


Abstract
Ribosome biogenesis, a central and essential cellular process, occurs through sequential association and mutual co-folding of protein–RNA constituents in a well-defined assembly pathway. Here, we construct a network of co-evolving nucleotide/amino acid residues within the ribosome and demonstrate that assembly constraints are strong predictors of co-evolutionary patterns. Predictors of co-evolution include a wide spectrum of structural reconstitution events, such as cooperativity phenomenon, protein-induced rRNA reconstitutions, molecular packing of different rRNA domains, protein–rRNA recognition, etc. A correlation between folding rate of small globular proteins and their topological features is known. We have introduced an analogous topological characteristic for co-evolutionary network of ribosome, which allows us to differentiate between rRNA regions subjected to rapid reconstitutions from those hindered by kinetic traps. Furthermore, co-evolutionary patterns provide a biological basis for deleterious mutation sites and further allow prediction of potential antibiotic targeting sites. Understanding assembly pathways of multicomponent macromolecules remains a key challenge in biophysics. Our study provides a ‘proof of concept’ that directly relates co-evolution to biophysical interactions during multicomponent assembly and suggests predictive power to identify candidates for critical functional interactions as well as for assembly-blocking antibiotic target sites.
INTRODUCTION
Macromolecular complexes are the functional units of most core cellular processes (1) such as transcription, translation, protein degradation, signal transduction. Consequently, biogenesis pathways of these molecular nanomachines are central to cellular metabolism. A comprehensive biophysical understanding of how the different or identical subunits co-assemble into complex structures is, therefore, a central issue in biology.
The components of a macromolecular complex self-assemble into a heterogeneous structure through a complex series of molecular recognitions and structural rearrangements. Despite their biological importance, such processes have proven difficult to resolve (2,3). Individual subunits can self-assemble into a complex in numerous possible paths (3–5). For instance, 21 monomers of the bacterial small ribosomal subunit (SSU) can assemble through millions of potential intermediate structures (3). However, only a few energetically favorable intermediates make a measurable contribution to the assembly (5,6). This indicates that biological macromolecules involve orchestrated physical interactions among the constituents (3). An ordered assembly is, therefore, fundamental to the biogenesis of macromolecular complexes and the assembly order is generally conserved in evolution (6,7).
Conserved assembly pathways are generally associated with conserved structures and sequences of the constituent monomers. The bacterial SSU, for which assembly mechanisms have been investigated for decades, is composed of a 16S rRNA and about 20 proteins (S2, S3, …, S21). The 16S rRNA is remarkably conserved in sequence (8,9), secondary structural elements and 3D structure (10); ribosomal proteins also exhibit highly conserved 3D architecture (11). Simultaneous conservation of assembly pathway, sequence and structure allow us to speculate that ordered assembly pathways have emerged through mutual evolutionary readjustments among the constituents (3). Co-evolution refers to coordinated evolutionary changes between two or more evolutionary units, such as amino acid positions within proteins, phenotypic characteristics etc. (12,13). Molecular co-evolution is generally observed among physically interacting proteins, interacting domains of a protein, proximal amino acids of the same or two interacting proteins (12–16) and even among the constituents of multicomponent complexes (17,18). Here, we seek to address whether patterns of co-evolution among the components of a complex are correlated with the biophysical mechanism (thermodynamics and kinetics) of its self-assembly process. This approach requires a macromolecular complex for which description of structural reconstitution events are known in molecular detail. The bacterial SSU is selected for this purpose.
The protein–rRNA constituents of the bacterial SSU can be efficiently assembled from purified components to the functional native structure in vitro (19). This has facilitated visualization of ribosome assembly at the molecular level. Ribosome assembly is organized in three independently folding domains of the 16 rRNA: 5′ domain, central domain and 3′ domain (20,21). The assembly nucleates concurrently from multiple self-folding sits of the 16S rRNA (22) and proceeds in a 5′-to-3′ direction (23). Protein binding follows a specific hierarchy (24,25), which depends on the temporal order of binding site availability (25,26). Before the initiation of protein binding, 16S rRNA is partially folded, having most of its secondary structural elements in place. Proteins rapidly associate with this partially folded rRNA, forming initial encounter complexes that lead to mutual co-folding of the two molecular species (22). This co-folding reorganizes the local rRNA conformation and progresses toward the native state. Finally, the three independently folding domains pack together, generating the functional APE sites.
Here, we take advantage of the extensive knowledge on ribosome structure and assembly mechanism, along with the wide-ranging bacterial genomic data to show that characteristic patterns of co-evolution are driven by the thermodynamics and kinetic constraints of ribosome assembly. We develop efficient computational methods and metrics, which allow us to extract functionally relevant information from co-evolutionary signals. Directly relating co-evolution to biophysical interactions and thermodynamic and kinetic constraints of folding reveals the underlying bases of known deleterious mutations and antibiotic binding sites. Our analysis places many previously documented experimental findings about the system into evolutionary context and it also provides predictions for further empirical studies to deepen our understanding of the structure and biophysics of the small ribosomal subunit.
MATERIALS AND METHODS
Dataset
Because ribosomal proteins and RNA (8–11) are highly conserved, we have collected a diverse set of ribosomal protein and 16S rRNA sequences from 280 species representing all 18 phyla of bacterial superkingdom. These species were selected to represent the sequence diversity of the entire phylum (27). Ribosomal protein and RNA sequences are collected from NCBI database (www.ncbi.nlm.nih.gov/) and Comparative RNA Website (28) respectively. A dataset of three high-resolution X-ray crystallographic structures and one low-resolution Cryo-Electron Microscopic structure of Escherichia coli small ribosomal subunit are used for structural analysis. The sequence and structure datasets are provided in Supplementary Dataset.
