expression was induced with 1-mM isopropyl-β-D-thiogalactopyranoside. Induction was carried out for 4 h for GST-APE1 and GST and for 2 h for Mia40 at 37°C and then cells were collected by centrifugation at 4000 × g for 20′ at 4°C. Cells were resuspended in Lysis buffer (20-mM Tris HCl (pH 8.0), 250-mM NaCl, 0.1% [v/v] Tween-20) with protease inhibitor cocktail (Sigma), sonicated five times for 30 s and then centrifuged at 16 000 × g for 30 min. Recombinant proteins were purified from the clarified extracts on an AKTA Prime FPLC system (GE Healthcare) using GSTrap HP columns (GE Healthcare). To remove the GST tag from GST–APE1, WT and mutant proteins were further hydrolyzed with factor Xa (five factor Xa units per milligram of recombinant GST-fused protein) for 4 h at room temperature (RT). The protease was then removed from the sample using a benzamidine HiTrap FF column (GE Healthcare), and the proteins were then purified on a GSTrap HP column to purify APE1 from the GST tag. The quality of purification was checked by SDS-PAGE analysis. Accurate quantification of all recombinant proteins was performed by colorimetric Bradford assays (Bio-Rad) and confirmed by SDS-PAGE (Supplementary Figures S1 and S2).
GST pull-down assay
For GST pull-down experiments, 15 pmol of GST-Mia40 was added together with 15 pmol of APE1 WT or APE1 C65S, C93S, C99S, C65/93S, C65/99S mutants, to 10 μl of glutathione-Sepharose 4B beads (GE Healtcare). Binding was performed in Binding buffer (1 X PBS, 50-mM Tris pH 8.0, 50% glycerol, 0.5% NP40 and protease inhibitor) for 3 h under rotation at 4°C. The beads were washed three times with Washing buffer (1 X PBS, 0.1% NP40, 10% glycerol and protease inhibitor). Proteins were eluted for 10 min with Elution buffer (50-mM Tris pH 8.0 and 10-mM GSH). Then, samples were dissolved in reducing lamely buffer (250-mM Tris-HCl pH 6.8, 8% SDS, 40%, Glycerol, 0.08% Bromophenol Blue, 20% β-mercaptoethanol) or non-reducing lamely buffer (250-mM Tris-HCl pH 6.8, 8% SDS, 40%, Glycerol, 0.08% Bromophenol Blue) and loaded onto 12% SDS-PAGE gels followed by western blot. The presence of APE1 and GST proteins was detected using anti-APE1 protein monoclonal antibody and GST monoclonal antibody, respectively.
Endonuclease assay
Endonuclease assay was performed by carrying out enzymatic reactions in a final volume of 10 μl. After mitochondrial purification, the protein content was quantified by Bradford and 25 ng of mitochondrial protein extract was dissolved in AP buffer (50-mM HEPES, 50-mM KCl, 10-mM MgCl2, 1-μg/ml BSA, 0.05% (w/v) Triton X-100, pH 7.5). Reactions were started by adding 100 nM of double-stranded abasic DNA substrate obtained by annealing a DY-782-labeled oligonucleotide [5′-AAT TCA CCG GTA CCF TCT AGA ATT CG-3′] (Eurofins MWG Operon), where F indicates a tetrahydrofuran residue, with the complementary sequence [5′-CGA ATT CTA GAG GGT ACC GGT GAA TT-3′] and incubated at 37°C for the reported amount of time. Reactions were halted by the addition of Stop solution (96% (v/v) formamide, 10-mM EDTA, Xylene cianolo and bromophenol blue), separated onto a 20% (w/v) denaturing polyacrylamide gel and analyzed on an Odyssey CLx scanner (Li-Cor Biosciences). mtAPE1 endonuclease activity of control and Mia40 siRNA was measured in all five biological replicates. The percentage of substrate converted to product was determined using the ImageStudio software (Li-Cor Biosciences). For each time point reported in the diagram of Figure 3C the percentage of product formation and SD were calculated as the average of the biological replicates.
