


Abstract
There are the two major pathways responsible for the repair of DNA double-strand breaks (DSBs): non-homologous end-joining (NHEJ) and homologous recombination (HR). NHEJ operates throughout the cell-cycle, while HR is primarily active in the S/G2 phases suggesting that there are cell cycle-specific mechanisms that regulate the balance between NHEJ and HR. Here we reported that CDK2 could phosphorylate RNF4 on T26 and T112 and enhance RNF4 E3 ligase activity, which is important for MDC1 degradation and proper HR repair during S phase. Mutation of the RNF4 phosphorylation sites results in MDC1 stabilization, which in turn compromised HR during S-phase. These results suggest that in addition to drive cell cycle progression, CDK also targets RNF4, which is involved in the regulatory network of DSBs repair.
INTRODUCTION
DNA damage response (DDR) is a series of signal transduction events triggered by DNA damage in cells (1), including the recruitment and activation of proteins involved in DNA damage sensing, checkpoint signaling, chromatin remodeling and DNA repair. The recruitment of DNA damage factors to DNA damage sites is complex and elaborate (2,3). Different DNA damage factors are recruited through distinct processes (2,3). The accumulation of DNA damage factors facilitates DNA repair (4). There are two prominent repair pathways that repair DSBs: non-homologous end joining (NHEJ) and homologous recombination (HR) (5). A homologous template is not required in NHEJ, the two broken ends of DNA are directly ligated resulting in quick, but error-prone, repair (6). Unlike NHEJ, an intact homologous DNA sequence is utilized in HR, which makes HR more accurate. Therefore, HR primarily operates in the S/G2 phases of the cell cycle in mammalian cells as it requires an intact sister chromatid (7). HR is reported to occur in several steps. The initial resection of the DNA ends is regulated by the MRN complex with CtIP to produce short 3′ overhangs (8–10). Then the 3′ overhangs are extended by further resection through Exo1 and Dna2 nucleases (11–13). The 3′ overhangs are recognized by the replication protein A (RPA) which is then replaced by Rad51 (radiation sensitive 51) with the assistance of other factors (14). The Rad51 bound ssDNA then moves into the homologous double-stranded DNA (dsDNA) template (strand invasion)(15). As the invading 3′ strand extend, Holliday junctions are formed, which will be resolved subsequently (16–18). Thus an error-free repair of the DSBs is completed (16–18). Although the process of HR and NHEJ are extensively studies, how the NHEJ and HR pathways cooperate to complete the repair of DSBs remains unclear.
Cyclin-dependent kinases (CDKs) is a family of serine/threonine kinases. Forming a complex with cyclins, CDKs tightly control the cell cycle (19,20). It is established that D-type cyclins form a complex with CDK4 and/or CDK6, which could phosphorylate Retinoblastoma protein (Rb) family early in the G1 phase (21,22). This leads to the activation of E2F transcription factors, which induce the expression of E2F targeting genes required for cell cycle progression (23,24). In the late G1 phase, CDK2/cyclin E complexes regulate the transition from G1 to S phase (21,22). Then CDK2/cyclin A complexes plays an important role in S phase progression. Finally CDK1/cyclin B complexes are involved in the progression of mitosis (25). However, when the interphase CDKs (CDK2, CDK3, CDK4 and CDK6) are absent, the CDK1 could compensate and drive the cell division and embryonic development in mice, indicating the CDKs have a significant plasticity in regulating cell cycle progression (26). It was reported that CDKs are also involved in other functions other than cell cycle regulation, such as DNA damage response (16,27,28). In yeast, CDK1 is required for the Mec1/Rad53-mediated checkpoint response following DSB and the Mre11-dependent DSB resection (29). Inhibition of CDK would abrogate the DSB resection, while a Sae2 (CtIP in human) S267E mutant mimicking a CDK phosphorylation site could alleviate the need of CDK activity (30). In Human, CDK mediated-phosphorylation of CtIP at Thr847 has also been shown to be important for DSB resection (31). Besides, there are many proteins involved in DDR are found to be CDK targets, such as BRCA1 and 2, Rad9, Crb2, and ATRIP, and these phosphorylation events have been shown to be important for proper DNA damage response (32–36). It was proposed that the DNA damage response is regulated by the overall CDK activity in mammalian cells (28).
In our previous study, we found that MDC1, although is required for the accumulation of DDR factors, needs to be sumoylated and removed from DSBs for proper HR. RNF4 regulates the degradation of sumoylated MDC1, and a defect in MDC1 sumoylation results in ineffective HR repair (4,37,38). In this study, we report that RNF4 could be phosphorylated by CDK2 in S-phase, and RNF4 phosphorylation is required for the degradation of MDC1 and proper HR repair following DNA damage. The inhibition of RNF4 phosphorylation results in sustained IR induced MDC1 foci and a defect in HR repair. These findings shed new light on the mechanism how HR repair is regulated in a cell cycle-dependent manner.
MATERIALS AND METHODS
Cell culture
U2OS cells were grown in McCoy's 5A (ATCC) supplemented with 10% fetal bovine serum (Gibco). HEK293T cells were grown in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco).
Plasmids and Cloning
RNF4 was cloned into pCMV-Myc (Clontech) and pCMV/myc/nuc (Invitrogen, modified to have S/FLAG/SBP instead of myc/nuc). Deletion and mutations were generated by site-directed mutagenesis (Stratagene).