Evolutionary analysis
A number of computational methods have been developed in the last decade to identify co-evolving residue pairs (12). The pioneering approach of McBASE (29) was followed by the CAPS method (30), which has successfully predicted protein interactions and 3D contacts. Both of these methods identify pattern of substitutions between site pairs and estimate their similarity using a pre-calculated amino-acid substitution matrix. Thus, these approaches are only applicable to protein sequences. Ancestral sequence prediction based methods have been proven accurate in predicting both protein and RNA tertiary contacts among closely related sequences (31,32). However, the performance of ancestral reconstitution based approaches in large-scale analysis remains to be tested (12). We have employed Mutual Information (MI) approach in our work, because this approach is applicable to a large and diverse dataset (33–37) and has been proven sensitive for inferring both protein and RNA 3D contacts (38,39), as well as contacts between protein and RNA (38,40). MI score between two residues a and b, located at i-th and j-th positions of the alignment are calculated as follows:
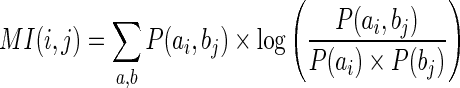 |
(1) |
where 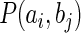 is the joint probability distribution and
is the joint probability distribution and 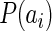 and
and 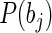 are the marginal probability distributions of residues a and b, located at i-th and j-th positions of the alignment respectively. Sole MI values are associated with false-positives, as we tend to identify the co-evolving pairs. Firstly, modeling studies showed that alignments should contain at least 125 sequences before Mutual Information ‘signal’ becomes apparent over random noise (37). Secondly, position pairs might have elevated MI due to the phylogenetic relationships of the organisms represented in the alignment (33). This may be minimized by excluding highly similar sequences from closely related species from the alignment (37,41). We have used a large sequence dataset of 280 diverse species that keeps the first two sources of false positives at minimum. Finally, positions with higher variability, or entropy, will tend to have higher levels of both random and non-random MI than positions of lower entropy (37,42). To counter this effect, each site pair score is weighed against the average score of its constituting sites; the Row-Column-Weighed score (34), termed as the
are the marginal probability distributions of residues a and b, located at i-th and j-th positions of the alignment respectively. Sole MI values are associated with false-positives, as we tend to identify the co-evolving pairs. Firstly, modeling studies showed that alignments should contain at least 125 sequences before Mutual Information ‘signal’ becomes apparent over random noise (37). Secondly, position pairs might have elevated MI due to the phylogenetic relationships of the organisms represented in the alignment (33). This may be minimized by excluding highly similar sequences from closely related species from the alignment (37,41). We have used a large sequence dataset of 280 diverse species that keeps the first two sources of false positives at minimum. Finally, positions with higher variability, or entropy, will tend to have higher levels of both random and non-random MI than positions of lower entropy (37,42). To counter this effect, each site pair score is weighed against the average score of its constituting sites; the Row-Column-Weighed score (34), termed as the 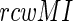 is defined as:
is defined as:
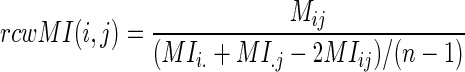 |
(2) |
where MIi. and MI.j are the summation of the MI values of residues i and j, respectively, to all other residues in the MSA. 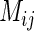 is the MI between residues i and j. Thus,
is the MI between residues i and j. Thus, 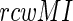 filtering is a simultaneous bi-dimensional optimization, which weights an existing matrix to identify the strong signals. A high MI indicates strong dependency between two sites, while a high
filtering is a simultaneous bi-dimensional optimization, which weights an existing matrix to identify the strong signals. A high MI indicates strong dependency between two sites, while a high 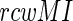 score indicates the pairs without the phylogenetic noise. To optimize between these two parameters (Extended Materials and Methods section), we have chosen only those pairs as co-evolving which simultaneously satisfy the two conditions (i) The MI values exceed the 99.9% confidence interval of the entire MI distribution spectrum and (ii) The
score indicates the pairs without the phylogenetic noise. To optimize between these two parameters (Extended Materials and Methods section), we have chosen only those pairs as co-evolving which simultaneously satisfy the two conditions (i) The MI values exceed the 99.9% confidence interval of the entire MI distribution spectrum and (ii) The 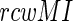 values exceed the 90% confidence interval of the entire
values exceed the 90% confidence interval of the entire 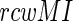 distribution spectrum. This filtering is particularly important for finding co-evolving nucleotide-amino acid pairs. While the same MI-treatment is applied to both protein and rRNA, random MI scores due to one or both invariable sites are the principle source of error (40). This is minimized through two steps. First, we use an alignment of a large number of diverse sequences in order to reduce the probability of random low MI due to invariable sites (<1%) (40). Second, we use only the top hits (exceeding 99.9% confidence interval) from the MI spectrum. Thus, our methodology incorporates only the correlated fashion of mutations to identify the co-evolving residue positions (no other constraints, such as distance between residue pairs in the 3D structure, are imposed). The CAPS method (30) is also used for protein sequence analysis, for validating the MI-based predictions. All the co-evolving pairs are available in the Supplementary Data and a statistics of the co-evolving pairs is provided in Supplementary Materials section 3.1.
distribution spectrum. This filtering is particularly important for finding co-evolving nucleotide-amino acid pairs. While the same MI-treatment is applied to both protein and rRNA, random MI scores due to one or both invariable sites are the principle source of error (40). This is minimized through two steps. First, we use an alignment of a large number of diverse sequences in order to reduce the probability of random low MI due to invariable sites (<1%) (40). Second, we use only the top hits (exceeding 99.9% confidence interval) from the MI spectrum. Thus, our methodology incorporates only the correlated fashion of mutations to identify the co-evolving residue positions (no other constraints, such as distance between residue pairs in the 3D structure, are imposed). The CAPS method (30) is also used for protein sequence analysis, for validating the MI-based predictions. All the co-evolving pairs are available in the Supplementary Data and a statistics of the co-evolving pairs is provided in Supplementary Materials section 3.1.