Proximity ligation assay (PLA)
To monitor the interaction between ectopic APE1 and endogenous Mia40 in living cells, the in situ Proximity Ligation Assay kit (Olink Bioscience) was used. HeLa cells were seated into a glass coverslip in the amount of 8 × 104 per 24 X multiwell plate, and then transiently transfected with a FLAG-tagged form of APE1 WT, or mutants. Twenty four hours after transfection, cells were fixed with 4% (w/v) paraformaldehyde, permeabilized for 5 min with Triton X-100 0.25% in PBS 1X and incubated with 5% normal fetal bovine serum in PBS-0.1% (v/v) Tween-20 (blocking solution) for 30 min, to block unspecific binding of the antibodies. Cells were then incubated with the mouse monoclonal anti-FLAG FITC conjugated antibody (Sigma Aldrich) at a final dilution of 1:500 for 1 h an RT, in a humid chamber. After washing three times with PBS-0.1% (vol/vol) Tween-20 (washing solution) for 5 min, cells were then incubated with a rabbit anti-Mia40 overnight at 4°C. PLA was performed following manufacturer's instructions. Technical controls, represented by the omission of the anti-Mia40 primary antibodies, resulted in the complete loss of PLA signal. Images were acquired using a Leica TCS SP laser-scanning confocal microscope (Leica Microsystems) equipped with a 488-nm argon laser, a 543-nm HeNe laser and a 63X oil objective (HCX PL APO 63X Leica). At least 15 randomly selected cells per condition were analyzed. PLA-spots present in each single cell were then scored using the BlobFinder software (Olink Bioscience). Anti-FLAG staining for APE1 was used to identify cell nuclei.
Cell viability assay
Cell viability was measured through the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer's instructions.
Statistical analyses
Statistical analyses on biological data were performed using the Microsoft Excel data analysis program for Student's t-test analysis. P < 0.05 was considered as statistically significant.
RESULTS
Cys55 of Mia40 forms a mixed disulfide bond with APE1
In order to assess the interaction between APE1 and Mia40 in an in vivo cell model, we transiently transfected HeLa cells with pCMV 5.1 plasmids expressing Mia40-HisTag, and APE1-FLAG (IP) or the empty vector as control (Mock). Then, APE1-FLAG was immunopurified and Mia40-HisTag protein was detected in the IP fraction (Figure 1A). To evaluate if the interaction between the two proteins involved the formation of a disulfide bond, recombinant Mia40 expressed in fusion with a GST tag at the N-terminus (rGST-Mia40) (Supplementary Figure S1A) and recombinant APE1 (rAPE1) (Supplementary Figure S2) were produced and the interaction quantified through GST pull-down assays. rGST-Mia40 was used as ‘bait’ and rAPE1 was used as ‘prey’, while GST protein alone was used as a negative control. GST pull-down samples separated in SDS-PAGE under reducing conditions demonstrated that APE1 was able to interact with Mia40 (Figure 1B, lane 2), and incubation with a reducing agent (DTT) led to an almost complete loss of APE1 signal in the pull-down fraction (lane 3). To evaluate the presence of a Mia40/APE1 complex, GST pull-down samples were also separated under non-reducing conditions and immunorecognition was performed with anti-APE1 anti-GST antibodies. In both cases was demonstrated the formation of a high-molecular weight band accounted for the presence of a complex between the two proteins (Figure 1B, lane 2) that was lost in samples treated with DTT (lane 3).
Data from the literature identify the Cys55 of Mia40 as the catalytically active residue responsible for the disulfide bond formation with its substrates (18). To determine if APE1 also formed a mixed disulfide bridge with Mia40 through this residue, GST pull-down assays were performed comparing APE1's binding activity of rGST-Mia40-WT with that of rGST-Mia40 C55S mutant form (Figure 1C). The mutant Mia40 protein showed a significant reduction of affinity with APE1 (40 ± 2%) but not a complete loss of binding as for other Mia40 substrates (22,27). In conclusion, our data demonstrate that Mia40 interacts with APE1 through the formation of a mixed disulfide bridge involving Mia40 Cys55. Moreover, the partial persistence of interaction even when Cys55 is mutated supports the hypothesis of a non-covalent contribution to the interactions between the two proteins.