Immunoprecipitation and western blotting
Cells were lysed with modified NETN lysis buffer (0.5% NP40, 250 mM NaCl, 50 mM Tris, 1 mM EDTA). To immunoprecipitate MDC1 and RNF4, cells were lysed in NETN containing 250 mM NaCl, 20 mM NEM, 1 mM iodoacetamide and protease inhibitors. Cell lysates were then diluted to 100 mM final concentration of NaCl. Diluted lysates were centrifuged and supernatants were subjected to immunoprecipitation with the indicated antibodies. The rabbit anit-MDC1 antibody was generated as described previously (39). Antibodies used in the studies are: anti-RNF4 (Sigma and Novus), anti-FLAG (Sigma), anti-HA (Covance), anti-Myc (Covance), anti-β-actin (Sigma), anti-Phospho-(Thr)MAPK/CDK substrate (Cell Signaling). For western blotting, antibodies were used at a concentration of 0.5 μg ml−1.
Immunofluorescence
To visualize ionizing radiation induced foci (IRIF), cells were cultured on coverslips and treated with 2 Gy IR followed by recovery as indicated. Cells were then washed in PBS, incubated in 3% paraformaldehyde for 15 min, and permeabilized in 0.5% Triton solution for 5 min at room temperature. Samples were blocked with 5% goat serum and then incubated with primary antibodies for 30 min. Samples were washed three times and incubated with secondary antibodies for 30 min. Cells were then stained with DAPI to visualize nuclear DNA. The coverslips were mounted onto glass slides with anti-fade solution and visualized using a Nikon eclipse 80i fluorescence microscope. The following antibodies were used in immunofluorescence: MDC1 (Millipore), Rad51 (GeneTex), Myc (Covance).
Chemicals and DNA-damaging agents
Ionizing radiation treatment was performed by using an X-ray machine (RS 2000, RadSource). When needed, double thymidine (Sigma) block (2 mM) was performed to synchronize the cells.
In vivo sumoylation assay
Transfected HEK293T cells were lysed in 120 μl of 62.5 mM Tris–HCl (pH 6.8), 2% SDS, 10% Glycerol, 20 mM N-ethylmaleimide (NEM), boiled for 15 min, diluted 10 times with NETN buffer containing protease inhibitors and 20 mM NEM, and centrifuged (16 000 g, 10 min, 4°C) to remove cell debris. With these conditions, the interaction between MDC1 and other possible sumoylated proteins should be disrupted. The lysates were immunoprecipitated with EZviewTM Red Anti-HA affinity gel (Sigma) for 1.5 h at 4°C with rocking and the precipitates were washed and eluted in SDS sample buffer. Western blots were analyzed with indicated antibodies. A covalent reaction but not an interaction with another SUMOylated protein should be detected.
In vitro kinase assay
HEK 293T cells were transfected with FLAG-CDK2 and V5-CyclinE. 48 h later, the cells were collected and the cell lysate were subjected to immunoprecipitated with anti-FLAG antibody. The protein conjugated to the beads were eluted with FLAG peptides and subjected to in vitro kinase assay. Or the active CDK2/CyclinE protein from Millipore was used as kinase in the assay. GST and GST-RNF4 expressed in bacterial were purified and subjected to kinase assay as substrates. The phosphorylation of RNF4 by CDK2 was examined using anti-CDKs substrate antibody.
In vitro sumoylation assay
SUMO modification reactions were typically performed in a total volume of 20 μl with 200 ng of SUMO activating enzyme (UBA2) (Boston Biochem), 100 ng of Ubc9 (Boston Biochem), 1 μg of SUMO1 (Boston Biochem), 100 ng of PIAS4 (Novus) and recombinant GST-MDC1 (aa1818–2089) bound to GST-sepharose (20 μg). The 10X reaction buffer (Boston Biochem) was added with 4 mM ATP-Mg. Reactions were incubated at 30°C for 1hr and stopped by addition of E1 stop buffer (Boston Biochem). The beads were washed with NETN and subjected to in vitro ubiquitination assay.
In vitro ubiquitination assay
The Myc-RNF4 WT or FLAG-RNF4 WT, 2TA, 2TE mutant were expressed in HEK 293T and purified by immunoprecipitation with anti-Myc or anti-FLAG antibody. The Myc-RNF4 or FLAG-RNF4 proteins were then eluted with Myc peptide (200 ng μl−1) (Sigma) or FLAG peptide (200 ng μl−1) (Sigma). Sumoylated GST-MDC1(aa1818–2089) bound to GST-sepharose were washed with Tris-HCl (50 mM, pH7.5) and then incubated with 200 ng of UbcH5a (Boston Biochem), 300 ng of ubiquitin activating enzyme (UBE1) (Boston Biochem), 2 μg of HA-ubiquitin (Boston Biochem), 3 μg Myc-RNF4 or FLAG-RNF4 in 40 μl of reaction buffer (50 mM Tris[pH7.5], 2.5 mM MgCl2, 2 mM ATP, 2 mM DTT). The mixture was incubated at 37°C for 40min, washed with NETN and boiled in SDS sample buffer.
In vivo ubiquitination assay
Transfected HEK293T cells were synchronized and then treated with MG132 (20 μg ml−1) for 2 h followed by irradiation (5 Gy). 4 h later, cells were lysed in 120 ul of 62.5 mM Tris–HCl (pH 6.8), 2% SDS, 10% Glycerol, 20 mM N-ethylmaleimide (NEM) and 1 mM iodoacetamide, boiled for 15 min, diluted 10 times with NETN buffer containing protease inhibitors, 20 mM NEM and 1 mM iodoacetamide and centrifuged (16 000 g, 10 min, 4°C) to remove cell debris. The lysates were immunoprecipitated with EZviewTM Red Anti-HA affinity gel (Sigma) for 1.5 h at 4°C with rocking and the precipitates were eluted in SDS sample buffer. Western blots were analyzed with indicated antibodies.
Homologous recombination assay
A HeLa clone with the integrated homologous recombination reporter DR-GFP was a gift from Dr Maria Jasin (Sloan Kettering). One day after transfection with control or RNF4 siRNA, HeLa-DR-GFP cells were co-transfected with vector, siRNA-resistant WT FLAG-RNF4, FLAG-RNF4-2TA or FLAG-RNF4-2TE and an I-SceI expression vector (pCBA-I-SceI). Cells were harvested two days after I-SceI transfection and subjected to flow cytometric analysis to examine recombination induced by I-SceI digestion. The parallel transfection with pEGFP-C1 was used to normalize for transfection efficiency. Results are normalized to control siRNA.