Network construction and analysis
Co-evolving residue pairs are transformed into an undirected, unweighted network, in which each node represents a residue position (amino acid/nucleotide) and two nodes are connected if they are co-evolving.
Network analysis at local-level
Two topological parameters are computed: degree and clustering coefficient. Degree or node connectivity is defined as the number of nodes adjacent to the node of interest. Clustering Coefficient (Cn) of a node n of an undirected network is the measurement of its cliquishness. High clustering coefficient of a node indicates a locally dense region of the network.
Network analysis at global-level
Assortative mixing and coefficient of assortativity
A network shows assortative mixing if the high degree nodes in the network tend to be connected with other high-degree nodes. This is quantified in the coefficient of assortativity (43):
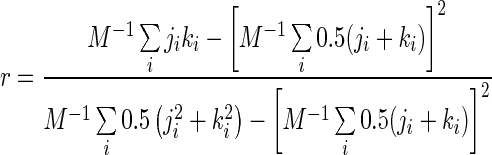 |
(3) |
Here, ji and ki are the degrees of the nodes at either ends of the ith edge and M is the total number of edges.
Co-evolution order
For a protein structure, contact order is defined as the average primary separation between two physically contacting amino acids (44). We have applied an analogous measure, termed ‘Co-Evolution Order’ (CEO), which measures the average primary chain separation of the two co-evolving residues. It is defined as:
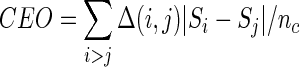 |
(4) |
where nc is the total number of CEPs, Si and Sj are the sequence position of the residues i and j, and Δ(i, j) selects the residues (i and j) that are in contact.
Module analysis
Module analysis locates the densely connected sub-networks exhibiting modular organization and finds whether it significantly differs (Z-statistics) from any random sample of nodes of the background network. Module analysis is performed by ClusterONE (45).
Structure analysis
Protein–protein and protein–rRNA interfaces and molecular contacts
An amino acid residue is defined as an interface residue if it loses >1 Å2 of its accessible surface area when passing from an uncomplexed state (free protein) to a complexed state (RNA-bound) (46). The same criterion is used to identify the rRNA nucleotides that physically recognize a protein. Surface Racer program (47) is used to calculate the surface areas with 1.4 Å probe radius. Intermolecular van der Waals contacts are considered for all atoms pairs separated by ≤5.0 Å distance. Hydrogen bonds are identified for D–A distance ≤3.35 Å, and D–H–A angle to be within 90–180°, where D stands for the donor group and A for the acceptor group.
All the statistical analyses were performed using PAST statistical software package (48). Molecular graphics were prepared using PyMOL Molecular Graphics System.
A detailed version of the Materials and Methods is provided as Extended Materials and Methods.
RESULTS AND DISCUSSION
The cooperativity phenomenon
Molecular basis of cooperativity in macromolecular assembly
At each stage of the assembly hierarchy, partially folded rRNA segments recognize groups of proteins and the assembled protein–rRNA components mutually co-fold (22,49). The pre-binding of early binder proteins and the rRNA structural reconstitutions they induce simultaneously enable recognition of the next set of proteins. This molecular event explains the increase in rRNA-binding affinity and accelerated binding rate of late binder proteins that follows the association of early binders (24,25). Thus, the assembling protein–rRNA components are energetically coupled. The coupling free energy (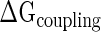 ) arises either from the direct physical interactions between two proteins, or through protein-mediated conformational changes of 16S rRNA structure or both (3).
) arises either from the direct physical interactions between two proteins, or through protein-mediated conformational changes of 16S rRNA structure or both (3). 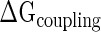 ensures that an intermediate sub-complex is more stable than its individual components, and this stability gain drives the assembly forward (3). This phenomenon is termed ‘cooperativity’ and it is relevant in other aspects of conformational transitions, such as protein folding (50) and allosterism (51).
ensures that an intermediate sub-complex is more stable than its individual components, and this stability gain drives the assembly forward (3). This phenomenon is termed ‘cooperativity’ and it is relevant in other aspects of conformational transitions, such as protein folding (50) and allosterism (51).
Characteristic patterns of co-evolution among cooperative protein pairs
The thermodynamic order of protein binding in SSU assembly was initially determined by Nomura (24) using a series of equilibrium reconstitution experiments and is summarized in the classic Nomura Assembly Map (Figure 1A). Energetic coupling among the cooperative protein pairs acts as a constraint of ribosome assembly (constraint at structure space). We investigated whether cooperative protein pairs are associated with stronger co-evolutionary constraints compared to any random pair (constraint at sequence space). We identify co-evolving amino acid/nucleotide pairs (CEPs) within the same molecules (intra-protein and intra-rRNA) and among different molecules (inter-protein and protein–rRNA) constituting the SSU. We define and measure three categories of CEPs among all possible protein pairs: (i) direct: individual residues of two proteins co-evolve; (ii) indirect: amino acid residues of two proteins co-evolve with the same rRNA nucleotide (structural reorganization induced by the early binder likely propagates through the rRNA); and (iii) third-party: rRNA nucleotides that physically recognize the two cooperative proteins co-evolve with each-other. We have investigated whether the amino acid pairs maintaining indirect co-evolutionary relationships also exhibit transitive co-evolution (if A co-evolves with both B and C, then B and C also co-evolve) with each other. Although the expected value under a null hypothesis of independence is 3.66%, almost 29.9% of amino acid pairs show transitive co-evolution. A visual illustration of the three categories of CEPs is provided in Figure 2 for S7–S13 cooperative pair. Since 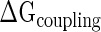 arises both from protein–protein physical contacts and protein induced conformational rearrangements of the rRNA, the three categories of CEPs support that both the protein and rRNA segments are constrained by energetic coupling.