Cys93 of APE1 is essential for disulfide bond formation with Mia40
GST pull-down experiments were performed to identify which of APE1 redox cysteine formed a mixed disulfide bridge with Mia40. We expressed and purified recombinant APE1 single and double cysteine to serine mutants (C65S, C93S, C99S, C65S+C93S, C65S+C99S) (Supplementary Figure S2). As reported in Figure 2A, the substitution of APE1 Cys93 and the substitution of both cysteine in position 65 and 93 with serine residues completely abolished the interaction of the two proteins confirming the residue Cys93 of APE1 to be primarily involved in the disulfide bond formation with Mia40. The substitution of Cys99 (single and double mutants with Cys65) did not alter the interaction with Mia40. Interestingly, the substitution of Cys65 alone determined a stronger interaction with Mia40 than the WT protein (Figure 2A). To further investigate in vivo the interaction between APE1 and Mia40, we performed PLA assay overexpressing APE1-FLAG WT, Cys65 and Cys93 to Ser mutants and quantified the interaction with endogenous Mia40 (Figure 2B). Presence of PLA signals demonstrated that ectopic APE1 interacted with endogenous Mia40 which is known to be present only in the mitochondria IMS. As reported in the histogram of Figure 2B, APE1 C65S had a stronger interaction with Mia40 than the WT protein, while the C93S mutant showed PLA signals similar to that of the control reaction (Ctrl) where the primary antibody for Mia40 was omitted. Interestingly, the stronger interaction of APE1 C65S was not accompanied by more efficient localization of APE1 in mitochondrial (6). This behavior is reminiscent of the conditional mutant mia40-4 in yeast that binds client proteins but is defective in the completion of their oxidative folding and ultimately their mitochondrial accumulation (16,25).
Mutation of APE1's Cys93 affects protein stability inducing mtDNA damage
We evaluated the effect of APE1's redox-active cysteine mutation on the stability of the protein, by transiently transfecting HeLa cells with pCMV 5.1 vectors expressing APE1 WT, Cys65, Cys93, Cys99, Cys65+Cys93 and Cys65+Cys99 to Ser mutants. Expression levels of ectopic flagged APE1's mRNA were analyzed by RT-PCR not showing any major significant differences between the WT and all mutant forms (Figure 3A). On the contrary, ectopic flagged APE1 protein levels in total cell extracts revealed a significant reduction of both the single C93S (33 ± 8%) and the double C65S+C93S (12 ± 8%) mutants, therefore supporting a pivotal role of Cys93 for the overall stability of the protein itself (Figure 3B). Next, we assessed the effect of the expression of APE1's C65S and C93S mutants on the levels of mtDNA damage. To down regulate endogenous APE1's expression, we transiently transfected HeLa cells with a pSuper vector coding for an shRNA sequence that specifically target APE1's mRNA, as well as a scramble sequence as a control, and re-expressed shRNA resistant APE1 WT, Cys 93 and Cys56 to Ser mutant forms (Figure 3C). Then, we evaluated the effect on the stability of the mtDNA of these mutants by measuring the levels of mtDNA damage through a Q-PCR assay that allowed us to calculate the relative number of mtDNA lesions (30). As reported in the histogram of Figure 3D, down regulation of APE1 expression led to increased levels of mtDNA damage, while the re-expression of APE1 WT rescued the phenotype. Remarkably, re-expression of the redox C65S mutant did not rescue the phenotype, while the C93S mutant showed even higher levels of mtDNA damage compared to silenced cells. In conclusion, we proved that APE1's Cys93 residue is necessary for the interaction with Mia40, and that the mutation of this residue determines the instability of the protein, inducing a mitochondrial stress condition that finally leads to the increase of mtDNA damage.
APE1 is required for the repair of mtDNA
To prove that APE1 is necessary for the maintenance of the mitochondrial genome stability HeLa cells where endogenous expression of APE1 was knocked-down by siRNA technology in a conditional manner through a doxycycline-responsive promoter were used (29). Control HeLa cells, obtained after stable transfection with a scrambled sequence (Scr-1), or silenced HeLa cells bearing the APE1 specific siRNA plasmid (shRNA), were treated with doxycycline for 10 days, mitochondria were isolated, and expression levels of APE1 were evaluated by western blotting analysis. As reported in Figure 4A, APE1 mitochondrial expression in the shRNA expressing clone was strongly reduced, with protein levels less than 10% compared to the control. The endonuclease activity was also significantly reduced (Figure 4B). The levels of mtDNA damage in the HeLa cell model upon APE1 knock down were then measured, and interestingly, lack of APE1 in shRNA expressing cells was sufficient to strongly increase the mtDNA damage under basal conditions (white bars) (Figure 4C). Moreover, when cells were treated with H2O2 (gray bars) the mtDNA damage was significantly increased in shRNA expressing clone with respect to control. Altogether, our data support the essential role of APE1 for the repair of oxidative lesions and the repair of mtDNA.