NHEJ assay
The in vivo end-joining reporter plasmid pEGFP-Pem1-Ad2 has been described (40). Prior to transfection, the pEGFP-Pem1-Ad2 plasmid was digested with HindIII (NEB) for 12 h. A pCherry plasmid (Clontech) was co-transfected with linearized pEGFP-Pem1-Ad2 as a control for transfection efficiency. The cell line under analysis was subcultured a day before transfection and was, 60–70% confluent for transfection. One day after transfection with control or RNF4 siRNA, digested pEGFP-Pem1-Ad2 and pCherry were co-transfected with control vector, siRNA-resistant WT FLAG-RNF4, FLAG-RNF4-2TA or FLAG-RNF4-2TE. Green (EGFP) and red (Cherry) fluorescence was measured by fluorescence-activated flow cytometry (FACS) 48 h later. For FACS analysis cells were harvested, washed in 1X PBS and fixed using 2% paraformaldehyde. FACS analysis was performed on a FACSCalibur instrument (BD Biosciences).
Statistical analysis
Student's t-test was performed. Error bars represent SEMs of three independent experiments.
RESULTS
DSB induced degradation of MDC1 happens more promptly in S-phase
HR is thought to be restricted to the S and G2 phases of the cell cycle when a sister chromatid is available as a homologous DNA template (41). It is more accurate comparing to NHEJ as NHEJ does not require a homologous template (30). NHEJ is not restricted to a certain phase of the cell cycle (42). Previously, we and other groups found that RNF4 regulates HR through ubiquitnating sumoylated MDC1 (4,37,38). To further study whether the RNF4-MDC1 pathway is regulated in a cell cycle dependent pathway, we examined the degradation of MDC1 following DNA damage in G1 and S phase. As shown in Figure 1A, ionizing radiation (IR) induced a decrease of MDC1 levels in S phase, but not in G1 phase. Consistent with this result, MDC1 foci are more sustained in G1 phase cells after IR compared with S phase cells, even though the initial MDC1 foci formation was comparable in those cells (Figure 1B). Since RNF4 only targets sumoylated MDC1, we next examined whether the sumoylation of MDC1 is regulated in a cell cycle dependent way. As shown in Figure 1C, the sumoylation of MDC1 was induced after DNA damage in both G1 and S phase cells. These results suggest that MDC1 sumoylation is regulated in a cell cycle-independent manner. We next hypothesized that RNF4 itself might be regulated in a cell cycle specific mechanism. When we examined the E3 ligase activity of RNF4 during G1 and S-phase by testing the autoubiquitination of RNF4 in vitro, we found that the E3 ligase activity of RNF4 is enhanced in S-phase compared with G1 phase (Figure 1D). On the other hand, DNA damage did not affect RNF4 activity (Figure 1D). These results suggest that RNF4 E3 ligase activity is upregulated in S phase.
Figure 1.
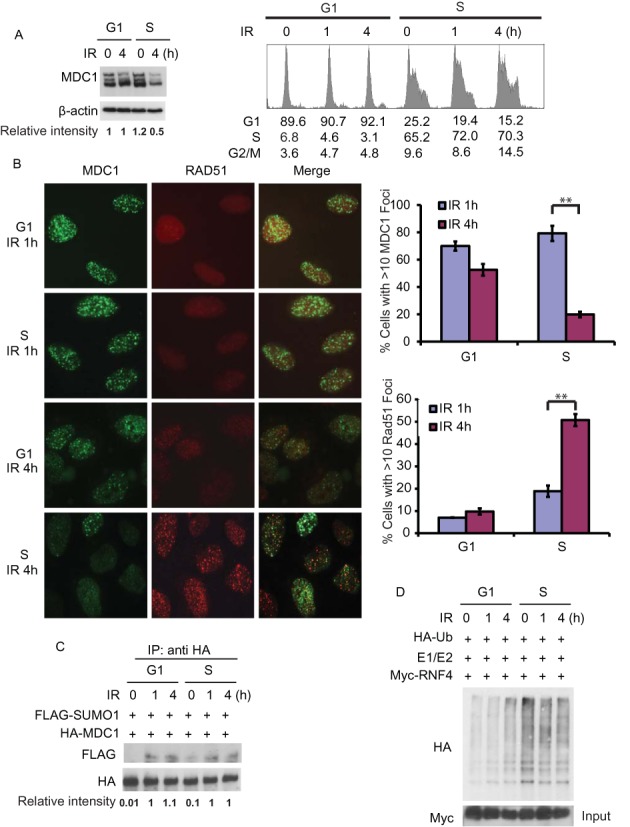
DSB induced degradation of MDC1 happens more promptly in S-phase. (A) HEK 293T cells were synchronized in G1 phase by double thymidine block or released into S phase. Then the cells were treated with or without irradiation (5 Gy) and harvested at indicated time points. Left panel: the level of endogenous MDC1 was examined with indicated antibodies by immunoblotting. Blots were quantified using ImageJ (National Institutes of Health). Right panel: the cell cycle profiles of the cells are analyzed by flow cytometry. (B) U2OS cells were synchronized in G1 phase by double thymidine block or released into S phase. Then the cells were treated with or without irradiation (2 Gy) and probed with indicated antibodies at the indicated time point. Representative images are shown in the left panels, the quantification of the percentage of cells displaying MDC1 (upper) or Rad51 (lower) foci are shown in right panels. Error bars represent SEM. from three independent experiments. **P < 0.01 two‐tailed Student's t‐test. >200 cells were counted per experiment. (C) HEK 293T cells transfected with HA-MDC1 and FLAG-SUMO1 were synchronized in G1 phase by double thymidine block or released into S phase. Then the cells were treated with or without irradiation (5 Gy) and harvested at indicated time points. MDC1 was then immunoprecipitated with anti-HA antibody and MDC1 sumoylation was examined using anti-FLAG antibody. Blots were quantified using ImageJ (National Institutes of Health). (D) HEK 293T cells transfected with Myc-RNF4 were synchronized by double thymidine block or released into S phase. The cells were irradiated (5 Gy) and then collected at indicated time points. RNF4 was then immunoprecipitated and subjected to in vitro ubiquitination reactions in the presence of HA-Ub, UbcH5a and UBE1. The ubiquitination of RNF4 were analyzed by immunoblot as indicated.