arises both from protein–protein physical contacts and protein induced conformational rearrangements of the rRNA, the three categories of CEPs support that both the protein and rRNA segments are constrained by energetic coupling.
Figure 1.
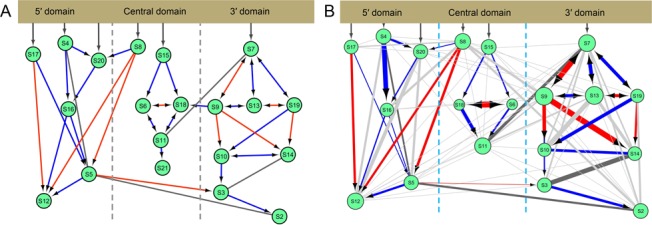
The Nomura Assembly Map. (A) The modified Nomura Assembly Map: 16S rRNA shown at the top, proteins are shown as green circles. Each line connecting two proteins indicates a directional cooperative relationship (the protein at the bottom depends on the pre-binding of the protein at the top); red arrows indicate simultaneous presence of physical interaction and kinetic cooperativity; blue arrows signify only kinetic cooperativity and black lines signify only physical interaction. The S19–S10 kinetic cooperativity refers to the prebinding experiments of ref. (25). (B) The edge-weighted inter-protein co-evolutionary network is projected onto the modified Nomura Assembly Map. Proteins are shown as green circles, circle size is proportional to the number of other proteins it co-evolves with. S21 is not shown (not analyzed as it is not universal in bacterial superkingdom). Gray colored edges represent the presence of co-evolutionary relationships that are not associated with direct biophysical constraints (some gray edges are associated with indirect cooperative relationships). Thickness of the lines connecting two proteins is proportional to PS.
Figure 2.
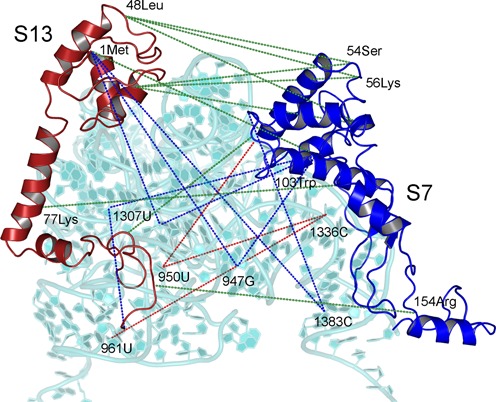
The three categories of co-evolutionary pattern between S7 and S13 cooperative pair are shown. The proteins and 16S rRNA are presented as cartoon views. Each dotted line represents co-evolution between two residues it connects. Green lines represent direct co-evolution (e.g. 54Ser of S7 co-evolves with 48Leu of S13), blue lines represent indirect co-evolution (e.g. 103Trp of S7 and 1Met of S13 co-evolve with 947G of 16S rRNA) and red lines signify third-party co-evolution (1336C co-evolves with 950U). Only some residue positions are highlighted to maintain the clarity of the image.
Cooperative protein pairs exhibit strong co-evolutionary preference
Next, we computed probabilities of CEPs between protein pairs and converted the probability values into entropy-like functions: termed co-evolutionary preference score (PS) (Supplementary Materials section 3.2.1). Considering individual proteins as nodes and PS scores as their edge-weights, we construct an edge-weighted network and project it onto the modified Nomura Assembly Map (Figure 1B). Cooperative protein pairs show a clear signal of elevated rates of co-evolution (high PS values compared to random protein pairs, Permutation Mann–Whitney (MW) U-test, P < 10−9). This result is consistent with the observation of strong structural constraints between such protein pairs.
Strength of co-evolutionary preference depends on the molecular nature of energetic coupling
The rRNA structural reconstitutions induced by primary binders (primary proteins binding independently with 16S rRNA) assist the subsequent association of secondary binders (secondary proteins depend on primary binders); but these reconstitutions generally dampen prior to tertiary binding (those depend on primary and secondary binders). For instance, S15 protein rearranges the relative orientations of a set of rRNA helices (H20, H21, H22 and H23) at the central domain, enabling the subsequent association of secondary S6–S18 heterodimer. However, these rRNA helices do not experience further significant reconstitutions during the succeeding binding of tertiary S11 protein (52). Therefore, the primary–tertiary indirect cooperativity mostly emerges from protein–protein physical interactions, which are essential to drive the assembly forward. This explains our observation that 18 out of 23 primary-tertiary indirect cooperativities are also associated with strong PS compared to random non-cooperative pair. However, the PS values among directly cooperative pairs exhibit slight elevation compared to those of the indirectly cooperative pairs (P < 0.05). When we include the indirectly cooperative proteins into the dataset and compare their PS scores with any random protein pair (Permutation MW U-test), the probability of no association decreases to P < 10−16.