Mia40 is required for mitochondrial translocation of APE1
Having demonstrated that APE1 interacts with Mia40, the next step was to test our hypothesis for an active role of this protein in controlling APE1's translocation into the mitochondrial IMS. First, Mia40 was overexpressed and the amount of APE1 within mitochondrial fractions was measured. A slight, but significant increase of the amount of mitochondrial APE1 form was observed (Figure 5A). Next, to prove the biological relevance of the MIA pathway in controlling APE1 mitochondrial translocation, HeLa cells were transiently transfected with specific siRNA against Mia40 (Mia40) and the relative scramble control (Ctrl). Seventy two hours upon transfection, cells were harvested, mitochondria and nuclei isolated and the amount of APE1 and Mia40 was evaluated through western blotting analyses. Mia40 levels were significantly reduced (up to 90%) and consequently, a significant reduction of APE1 levels (35 ± 7%) in mitochondria was observed, suggesting a direct role of Mia40 in controlling APE1's mitochondrial translocation. On the contrary, no significant variation in APE1's nuclear contents was observed (Figure 5B). Then, to verify if the decrease of APE1 content resulted in a reduction of mitochondrial endonuclease activity, APE1's enzymatic activity on abasic DNA was measured. A tetrahydrofuran (THF) containing dsDNA probe was incubated with equal amounts of mitochondrial extracts from Ctrl and Mia40 siRNA-transfected cells for the reported times. A significant decrease of endonuclease activity was apparent upon Mia40 knock down due to the reduction of APE1 amount in the mitochondrial fraction (Figure 5C). To further support the physiological relevance of Mia40 in controlling APE1 trafficking, we measured the level of mtDNA damage in Mia40 siRNA-transfected cells, and under basal conditions, loss of Mia40 expression did not significantly affect mtDNA damage levels. However, when cells were treated with H2O2, the degree of damage was significantly increased, suggesting that the reduction in mitochondrial APE1 amount, due to the loss of Mia40, affected the mtDNA repair capability and thus sensitizing cells to oxidative damaging agents (Figure 5D). The same experiment was performed on the human hepatocellular carcinoma (HCC) cell line JHH6, by measuring the level of mtDNA damage in Mia40 siRNA-transfected cells upon H2O2 treatment. The data obtained were consistent with the previous results on the HeLa cell line, therefore supporting the generality of the mechanism (Figure 5D). Overall, these data demonstrate, for the first time, a direct role of the Mia40 protein in the import of APE1 into the mitochondrial compartment and its indirect role in controlling the stability of mtDNA upon oxidative damage.
Loss of Mia40 expression sensitizes JHH6 hepatic cells to MMS and H2O2 treatments
APE1's extra-nuclear localization is a marker of poor prognosis in different cancers, such as hepatocarcinoma (31). However, up to now, the role of this aberrant localization in tumor progression has been unknown. In order to determine whether a correlation exists between Mia40 expression levels and the amount of mitochondrial APE1 protein form in tumor cells, subcellular fractionation was performed on two hepatoma cell lines in which we previously demonstrated that the expression levels of APE1 were significantly different, particularly in the cytoplasmic compartment (31): Huh7 (well-differentiated HCC) and JHH6 (poorly differentiated HCC). Western blot analyses highlighted the existence of a direct correlation between the expression levels of Mia40 and the amount of APE1 in the mitochondrial fraction of JHH6 cell extracts (Figure 6A). To ascertain that the increased mitochondrial localization of APE1 was associated with increased resistance to genotoxic damage, both cell lines were treated with increasing amounts of the DNA alkylating agent MMS and cell viability was measured upon 8 h of treatment. As reported in Figure 6B, JHH6 cells were significantly more resistant to the MMS-treatment than Huh7 cells. The contribution of Mia40 to the increased resistance observed in JHH6 cells was verified. Down regulation of Mia40 expression by siRNA transfection in JHH6 cells caused a significant reduction of mitochondrial APE1's levels (Figure 6C) and a concomitant increased sensitivity to MMS and H2O2 treatments than seen in the control cells (Figure 6D). In conclusion, our data support the general hypothesis for an unexpected role of the MIA pathway in chemoresistance by directly controlling APE1 translocation into mitochondria and therefore influencing the stability of mtDNA upon genotoxic damage.