CDK2 phosphorylates RNF4 on T26 and T112 during S-phase
As CDKs are a family of protein kinases playing important roles in regulating cell cycle (19,20) and RNF4 is regulated in a cell cycle dependent manner, we utilized a phospho-CDK substrates antibody to examine whether RNF4 is a CDKs substrate. As shown in Figure 2A, after cells were released from double thymidine block, there was increased RNF4 phosphorylation as cells entered S phase. To confirm this result, we treated the samples with λ phosphatase, and the phosphorylation of RNF4 was not evident after the treatment (Figure 2B). On the other hand, when we treated cells with IR, the phosphorylation on RNF4 did not change (Figure 2C). These results suggested that RNF4 might be phosphorylated by CDK in S phase.
Figure 2.
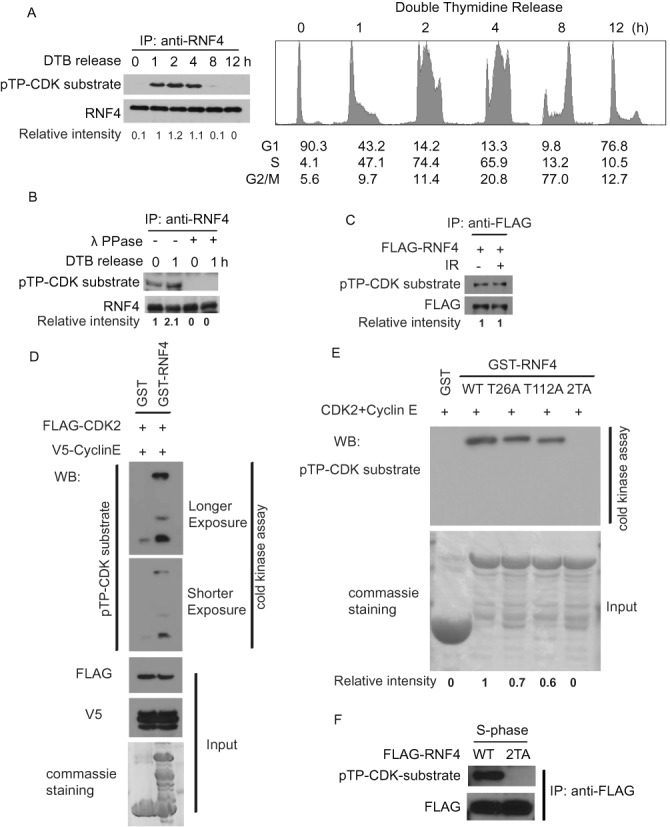
CDK2 phosphorylates RNF4 on T26 and T112 during S-phase. (A) HEK 293T cells were synchronized by double thymidine block and then released. Cells were collected at indicated time points. Left panel: RNF4 was then immunoprecipitated with anti-RNF4 antibody and the phosphorylation of RNF4 by CDKs were examined with indicated antibodies. Blots were quantified using ImageJ (National Institutes of Health). Right panel: the cell cycle profiles of the cells are analyzed by flow cytometry. (B) HEK 293T cells were synchronized by double thymidine block and then released. Cells were collected at indicated time points. RNF4 was then immunoprecipitated with anti-RNF4 antibody and treated with or without λ phosphatase. The phosphorylation of RNF4 by CDKs was examined with indicated antibodies. Blots were quantified using ImageJ (National Institutes of Health). (C) HEK 293T cells transfected with FLAG-RNF4 were synchronized by double thymidine block and then released into S phase. Then the cells were treated with or without irradiation (5 Gy). The cells were collected and the cell lysate were subjected to immunoprecipitated with anti-FLAG antibody. The phosphorylation of RNF4 by CDKs was examined with indicated antibodies. Blots were quantified using ImageJ (National Institutes of Health). (D) HEK 293T cells were transfected with FLAG-CDK2 and V5-CyclinE. 48 hr later, the cells were collected and the cell lysate were subjected to immunoprecipitated with anti-FLAG antibody. The protein conjugated to the beads were eluted with FLAG peptides and subjected to in vitro kinase assay. GST and GST-RNF4 expressed in bacterial were purified and subjected to kinase assay as substrates. The phosphorylation of RNF4 by CDK2 was examined with indicated antibodies. (E) GST and GST-RNF4 WT or mutants expressed in bacterial were purified and subjected to in vitro kinase assay with CDK2+Cyclin E (Millipore). The phosphorylation of RNF4 by CDK2 was examined using anti-phospho-CDKs substrate antibody. Blots were quantified using ImageJ (National Institutes of Health). (F) HEK 293T cells transfected with FLAG-RNF4 WT or 2TA mutant were synchronized by double thymidine block and then released into S phase. RNF4 was then immunoprecipitated with anti-FLAG antibody. The phosphorylation of RNF4 by CDKs was examined with indicated antibodies.