Proteins assisting inter-domain packing exhibit strong co-evolutionary preference
Each 16S rRNA domain can assemble independently (20,21) and their accurate fabrication is essential for biological functionality. This fabrication is achieved by both intra-rRNA molecular packing and physical interactions among proteins from different domains (53). Proteins involved in inter-domain fabrication (e.g. S7–S11, S5–S3, S8–S12, etc.) do not alter the binding kinetics of each other (25) but they appear at the thermodynamic dependency map (24). Significantly elevated PS values between the proteins involved in inter-domain fabrication compared to random inter-domain protein pairs (P < 10−4) indicates evolutionary conservation of these protein–protein interfaces.
Co-evolutionary preference can capture distantly placed cooperative protein pairs
Direct physical interaction is not the only molecular mechanism for energetic coupling, since many proteins maintain their cooperative relationships by reconstituting the rRNA conformation. For example, S15 induces a structural rearrangement at the 3-Way-Junction of the central domain, enabling the subsequent association of S6–S18 heterodimer about 35 Å away from S15 (52). Long distance, as well as steric, constraints are therefore crucial in ribosome assembly. To test for CEPs that are mutually dependent, but structurally remote, we limited comparisons to those that satisfy two criteria: (i) protein pairs are at least 10 Å apart from one another and (ii) CEPs are separated by minimum 30 Å in the 3D structure. We found strong associations (P < 10−4) between assembly interaction and co-evolution despite a considerable reduction in sample size. This approach revealed compelling candidates for cooperative pairs that do not physically interact; the candidates include the recently reported S19–S10 cooperative pair (25).
The 41 remaining co-evolutionary relationships are generally very weak and they serve the purpose of ‘background data’. However, among these 41 pairs, five pairs are associated with strong co-evolutionary preferences (S8–S2, S8–S9, S9–S11, S9–S12 and S13–S11), which cannot be explained by current knowledge. We predict that further experimental studies will reveal biophysical interactions for most of these cases. In summary, these results show that comparative sequence analysis can provide compelling candidates for cooperativity in macromolecular self-assembly.
Protein-induced rRNA helix rearrangements
Protein-induced rRNA structural rearrangements are the molecular mechanisms that establish inter-protein cooperative relationships. Although our knowledge regarding protein-induced rRNA helix rearrangements is limited, two such important cases have been studied extensively: the 5-Way Junction (5WJ) rearrangement at 5′-domain (49,54) induced by S4, and the 3WJ rearrangement at central domain (52) induced by S15.
The 5WJ rearrangement
S4 contacts the 5WJ (composed of helices H3, H4, H16, H17 and H18) of the 16S rRNA (Figure 3A) with its intrinsically disordered N-terminal domain and forms an initial encounter complex (55) with H16–H17 (22). The initial encounter is followed by a subsequent protein–rRNA co-folding (56). Helix H16 is one of the strongest signatures in the bacterial 16S rRNA (57), which is accompanied by (and co-evolves with) a 12 amino acid long bacterial-specific insertion in S4 (10). Numerous non-native contacts between this S4-insertion and the bacteria-specific tip of H16 (positions 408–434, E. coli numbering) are formed at the first 100 ms of protein–rRNA contact (22). We observe co-evolution between H16–H17 and the disordered N-terminal (Gln36-U421) and C-terminal (Ala158-U421) segments of S4, which indicates the essentiality of this initial encounter. The H17 segment 447–487 is another bacterial-specific structural signature (10), which participates in the initial encounter with S4 (22). We have identified several CEPs between the intrinsically disordered C-terminal loop (Lys8-A468, His41-A468) and N-terminal segment (Thr181-A460, Thr181-U464, Thr181-U476) of S4 and this signature site (Figure 3B).
Figure 3.
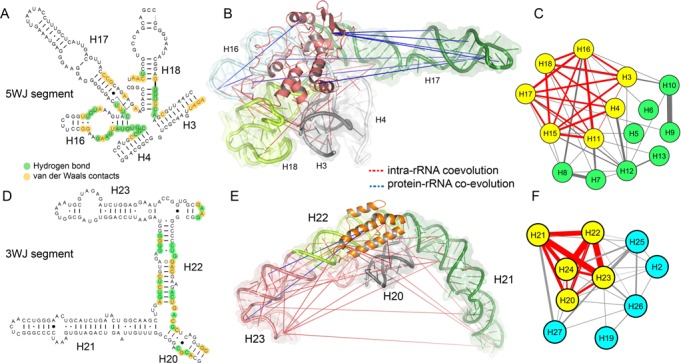
Co-evolutionary patterns at 5WJ and 3WJ rearrangement sites. (A) The native molecular contacts (green: H-bonds, yellow: van der Waals contacts) between S4 and 5WJ region of the 16S rRNA are highlighted. (B) The protein–rRNA and intra-rRNA co-evolutionary patterns at the 5WJ segment are shown (RNA: cartoon plus surface, S4: salmon cartoon). Each helix in the 5WJ segment is highlighted. (C) The inter-helix co-evolutionary network of 5′ domain is shown; each node represents one rRNA helix, while edge thickness is proportional to co-evolutionary preference scores. The 5WJ helices cluster as a dense-weighted module (yellow nodes, red edges). (D) Native molecular contacts between S15 and 3WJ region (green: H-bonds, yellow: van der Waals contacts) of the 16S rRNA. (E) The intra-rRNA co-evolutionary pattern at the 3WJ segment (RNA: cartoon plus surface, S15: orange cartoon). Each helix in the 3WJ segment is highlighted. (F) The inter-helix co-evolutionary network of central domain is shown; the 3WJ helices cluster as a dense-weighted module.