DISCUSSION
In this study, we investigated the role played by the interaction between Mia40 and the DNA repair protein APE1: specifically its control over the translocation into the mitochondrial compartment and the involvement of the protein transport pathway, MIA, in the maintenance of the mitochondrial genome stability. Previously, altered expression levels and aberrant localization have been demonstrated to correlate with tumor aggressiveness and chemoresistance (32–38) but the mechanisms responsible for the unusual phenotype have never been investigated in detail. As a consequence of its abnormal expression and localization, and due to its essential role in repairing DNA lesions generated by chemotherapeutic agents, APE1 is an intriguing candidate for novel therapeutic strategies aimed at inhibiting DNA repair as an effective adjuvant treatment in cancer therapy. Data reported in the literature concerning APE1 trafficking are still scanty and in some cases contradictory. In this work, we filled this gap and depicted a new model involving the MIA pathway that may explain how APE1 is translocated into the mitochondrial IMS. Several data reported in the literature demonstrate that APE1 is present within the mitochondria of mammalian cells in its full-length form (6,8,9), therefore excluding the hypothesis that involvement of a proteolytic process is responsible for the NLS removal and the re-directioning of APE1 into the mitochondrion (7). The mitochondrial targeting signal of APE1 has been identified in the region spanning the residues 289–318 where Lys299 and Arg301 are the critical sites so that, if mutated, they completely abolish APE1 mitochondrial translocation under oxidative stress conditions (9). Previous studies have also shown that APE1 translocates into the mitochondria IMS, mainly through the TOM-dependent pathway, but detailed mechanisms have not yet been proposed (9). Our data demonstrate that upon passage through the TOM channel, Mia40 is able to interact and bind APE1 by forming a disulfide bridge between APE1's Cys93 and Mia40's Cys55 residues (Figure 1 B and C). Through GST pull-down (Figure 2A) and PLA (Figure 2B) experiments, we demonstrated that mutation of Cys93 completely abolished the interaction with Mia40. Moreover, while mutating Cys65 to Ser, we observed an increased interaction between the two proteins. This could be explained when taking into account that when Cys65 alone is mutated, Mia40 is able to bind APE1's Cys93 residue but cannot complete the reaction because of the lack of the second cysteine (Cys65), necessary for the disulfide bond formation. To further evaluate the relevance of these two cysteine residues, we substituted endogenous APE1 through RNAi technology, re-expressed APE1 WT or Cys65 and Cys93 to Ser mutants (Figure 3C), and measured mtDNA damage. The WT form was able to rescue the phenotype of APE1's silenced cells, while no rescue was observed with the C65S mutant. Remarkably, expression of C93S mutant exerted a dominant negative effect leading to even higher levels of mtDNA damage (Figure 3D).
While the C-term DNA-repair domain of APE1 is highly conserved among different species, the N-term redox domain is unique to mammals and has been acquired during phylogenetic evolution (39). Through its redox domain, APE1 exerts its activity by reducing key cysteine residues of a number of important transcription factors involved in both cell survival processes and apoptosis induction. In the N-term domain three cysteine residues (65, 93 and 99) are present. The current crystallographic structure was obtained in 1997 using a recombinant protein missing the first 35 N-terminal amino acids (40). The Cys65 is absolutely required for redox activity on nuclear transcriptional factors but, in this model, it appears as a buried residue, and therefore APE1 should undergo a local or a more extensive remodeling process to expose the Cys65 to the surface for the interaction with TFs (41). Cys99 is exposed on the surface of the protein but its substitution has no effect on redox activity. Cys93 is also a buried residue and before the determination of the crystal structure of APE1, it was proposed that Cys65 and 93 may form a disulfide bond (42). Indeed, in the crystallographic structure of APE1, Cys65 and Cys93 are positioned on opposite sides of a β sheet with a distance of >8 Å apart and therefore a substantial conformational change in the structure of the protein would be required for the occurrence of a proper disulfide bond formation between these residues (42). Future studies are required to investigate APE1 structure and structural changes but it is likely that APE1 is a multifunctional dynamic protein that, in vivo, may adopt different structural conformations to fulfill its activities.
In a previous work, we demonstrated that the majority of APE1 protein accumulates within the IMS and that mutation of Cys65 residues leads to reduced mitochondrial APE1 localization (6). Here, we showed that Mia40 is involved in the transport of APE1 into the IMS. However, a portion of APE1 is further translocated, through a still unidentified mechanism, to the matrix where the mtDNA resides.
This mode of translocation was shown to exist for Mrp10, a subunit of the mitochondrial ribosome, which is located in the matrix and crosses the IMS interacting with Mia40 (43). This protein presents conserved cysteine residues that are oxidized by Mia40 during import. The semifolded protein with one disulfide bond is able to pass the inner membrane to localize in the matrix. Conversely, if Mia40 is silenced the protein in the matrix is only partially folded and degraded because it is not able to be assembled in the ribosomes (43).