The CDKs family consists of many different members and different CDK functions in different cell cycle regulations (21–24). It has been reported that CDK2 plays an important role in the S-phase (21,22). We next examined whether RNF4 is phosphorylated by CDK2. As shown in Figure 2D, CDK2 could directly phosphorylate RNF4 in vitro. The consensus sequence of a CDK substrate is [S/T*]PX[R/K]. When we examined the sequence of RNF4, we found there are two candidate sites Thr26 and Thr112. As shown in Figure 2E, mutating Thr26 or Thr112 to Ala partially decreased CDK2-mediated phosphorylation of RNF4 in vitro, while mutating both sites (2TA) totally abolished RNF4 phosphorylation, suggesting CDK2 phosphorylates T26 and T112 of RNF4 in vitro. Furthermore, we studied the phosphorylation on RNF4 in vivo. Consistent with the in vitro result, the 2TA mutant from S phase cells cannot be recognized by anti-phospho-CDK substrates antibody (Figure 2F). Collectively, these data suggest that CDK2 phosphorylates RNF4 on T26 and T112 in S-phase.
The phosphorylation of RNF4 by CDK regulates the HR pathway through targeting sumoylated MDC1
Because DSB induced degradation of MDC1 by RNF4 and upregulation of RNF4 E3 activity occurs in S-phase, we next determined if the RNF4-MDC1 pathway is regulated by the phosphorylation on RNF4. First, we investigated whether phosphorylation on RNF4 affects its activity. As shown in Figure 3A, in G1 phase, the E3 ligase activity (as determined by autoubiquitination of RNF4) of WT RNF4 and the 2TA mutant was comparable. However, while the E3 ligase activity of WT RNF4 was enhanced in S phase, no upregulation of E3 ligase activity was observed for the 2TA mutant. On the other hand, the phospho-mimic mutant (RNF4-2TE) showed enhanced activity even in G1 phase (Figure 3A), suggesting that the phosphorylation on RNF4 by CDK is important for its increased activity in S phase. Furthermore, we confirmed the ubiquitination of MDC1 is affected by the phosphorylation on RNF4 with an in vitro ubiquitination assay. GST-MDC1 (aa1818–2089) is sumoylated in vitro (4) and then subjected to the in vitro ubiquitination assay with RNF4 purified from cells at different cell cycle stages. As shown in Figure 3B, the ubiquitination of GST-MDC1 (aa1818–2089) was significantly increased by WT RNF4 from S phase cells compared with RNF4 from G1 phase cells (lanes 2 and 3). RNF4 2TA showed low activity for MDC1 ubiquitination, no matter it was from G1 or S phase (lanes 4 and 5). On the other hand, the phospho-mimic mutant of RNF4, 2TE, showed higher activity toward MDC1 independent of cell cycle phases (lanes 6 and 7). Furthermore, consistent with the in vitro results, expression of the 2TA mutant resulted in decreased MDC1 polyubiquitination, while expression of the 2TE mutant enhanced MDC1 polyubiquitination even in G1 phase (Figure 3C). These results established that the phosphorylation on RNF4 by CDK promotes the E3 ligase activity of RNF4.
Figure 3.
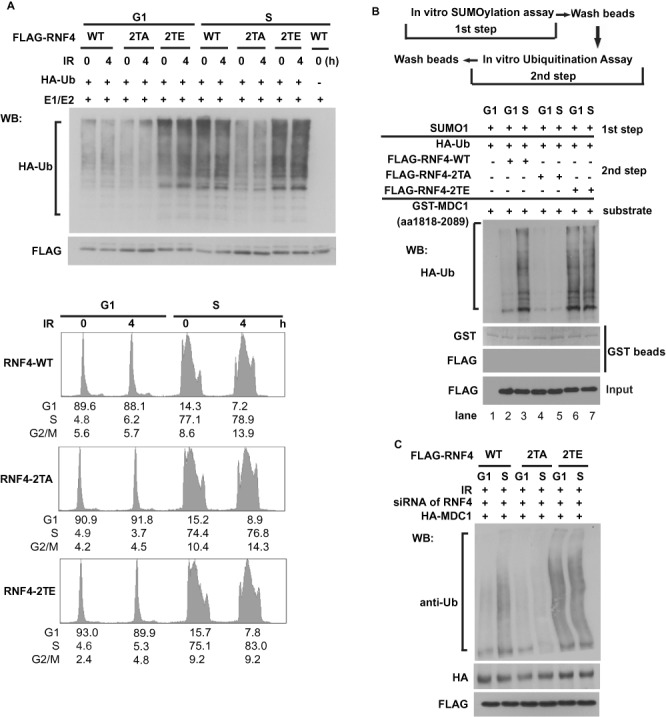
The phosphorylation on RNF4 affects its activity. (A) Upper panel: HEK 293T cells transfected with FLAG-RNF4 WT, 2TA or 2TE mutant were synchronized in G1 phase by double thymidine block or released into S phase. The cells were irradiated (5 Gy) and then collected at indicated time points. RNF4 was then immunoprecipitated and subjected to in vitro ubiquitination reactions in the presence of HA-Ub, UbcH5a and UBE1. The ubiquitination of RNF4 were analyzed by immunoblot as indicated. Lower panel: the cell cycle profiles of the cells were analyzed by flow cytometry. (B) Upper panel: schematic of the combined in vitro sumoylation and ubiquitination assay procedures. Lower panel: bacterially expressed GST‐MDC1 (aa 1818–2089) bound to GST‐sepharose were subjected to combined in vitro sumoylation and ubiquitination reactions as shown in the upper panel. The GST‐MDC1(aa 1818–2089) was subjected to in vitro sumoylation reactions in the presence of SUMO1 and then subjected to in vitro ubiquitination reactions in the presence or absence of HA‐Ub, RNF4‐WT, RNF4‐2TA, or RNF4-2TE mutant as indicated. The ubiquitination of GST‐sepharose‐bound protein were analyzed by immunoblot as indicated. (C) Cells depleted RNF4 with siRNA were cotransfected with HA-MDC1 and FLAG-RNF4-WT, 2TA or 2TE mutant. The cells were synchronized in G1 phase by double thymidine block or released into S phase and then irradiated (5 Gy). The ubiquitination of HA-MDC1 was then analyzed with indicated antibodies.