The initial encounter is followed by protein-induced structural rearrangements of H18 pseudoknot (H17–H18 junction), the 430-loop of H16 and a right-angle motif between H3, H4 and H18 (54). Surprisingly, we do not observe any protein–rRNA CEPs between S4 and H3–H4–H18; this indicates that S4 is required, but may not be indispensable for further reconstitutions of 5WJ. Molecular Dynamic simulation studies support our hypothesis by reporting that 5WJ segment has an inherent self-folding capability. S4 protein only slows down the rRNA fluctuations and induces anisotropic motions between the intermediate and the native complex (49). The disordered N-terminal loop of S4 likely plays the key role in guiding the rRNA folding (49), but the folding itself emerges from the inherent rRNA plasticity. Therefore, evolutionary signatures of this biophysical process are likely to be found in intra-rRNA co-evolutionary pattern, rather than protein–rRNA co-evolution.
To gain further insight, we transform the intra-rRNA CEPs from each of the 16S rRNA domains into three independent edge-weighted networks where each rRNA helix represents a node and PS values between helix pairs are the edge-weights (Supplementary Materials section 3.3). We perform module analysis (simultaneously considering the local density and edge-weights) on these networks to identify the subset of helices exhibiting stronger tendency of having CEPs.
The dense-weighted module at 5′-domain includes the 5WJ helices (H3, H4, H16, H17 and H18), along with H11 and H15 (Figure 3C, Supplementary Table S1). The residue-level co-evolutionary pattern at the 5WJ is shown in Figure 3B. Although protein–rRNA co-evolution is limited only between S4-H16 and S4-H17, strong intra-rRNA co-evolution is observed among these helices. This is evident from the fact that 5WJ helices exhibit significantly higher PS than rest of the 5′-domain helices (P < 10−3). The energetic coupling among the helices emerging from their protein-guided folding might be the physical origin of this co-evolution. S4 binds at 5WJ, stabilizes the 530 loop (H18) of the decoding center and increases flexibility of H3 (54). In the next step, S16 protein, recognized by H15, drives a conformational change at H3 that stabilizes pseudoknots at H18 in the decoding center (58). This functional constraint between H3 and H18 is reflected in their high PS (strongest among 5WJ helices). S16 is cooperative to S4 and S20 proteins; S20 is recognized at H11 and both H15 and H11 are tightly packed with the 5WJ helices in the native structure. This is likely the reason that they appear at the same dense-weighted module with the 5WJ helices (Figure 3C).
The 3WJ rearrangement
The 3WJ rearrangement induced by S15 protein nucleates the central domain assembly (52). The thermodynamic cycle of 3WJ rearrangement (21) shows how inter-protein energetic coupling is maintained by helix rearrangements. S15 binds with the 3WJ RNA (Figure 3D) with high affinity and is accompanied by a conformational change in the rRNA, by which H20 and H22 form an acute angle between them, and H21 and H22 are coaxially stacked (Figure 3E). At the absence of S15, the succeeding S6-S18 heterodimer binds weakly (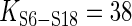 nM,
nM, 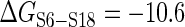 kcal/mol) at the junction of H22, H23a and H23b. But S15-induced 3WJ rearrangement dramatically increases the affinity of S6-S18 (
kcal/mol) at the junction of H22, H23a and H23b. But S15-induced 3WJ rearrangement dramatically increases the affinity of S6-S18 (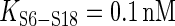 ,
, 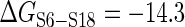 kcal/mol), causing –3.7 kcal/mol coupling energy between S15 and S6–S18. This stabilizes the folded rRNA structure to its native state (21,25) and accelerates the S6–S18 association about 120 fold (25). Our analysis shows that S15 protein co-evolves with only two nucleotide positions of H23 (Ile32-U701 and Ile32-G711), while S6–S18 heterodimer co-evolves with few nucleotide positions at H22 and H23 (G664, C679 and A687). However, numerous intra-rRNA CEPs are identified among the 3WJ helices (Figure 3E); the dense-weighted module of central domain includes the 3WJ helices and helix H24 (Figure 3F). Therefore, similar to 5WJ scenario, this result supports the fact that 3WJ rearrangement is mostly determined by inherent rRNA plasticity (59). Helices H21 and H24 recognize another primary binder S8 (binds before S15) and helix H24 is involved in constructing the E-site of ribosome as well. This might be the biological reason that H24 preferably co-evolves with 3WJ helices.
kcal/mol), causing –3.7 kcal/mol coupling energy between S15 and S6–S18. This stabilizes the folded rRNA structure to its native state (21,25) and accelerates the S6–S18 association about 120 fold (25). Our analysis shows that S15 protein co-evolves with only two nucleotide positions of H23 (Ile32-U701 and Ile32-G711), while S6–S18 heterodimer co-evolves with few nucleotide positions at H22 and H23 (G664, C679 and A687). However, numerous intra-rRNA CEPs are identified among the 3WJ helices (Figure 3E); the dense-weighted module of central domain includes the 3WJ helices and helix H24 (Figure 3F). Therefore, similar to 5WJ scenario, this result supports the fact that 3WJ rearrangement is mostly determined by inherent rRNA plasticity (59). Helices H21 and H24 recognize another primary binder S8 (binds before S15) and helix H24 is involved in constructing the E-site of ribosome as well. This might be the biological reason that H24 preferably co-evolves with 3WJ helices.
Kinetic trap: slow structural reconstitutions at the 3′ domain
Slow refolding observed at the 3′ domain (23) may result from a domain-wide kinetic trap (23,25). Kinetic trap refers to a state in the assembly in which further conformational transitions from an intermediate sub-complex become difficult due to its thermodynamic stability. If an assembly is arrested in a kinetic trap, the accumulated intermediate can proceed only slowly or not at all on the assembly.