To assess the physiological relevance of the proposed import mechanism, we determined the contribution of the MIA pathway in the stability of mtDNA by correlating the expression levels of Mia40 with the mitochondrial APE1 content. Data obtained highlighted that overexpression of Mia40 in HeLa cells led to increased levels of mtAPE1 (Figure 5A). Conversely, the loss of Mia40 expression led to a significant reduction of mtAPE1 form (Figure 5B) that subsequently led to accumulation of mtDNA damage as a consequence of oxidative stress induction (Figure 5D). Therefore, our data clearly support a direct involvement of Mia40 in controlling APE1 translocation into the mitochondria and, consequently, in the stability of mtDNA. Also the tumor suppressor protein p53 was found to localize within human mitochondria through the MIA pathway. Authors demonstrated that the import is an active process activated by oxidative stress, respiration-dependent, and that it contributes to the maintenance of mtDNA integrity (28).
Cytoplasmic relocalization of APE1 has been found to be associated with a higher tumor aggressiveness and a poorer prognosis for the patient in HCC (31). Because Mia40 was also found to be upregulated in some tumors (44), we decided to analyze APE1 and Mia40 expression levels in two HCC cell lines with different degree of differentiation. Higher expression levels of Mia40 in the HCC poorly differentiated JHH6 cell line correlated positively with augmented levels of mitochondrial APE1 (Figure 6A) and, consequently, were associated with an increased resistance to alkylating agent -treatment with respect to the well-differentiated Huh7 cell line (Figure 6B). Also in this case, alteration of Mia40 expression and activity in JHH6 cells upon silencing determined a reduction of mitochondrial APE1 content which resulted in an increased cellular sensitivity to alkylating and oxidative stress (Figure 6D). Alkylating antineoplastic agents are lipophilic bearing positive charges and therefore tend to accumulate within the mitochondria and generate alkylation lesions on mtDNA with a 10-fold higher ratio with respect to nDNA (45). APE1 translocates into mitochondria increasing mtDNA repair rate and therefore, in our model, increased expression of Mia40 could lead to the promotion of cell survival.
In summary, our data strongly support the hypothesis for a redox-assisted mechanism dependent on Mia40 in controlling APE1 translocation within mitochondria. Several questions, in particular how the protein is translocated to the matrix compartment, are still open and will require further studies. However, our findings open new translational perspectives for cancer treatment based on the use of inhibitors of APE1/Mia40 interaction as adjuvant to DNA damaging chemotherapeutic agents.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Associazione Italiana per la Ricerca sul Cancro [IG14038 to G.T.]; the Crossborder Cooperation Program Italy–Slovenia 2007–2013, the European Regional Development Fund (ERDF) and the National Funds, the Autonomous Region Friuli Venezia Giulia in quality of Managing Authority [to G.T.]; the National Science Centre, Poland [2012/05/B/NZ3/00781 to M.W.]; the EMBO Installation Grant [to A.C.]. Funding for open access charge: the Associazione Italiana per la Ricerca sul Cancro [IG14038]; Crossborder Cooperation Program Italy–Slovenia 2007– 2013, the European Regional Development Fund (ERDF) and the National Funds, the Autonomous Region Friuli Venezia Giulia in quality of Managing Authority; National Science Centre, Poland [2012/05/B/NZ3/00781].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bapat A., Fishel M.L., Kelley M.R. Going ape as an approach to cancer therapeutics. Antioxid. Redox Signal. 2009;11:651–668. doi: 10.1089/ars.2008.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tell G., Damante G., Caldwell D., Kelley M.R. The intracellular localization of Ape1/Ref-1: more than a passive phenomenon. Antioxid. Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 3.Xanthoudakis S., Miao G.G., Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. U.S.A. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung H., Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 5.Stuart J.A., Hashiguchi K., Wilson D.M. III, Copeland W.C., Souza-Pinto N.C., Bohr V.A. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res. 2004;32:2181–2192. doi: 10.1093/nar/gkh533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vascotto C., Bisetto E., Li M., Zeef L.A., D'Ambrosio C., Domenis R., Comelli M., Delneri D., Scaloni A., Altieri F., et al. Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol. Biol. Cell. 2011;22:3887–3901. doi: 10.1091/mbc.E11-05-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chattopadhyay R., Wiederhold L., Szczesny B., Boldogh I., Hazra T.K., Izumi T., Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tell G., Crivellato E., Pines A., Paron I., Pucillo C., Manzini G., Bandiera A., Kelley M.R., Di Loreto C., Damante G. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutat. Res. 2001;485:143–152. doi: 10.1016/s0921-8777(00)00068-9. [DOI] [PubMed] [Google Scholar]