We next examined MDC1 foci in cells expressing WT RNF4, 2TA, or 2TE mutant in G1 or S phase. As shown in Figure 4A and Supplementary Figure S1A, in cells expressing RNF4 WT or 2TA, MDC1 showed similar pattern of focus formation following DNA damage in G1 phase. However, in S phase, MDC1 foci in cells expressing RNF4 2TA were more sustained compared to those in cells expressing WT RNF4. In cells expressing the 2TE mutant, MDC1 foci disappeared more promptly both in G1 and S phase. Because MDC1 need to be removed from the DNA damage sites before the loading of Rad51 (4), we next examined the Rad51 foci in the cells expressing WT RNF4, RNF4 2TA or RNF4 2TE. As shown in Figure 4B, Rad51 foci were hardly detected in all three lines in G1 phase, even though MDC1 is removed in cells expressing the 2TE mutant. Therefore, MDC1 removal is required but not sufficient for Rad51 loading. This might be due to other factors needed for Rad51 are not active in G1 phase, such as BRCA1 and CtIP (9,30,31,43–46). In S phase, Rad51 foci were readily detected in cells expressing WT RNF4 and RNF4 2TE, consistent with decreased MDC1 foci in these cells. However, Rad51 focus formation was greatly compromised in cells expressing RNF4 2TA, correlating with sustained MDC1 foci in these cells. These results suggest that RNF4 phosphorylation is important for MDC1 removal and Rad51 loading in S phase. We next investigated how RNF4 phosphorylation would affect HR. Using a reporter assay for HR, we found that there was a HR defect in cells expressing RNF4 2TA mutant which is similar to the cells knockdown endogenous RNF4 (Figure 4C and Supplementary Figure S1B). Cells expressing the 2TE mutant have moderately more HR capability. On the other hand, the phosphorylation status of RNF4 did not affect the NHEJ efficiency (Figure 4D and Supplementary Figure S1C). Consistently with these results, when we examined the disappearance of γH2AX foci as a readout for DNA repair capability in cells expressing RNF4 WT or 2TA mutant, the RNF4 2TA mutant causes a delay in the disappearance of γH2AX foci in S phase but not in G1 phase (Figure 4E and F). These results suggest that the phosphorylation of RNF4 by CDK regulates the HR pathway through targeting sumoylated MDC1.
Figure 4.
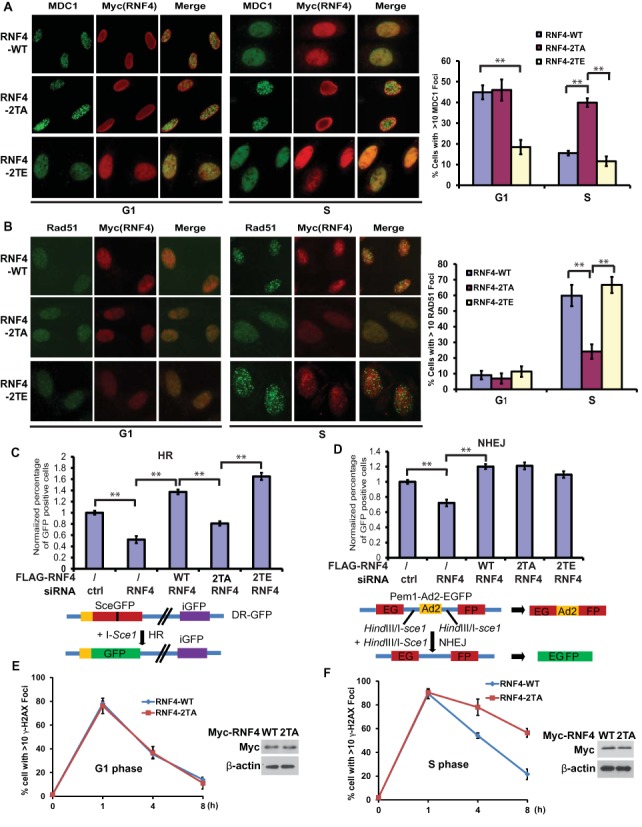
The phosphorylation of RNF4 by CDK2 regulates the HR pathway through targeting sumoylated MDC1. (A and B) U2OS cells transfected with Myc-RNF4 WT, 2TA or 2TE mutant were synchronized in G1 phase by double thymidine block or released into S phase. Then the cells were treated with irradiation (2 Gy) and probed with indicated antibodies 4 h later. Representative images are shown in the left panels, the quantification of the percentage of cells displaying MDC1 (A) or Rad51 (B) foci are shown in right panels. Error bars represent SEM. from three independent experiments. **P < 0.01 two‐tailed Student's t‐test. >200 cells were counted per experiment. (C and D) Cells depleted RNF4 with siRNA were reconstituted with FLAG-RNF4-WT, 2TA or 2TE mutant. Then the cells were subjected to HR (C) or NHEJ (D) assay as described in method. Error bars represent SEM. from three independent experiments. **P < 0.01 two‐tailed Student's t‐test. Lower panels: schematic of the reporters used in the assay. (E and F) U2OS cells transfected with Myc-RNF4 WT or 2TA mutant were synchronized in G1 phase by double thymidine block or released into S phase. Then the cells were treated with or without irradiation (2 Gy) and probed with anti-γH2AX, anti-Myc antibodies at indicated time point. The quantifications of the percentage of cells displaying γH2AX foci in G1 phase (E) or S phase (F) are shown. >200 cells were counted per experiment. Expression of Myc-RNF4 WT and 2TA mutant were examined using indicated antibodies (E and F, right panels).