The 3′ domain assembly faces substantial kinetic barriers after S7 binding, which is evident from the fact that S7 only mildly accelerates secondary protein (S9–S13–S19) bindings (25). The slow reconstitutions likely include mutual co-folding of the protein and rRNA components, which can be inferred from the observable numerous protein–rRNA CEPs in this domain. Furthermore, the 3′ domain edge-weighted co-evolutionary network includes a large dense-weighted module composed of 10 helices (H28, H29, H30, H31, H34, H35, H36, H39, H43 and H44) (Supplementary Table S1). This result differs from the 5′ and central domain scenario, where only a few helices constitute the dense-weighted module. 3′ domain is composed of 8 of the 20 proteins of SSU and the rRNA segment is structured into 16 helices, each making native contacts with proteins. So, we cannot explain the highly preferable co-evolution among these helices only by the maintenance of cooperative relationships. But H28, H29, H30, H31, H34, H43 and H44 are involved in constituting the tRNA-movement tunnel (60), which is constituted at the final stages of the assembly. Therefore, there is a strong probability that these helices might be arrested in a kinetic trap. Escape from kinetic trap involves an ensemble of infinitesimal reconstitution steps, for which the trapped helices face strong assembly constraints. This assembly constraint likely drives the preferable co-evolution among the intra-module helices.
The results above demonstrate the power of co-evolutionary patterns to predict the physical involvement of protein and rRNA components in the local folding process. In the next section, we test whether assembly kinetics also drive co-evolution.
Relative folding rates of 16S rRNA domains
In complex systems such as the ribosome, folding kinetics has been proven very difficult to resolve (3). However, available experimental data shows that the three domains of 16S rRNA fold independently and assemble at different rates (25): the central domain folds the quickest, the 3′ domain folds the slowest and the 5′-domain folds at an intermediate rate.
Folding rates of simple proteins are independent of their structural stabilities (folding free energy) and depend on the ‘topology’ of the native state (44,61). Contact order, a representative of protein ‘topology’, is defined as the average primary chain separation of residues in 3D contact. Higher values of contact order indicate numerous long-range interactions at the native state and such interactions require a long time to achieve during folding (44). Conversely, protein contact networks (networks of amino acids as nodes and non-bonded interactions among them as edges) with high assortative mixing (nodes tend to connect with nodes of similar degree) facilitate protein-folding rate, likely by allowing the rapid propagation of perturbation waves of conformational transitions (61). A non-covalent tertiary contact implies a steric constraint between two residues at the native state. Based on the strong statistical associations between assembly constraints and co-evolution, we expect that local patterns of co-evolution at different domains of 16S rRNA can vary according to their folding rates.
We introduce a parameter analogous to Contact Order, termed CEO, which measures the average primary chain separation of co-evolving pairs; the assortative mixing of the co-evolutionary network is computed by the ‘Coefficient of Assortativity’ (CoA). The lower CEO (97.50) and higher CoA (0.87) of the central domain (compared to 5′ and 3′ domains, Table 1) suggest relatively quicker folding, which is consistent with experimental evidence that central domain assembly faces few kinetic barriers after stabilization of the native rRNA conformation by S15 binding (21,25). Conversely, in 5′- and 3′-domains, kinetic barriers hinder reconstitution steps and folding is arrested in non-native stable conformations (25). The 3'-domain has a strong tendency to fall into a domain-wide kinetic trap that slows down its structural reconstitutions (23,25); consequently, 3' domain exhibits the highest CEO (192.75) and lowest CoA (0.62). The 5'-domain folds at an intermediate rate and exhibits intermediate CEO (136.05) and intermediate CoA (0.78).
Table 1. Co-Evolution Order (CEO) and Coefficient of Assortativity (CoA) at three different domains of 16S rRNA.
| 16S rRNA domain | CEO (all CEPs) | CEO (non-steric CEPs) | CoA (all CEPs) | CoA (non-steric CEPs) |
|---|---|---|---|---|
| 5′-Domain | 136.05 | 60.08 | 0.785 (10E−54) | 0.591 (10E−63) |
| Central domain | 97.50 | 32.45 | 0.871 (10E−43) | 0.708 (10E−35) |
| 3′-Domain | 192.75 | 159.57 | 0.615 (10E−32) | 0.408 (10E−21) |
The values in the parenthesis indicate the statistical significance of CoA value. Our results show that co-evolutionary patterns within SSU mimic the topology of small proteins according to local folding rates.
This analysis shows that co-evolutionary patterns mimic the topology of simple proteins according to how quickly the protein–rRNA constituents mutually co-fold into the native structure. Since two constrained sites are often structurally remote, we have investigated whether the assortative topology of the network persists if we remove the CEPs within steric interaction range. Remarkably, the association between local folding rates and the order of assortative mixing remains unaltered when considering only non-steric CEPs; similar results were obtained for CEO (Table 1). These results suggest a novel characteristic of a multidomain-multi-component system, where the topological features of a network of co-evolving residues at different domains are associated with their local folding rates (kinetics of the assembly).