- 9.Li M., Zhong Z., Zhu J., Xiang D., Dai N., Cao X., Qing Y., Yang Z., Xie J., Li Z., et al. Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 2010;285:14871–14881. doi: 10.1074/jbc.M109.069591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Druzhyna N.M., Wilson G.L., LeDoux S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter C., Park J.W., Ames B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. U.S.A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohr V.A. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic. Biol. Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 13.Bohr V.A., Stevnsner T., de Souza-Pinto N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 14.Grishko V.I., Rachek L.I., Spitz D.R., Wilson G.L., LeDoux S.P. Contribution of mitochondrial DNA repair to cell resistance from oxidative stress. J. Biol. Chem. 2005;280:8901–8905. doi: 10.1074/jbc.M413022200. [DOI] [PubMed] [Google Scholar]
- 15.Sokol A.M., Sztolsztener M.E., Wasilewski M., Heinz E., Chacinska A. Mitochondrial protein translocases for survival and wellbeing. FEBS Lett. 2014;588:2484–2495. doi: 10.1016/j.febslet.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 16.Chacinska A., Pfannschmidt S., Wiedemann N., Kozjak V., Sanjuán Szklarz L.K., Schulze-Specking A., Truscott K.N., Guiard B., Meisinger C., Pfanner N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bien M., Longen S., Wagener N., Chwalla I., Herrmann J.M., Riemer J. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol. Cell. 2010;37:516–528. doi: 10.1016/j.molcel.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Banci L., Bertini I., Calderone V., Cefaro C., Ciofi-Baffoni S., Gallo A., Kallergi E., Lionaki E., Pozidis C., Tokatlidis K. Molecular recognition and substrate mimicry drive the electron-transfer process between MIA40 and ALR. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4811–4816. doi: 10.1073/pnas.1014542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banci L., Bertini I., Calderone V., Cefaro C., Ciofi-Baffoni S., Gallo A., Tokatlidis K. An electron-transfer path through an extended disulfide relay system: the case of the redox protein ALR. J. Am. Chem. Soc. 2012;134:1442–1445. doi: 10.1021/ja209881f. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann S., Rothbauer U., Mühlenbein N., Baiker K., Hell K., Bauer M.F. Functional and mutational characterization of human MIA40 acting during import into the mitochondrial intermembrane space. J. Mol. Biol. 2005;353:517–528. doi: 10.1016/j.jmb.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 21.Yang J., Staples O., Thomas L.W., Briston T., Robson M., Poon E., Simoes M.L., El-Emir E., Buffa F.M., Ahmed A., et al. Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J. Clin. Invest. 2012;122:600–611. doi: 10.1172/JCI58780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banci L., Bertini I., Cefaro C., Cenacchi L., Ciofi-Baffoni S., Felli I.C., Gallo A., Gonnelli L., Luchinat E., Sideris D., et al. Molecular chaperone function of Mia40 triggers consecutive induced folding steps of the substrate in mitochondrial protein import. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20190–20195. doi: 10.1073/pnas.1010095107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sideris D.P., Tokatlidis K. Oxidative folding of small tims is mediated by site-specific docking onto Mia40 in the mitochondrial intermembrane space. Mol. Microbiol. 2007;65:1360–1373. doi: 10.1111/j.1365-2958.2007.05880.x. [DOI] [PubMed] [Google Scholar]
- 24.Grumbt B., Stroobant V., Terziyska N., Israel L., Hell K. Functional characterization of Mia40p, the central component of the disulfide relay system of the mitochondrial intermembrane space. J. Biol. Chem. 2007;282:37461–37470. doi: 10.1074/jbc.M707439200. [DOI] [PubMed] [Google Scholar]