DISCUSSION
CDKs play an important role in cell cycle progression through regulating cell cycle related proteins. Recent studies indicated that CDKs are involved in cell cycle independent functions such as DNA damage repair (16,27). It was found that CDK not only phosphorylates the key components of cell division and DNA replication machineries (47), but also proteins that function primarily in DNA damage repair (32,33,35,48), suggesting a role of CDK in DNA repair. NBS1 was identified as one of the CDK2 substrates and cells with mutant forms of NBS1 that cannot be modified by CDK2 are more sensitive to irradiation (49). In addition, CtIP was found to be phosphorylated by CDK2, which are essential for its interaction with BRCA1 (44–46) and its recruitment to DSBs (9,30,31,43). In Cdk2−/− cells, DNA repair was delayed and impaired (20,50). In addition, the Cdk2−/− mice are more sensitive to irradiation and died earlier than wild type mice (20,50). It was reported that the deficiency of CDK2 would impair the homologous recombination in mammalian cells (48,51). It was also reported that the MEFs depleted interphase CDKs are not hypersensitive to DNA-damaging agents and could repair efficiently, which is dependent on the functional CDK1 (28). It showed that there are overlaps among CDKs functions in DNA damage response (28). Although how CDKs modulates the responses to DNA damage in mammalian cells remains unclear, all these evidences revealed that CDK is not only important in the cell cycle signaling, but also in the repair pathway activated by DSB. Our results indicated that CDK also plays a role in regulating the DNA damage response factor MDC1 through RNF4, which provides evidence that CDK did play an important role in the DNA damage repair.
It has been reported that direct competition between HR and NHEJ factors for DSBs is important for the choice between the two pathways (52). HR is primarily active in S phase and early G2 phase, however, the regulation of HR by CDKs remains incomplete (27,53,54). In this study we investigated a new mechanism by which HR repair is promoted during S-phase. Our results showed that RNF4 would be phosphorylated by CDK2 during S-phase, which promotes its activity to ubiquitinate MDC1. Previous studies suggest that removing MDC1 from the DNA damage sites promptly is important for the HR repair (4,37,38). Therefore, CDK is not only important for the recruitment of CtIP, but also regulates the removal of MDC1 to promote HR. Our studies further suggest that timely and dynamic regulation of DDR factors at the sites of DNA damage is critical for proper DNA repair in specific cell phases. How CDK-mediated phosphorylation regulates RNF4 activity remains unclear. The two phosphorylation sites of RNF4 are outside the RING domain of RNF4. It is possible that CDK-mediated phosphorylation allosterically activates RNF4. Although our study indicated that CDK2 is the major contributor of the phosphorylation on RNF4 in S-phase, we cannot exclude the possibility that other CDKs are involved in the regulation. Previous study showed that in the absence of interphase CDKs, CDK1 could drive cell cycle in mouse (26). It was also reported that the MEFs depleted interphase CDKs can repair DNA efficiently (28). These studies suggested that CDKs have overlaps in their functions, especially that CDK1 could compensate CDK functions in the absence of interphase CDKs (26,28). Besides, HR repair is also active in early G2 when CDK1 plays a key role. It is possible that RNF4 is also regulated by CDK1. Understanding whether other CDKs contribute to the phosphorylation on RNF4 will require additional work.
Although RNF4 has been shown to regulate both NHEJ and HR (4,37), its role in NHEJ remains unclear. A recent study suggests that RNF4 is important for 53BP1 foci formation at telomeres (55) that requires its SIM domain and DNA binding motifs (55), whether or not its E3 ligase activity is important for this regulation is not determined. We showed that RNF4 phosphorylation is important for HR but not NHEJ. Since NHEJ is predominant in G1 and G2 phase, how RNF4 regulates NHEJ with lower activity in G1/G2? One possibility is that the basal level of RNF4's activity is sufficient for the regulation of NHEJ in G1/G2 phase while higher activity of RNF4 is important for HR in S-phase. Another possibility is that RNF4 promotes NHEJ independent of its E3 ligase activity. In this study, we focus on how RNF4 contributes to the cell cycle dependent mechanism that regulates the HR repair. How RNF4 regulates NHEJ in other cell cycle phase will be another important question to be addressed by future studies.
In summary, our studies revealed a novel pathway, CDK-RNF4-MDC1, to regulate the HR repair during S-phase, which provides an insight into the cell cycle dependent preference of different DSB repair pathway.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Haojie Huang (Mayo Clinic) for providing CDK2 and CyclinE constructs. We also thank Dr Maria Jasin for providing DR-GFP reporter and Dr Eric A. Hendrickson for providing pEGFP-Pem1-Ad2 plasmid.
FUNDING
National Basic Research Program of China [973 Program, 2013CB530700]; National Natural Science Foundation of China [31371367, 81322031, 81222029]; Program for New Century Excellent Talents in University [NCET-12-0411 to K.L.]; National Institutes of Health [CA148940 to Z.L., CA130996 to Z.L., CA189666 to Z.L.]. Funding for open access charge: National Basic Research Program of China [973 Program, 2013CB530700].
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhou B.B., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Lukas J., Lukas C., Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 3.Huen M.S., Chen J. Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem. Sci. 2010;35:101–108. doi: 10.1016/j.tibs.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo K., Zhang H., Wang L., Yuan J., Lou Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012;31:3008–3019. doi: 10.1038/emboj.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warmerdam D.O., Kanaar R. Dealing with DNA damage: relationships between checkpoint and repair pathways. Mutat. Res. 2010;704:2–11. doi: 10.1016/j.mrrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Lieber M.R., Ma Y., Pannicke U., Schwarz K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst) 2004;3:817–826. doi: 10.1016/j.dnarep.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Thompson L.H., Schild D. Recombinational DNA repair and human disease. Mutat. Res. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 8.Paull T.T., Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 9.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda S., Nakamura K., Taniguchi Y., Paull T.T. Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol. Cell. 2007;28:351–352. doi: 10.1016/j.molcel.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Mimitou E.P., Symington L.S. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Nimonkar A.V., Genschel J., Kinoshita E., Polaczek P., Campbell J.L., Wyman C., Modrich P., Kowalczykowski S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.San Filippo J., Sung P., Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 15.West S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell. Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 16.West S.C. The search for a human Holliday junction resolvase. Biochem. Soc. Trans. 2009;37:519–526. doi: 10.1042/BST0370519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ip S.C., Rass U., Blanco M.G., Flynn H.R., Skehel J.M., West S.C. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- 18.Constantinou A., Chen X.B., McGowan C.H., West S.C. Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J. 2002;21:5577–5585. doi: 10.1093/emboj/cdf554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan D.O. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 20.Satyanarayana A., Kaldis P. A dual role of Cdk2 in DNA damage response. Cell Div. 2009;4:9. doi: 10.1186/1747-1028-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherr C.J., Roberts J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 22.Sherr C.J., Roberts J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 23.Weinberg R.A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 24.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 25.Riabowol K., Draetta G., Brizuela L., Vandre D., Beach D. The cdc2 kinase is a nuclear protein that is essential for mitosis in mammalian cells. Cell. 1989;57:393–401. doi: 10.1016/0092-8674(89)90914-8. [DOI] [PubMed] [Google Scholar]
- 26.Santamaria D., Barriere C., Cerqueira A., Hunt S., Tardy C., Newton K., Caceres J.F., Dubus P., Malumbres M., Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 27.Wohlbold L., Fisher R.P. Behind the wheel and under the hood: functions of cyclin-dependent kinases in response to DNA damage. DNA Repair. 2009;8:1018–1024. doi: 10.1016/j.dnarep.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerqueira A., Santamaria D., Martinez-Pastor B., Cuadrado M., Fernandez-Capetillo O., Barbacid M. Overall Cdk activity modulates the DNA damage response in mammalian cells. J. Cell Biol. 2009;187:773–780. doi: 10.1083/jcb.200903033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N.M., et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huertas P., Cortes-Ledesma F., Sartori A.A., Aguilera A., Jackson S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huertas P., Jackson S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruffner H., Jiang W., Craig A.G., Hunter T., Verma I.M. BRCA1 is phosphorylated at serine 1497 in vivo at a cyclin-dependent kinase 2 phosphorylation site. Mol. Cell. Biol. 1999;19:4843–4854. doi: 10.1128/mcb.19.7.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esashi F., Christ N., Gannon J., Liu Y., Hunt T., Jasin M., West S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 34.Caspari T., Murray J.M., Carr A.M. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002;16:1195–1208. doi: 10.1101/gad.221402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers J.S., Zhao R., Xu X., Ham A.J., Cortez D. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res. 2007;67:6685–6690. doi: 10.1158/0008-5472.CAN-07-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onge R.P., Besley B.D., Pelley J.L., Davey S. A role for the phosphorylation of hRad9 in checkpoint signaling. J. Biol. Chem. 2003;278:26620–26628. doi: 10.1074/jbc.M303134200. [DOI] [PubMed] [Google Scholar]
- 37.Galanty Y., Belotserkovskaya R., Coates J., Jackson S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012;26:1179–1195. doi: 10.1101/gad.188284.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin Y., Seifert A., Chua J.S., Maure J.F., Golebiowski F., Hay R.T. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012;26:1196–1208. doi: 10.1101/gad.189274.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lou Z., Minter-Dykhouse K., Wu X., Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 40.Fattah F., Lee E.H., Weisensel N., Wang Y., Lichter N., Hendrickson E.A. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 42.Shrivastav M., De Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 43.Buis J., Stoneham T., Spehalski E., Ferguson D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012;19:246–252. doi: 10.1038/nsmb.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yun M.H., Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L., Nievera C.J., Lee A.Y., Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 46.Yu X., Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malumbres M., Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Muller-Tidow C., Ji P., Diederichs S., Potratz J., Baumer N., Kohler G., Cauvet T., Choudary C., van der Meer T., Chan W.Y., et al. The cyclin A1-CDK2 complex regulates DNA double-strand break repair. Mol. Cell. Biol. 2004;24:8917–8928. doi: 10.1128/MCB.24.20.8917-8928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wohlbold L., Merrick K.A., De S., Amat R., Kim J.H., Larochelle S., Allen J.J., Zhang C., Shokat K.M., Petrini J.H., et al. Chemical genetics reveals a specific requirement for Cdk2 activity in the DNA damage response and identifies Nbs1 as a Cdk2 substrate in human cells. PLoS Genet. 2012;8:e1002935. doi: 10.1371/journal.pgen.1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satyanarayana A., Hilton M.B., Kaldis P. p21 Inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol. Biol. Cell. 2008;19:65–77. doi: 10.1091/mbc.E07-06-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deans A.J., Khanna K.K., McNees C.J., Mercurio C., Heierhorst J., McArthur G.A. Cyclin-dependent kinase 2 functions in normal DNA repair and is a therapeutic target in BRCA1-deficient cancers. Cancer Res. 2006;66:8219–8226. doi: 10.1158/0008-5472.CAN-05-3945. [DOI] [PubMed] [Google Scholar]
- 52.Thompson L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 53.Karanam K., Kafri R., Loewer A., Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sonoda E., Hochegger H., Saberi A., Taniguchi Y., Takeda S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair. 2006;5:1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 55.Groocock L.M., Nie M., Prudden J., Moiani D., Wang T., Cheltsov A., Rambo R.P., Arvai A.S., Hitomi C., Tainer J.A., et al. RNF4 interacts with both SUMO and nucleosomes to promote the DNA damage response. EMBO Rep. 2014;15:601–608. doi: 10.1002/embr.201338369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.