- 25.Böttinger L., Gornicka A., Czerwik T., Bragoszewski P., Loniewska-Lwowska A., Schulze-Specking A., Truscott K.N., Guiard B., Milenkovic D., Chacinska A. In vivo evidence for cooperation of Mia40 and Erv1 in the oxidation of mitochondrial proteins. Mol. Biol. Cell. 2012;23:3957–3969. doi: 10.1091/mbc.E12-05-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer M., Riemer J. The mitochondrial disulfide relay system: roles in oxidative protein folding and beyond. Int. J. Cell Biol. 2013:742923. doi: 10.1155/2013/742923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabriel K., Milenkovic D., Chacinska A., Müller J., Guiard B., Pfanner N., Meisinger C. Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J. Mol. Biol. 2007;365:612–620. doi: 10.1016/j.jmb.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang J., Wang P.Y., Huang X., Chen X., Kang J.G., Hwang P.M. Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc. Natl. Acad. Sci. U.S.A. 2013;110:17356–17361. doi: 10.1073/pnas.1310908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vascotto C., Cesaratto L., Zeef L.A., Deganuto M., D'Ambrosio C., Scaloni A., Romanello M., Damante G., Taglialatela G., Delneri D., et al. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics. 2009;9:1058–1074. doi: 10.1002/pmic.200800638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furda A.M., Bess A., Meyer J.N., Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Maso V., Avellini C., Crocè L.S., Rosso N., Quadrifoglio F., Cesaratto L., Codarin E., Bedogni G., Beltrami C.A., Tell G., et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol. Med. 2007;13:89–96. doi: 10.2119/2006-00084.DiMaso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudhakar J., Khetan V., Madhusudan S., Krishnakumar S. Dysregulation of human apurinic/apyrimidinic endonuclease 1 (APE1) expression in advanced retinoblastoma. Br. J. Ophthalmol. 2014;98:402–407. doi: 10.1136/bjophthalmol-2013-304166. [DOI] [PubMed] [Google Scholar]
- 33.Li M.X., Shan J.L., Wang D., He Y., Zhou Q., Xia L., Zeng L.L., Li Z.P., Wang G., Yang Z.Z. Human apurinic/apyrimidinic endonuclease 1 translocalizes to mitochondria after photodynamic therapy and protects cells from apoptosis. Cancer Sci. 2012;103:882–888. doi: 10.1111/j.1349-7006.2012.02239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheng Q., Zhang Y., Wang R., Zhang J., Chen B., Wang J., Zhang W., Xin X. Prognostic significance of APE1 cytoplasmic localization in human epithelial ovarian cancer. Med. Oncol. 2012;29:1265–1271. doi: 10.1007/s12032-011-9931-y. [DOI] [PubMed] [Google Scholar]
- 35.Puglisi F., Aprile G., Minisini A.M., Barbone F., Cataldi P., Tell G., Kelley M.R., Damante G., Beltrami C.A., Di Loreto C. Prognostic significance of Ape1/ref-1 subcellular localization in non-small cell lung carcinomas. Anticancer Res. 2001;21:4041–4049. [PubMed] [Google Scholar]
- 36.Xie J., Zhang L., Li M., Du J., Zhou L., Yang S., Zeng L., Li Z., Wang G., Wang D. Functional analysis of the involvement of apurinic/apyrimidinic endonuclease 1 in the resistance to melphalan in multiple myeloma. BMC Cancer. 2014;14:11. doi: 10.1186/1471-2407-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Wang J., Xiang D., Wang D., Xin X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. Int. J. Oncol. 2009;35:1069–1079. [PubMed] [Google Scholar]
- 38.Wang D., Xiang D.B., Yang X.Q., Chen L.S., Li M.X., Zhong Z.Y., Zhang Y.S. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer. 2009;66:298–304. doi: 10.1016/j.lungcan.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 39.Georgiadis M.M., Luo M., Gaur R.K., Delaplane S., Li X., Kelley M.R. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorman M.A., Morera S., Rothwell D.G., de La Fortelle E., Mol C.D., Tainer J.A., Hickson I.D., Freemont P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo M., He H., Kelley M.R., Georgiadis M.M. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid. Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker L.J., Robson C.N., Black E., Gillespie D., Hickson I.D. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longen S., Woellhaf M.W., Petrungaro C., Riemer J., Herrmann J.M. The disulfide relay of the intermembrane space oxidizes the ribosomal subunit mrp10 on its transit into the mitochondrial matrix. Dev. Cell. 2014;28:30–42. doi: 10.1016/j.devcel.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Yang J., Staples O., Thomas L.W., Briston T., Robson M., Poon E., Simões M.L., El-Emir E., Buffa F.M., Ahmed A., et al. Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J. Clin. Invest. 2012;122:600–611. doi: 10.1172/JCI58780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bandy B., Davison A.J. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging. Free Radic. Biol. Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.