Nucleotides essential for protein recognition
We have investigated whether residues playing key roles in protein–rRNA recognition can be identified from their co-evolutionary patterns. A nucleotide is considered as protein-recognizing if it loses >1 Å2 of its accessible surface area when passing from uncomplexed state to complexed state. For each protein, we look for two categories of nucleotides among those physically recognize the protein: (i) conserved sites (conserved group: Gcon) and (ii) sites in the co-evolutionary network. We compute the degree and clustering coefficient values of the co-evolving nucleotides and based on both the parameters, they are clustered into several groups. This clustering approach allows us to isolate a small group of nucleotides (Gcoevol) associated with highest clustering scores compared to rest of the population (Supplementary Materials section 3.4). Interestingly, nucleotide positions experimentally identified to be important for recognizing a particular protein are always associated with 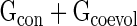 group (Supplementary Table S2). Nucleotides included in the Gcoevol group always co-evolve with the neighboring protein-recognizing rRNA stalk. For example, at S15 binding site (nucleates central domain assembly) we have isolated a group of five nucleotide positions (C582, C736, U740, G741 and U751). All these positions are experimentally identified as essential for recognizing S15 and phenotypic effects of their mutations are lethal (62,63). This co-evolutionary pattern suggests that protein-recognizing rRNA segments (experiencing protein-guided reconstitutions) are thermodynamically constrained to the nucleotides essential for protein recognition. This method may help to identify residue positions critical for ligand binding.
group (Supplementary Table S2). Nucleotides included in the Gcoevol group always co-evolve with the neighboring protein-recognizing rRNA stalk. For example, at S15 binding site (nucleates central domain assembly) we have isolated a group of five nucleotide positions (C582, C736, U740, G741 and U751). All these positions are experimentally identified as essential for recognizing S15 and phenotypic effects of their mutations are lethal (62,63). This co-evolutionary pattern suggests that protein-recognizing rRNA segments (experiencing protein-guided reconstitutions) are thermodynamically constrained to the nucleotides essential for protein recognition. This method may help to identify residue positions critical for ligand binding.
Global constraints in ribosome assembly: inter-domain packing
Ribosomal proteins involved in inter-domain packing exhibit numerous CEPs with their binding partners
A fundamental attribute of multidomain protein folding is the molecular packing of domains to stabilize the final native structure (64). The neighboring domains of the SSU exhibit intra-rRNA and protein–rRNA molecular contacts with each other, stabilizing their quaternary orientations in 3D space. Some proteins (S2, S2, S3, S5, S12 and S20) contact different 16S rRNA domains and proteins from other domains to ensure inter-domain packing (53). S20 stabilizes the quaternary orientation of helix H44; S2–S3–S5–S12 proteins predominantly bind to the central pseudoknots of 16S rRNA (near to H2 and H19), physically contacting multiple rRNA domains (53). Interestingly, these proteins exhibit significantly numerous (P < 0.05) protein–rRNA CEPs (∼72 CEPs/protein) compared to proteins connecting only one rRNA domain (∼11 CEPs/protein). Similarly, we observe strong PS among protein pairs maintaining inter-domain packing through physical interaction (S7–S11, S5–S3, S8–S12) compared to any random inter-domain protein pair.
Co-evolution among rRNA helices are jointly driven by local and global assembly constraints
Inter-domain packing is mainly mediated by numerous tertiary intra-rRNA molecular contacts. Helices around the neck and shoulder regions (53) of the SSU construct the tRNA-movement path (60) and the functional APE sites (Figure 4A). We generate an inter-helix edge-weighted contact network (rRNA helices are nodes, an edge represents molecular contact between them, edge-weight represents number of contacts) to identify the rRNA helices involved in inter-domain packing (Figure 5). For 5' and central domain packing these physically interacting helices are H1, H2, H3, H4, H7, H11, H12, H19, H20, H21, H27; for 5' and 3' domain packing, H2, H13, H18, H34, H36 and H44 play the key roles; and helices H20, H24, H25, H26, H28, H29, H36, H44 and H45 are essential for central and 3' domain packing.
Figure 4.
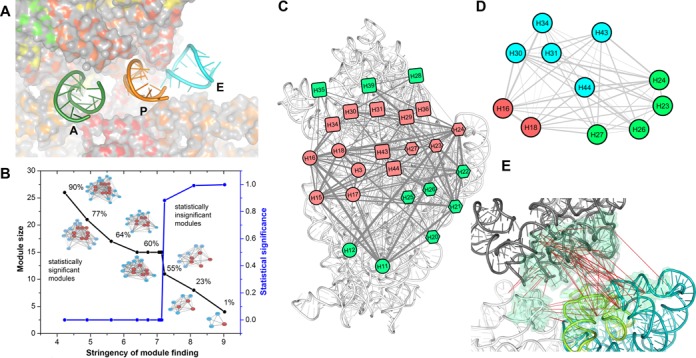
Co-evolutionary pattern at the APE-neighboring region of the 16S rRNA. (A) The tRNA movement tunnel: the A, P and E site tRNAs are shown as cartoon view (nt 28–40 segments of their anticodon arms), while 16S rRNA is shown as a colored surface. The tRNA-movement tunnel is highlighted by gradually increasing the surface transparency with surface depth. (B) The modules found (as computational stringency to finding a module is gradually reduced) within the inter-helix edge-weighted co-evolutionary network of 16S rRNA; black curve shows growth of module size, while blue curve indicates statistical significance of the module. Reduction of stringency is presented in a linear scale. Salmon colored nodes signify APE-neighboring helices. The percentages indicate how many known 16S rRNA point mutation sites reside within the module-helices. (C) The module including 90% of all reported mutation sites is projected onto the 16S rRNA (white cartoon). (D) The co-evolution among the APE-neighboring helices is shown. Helices from each domain are colored differently: red (5′ domain), green (central domain) and cyan (3′ domain). (E) The CEPs at the APE-neighboring region are shown. Many CEPs are identified between central and 3′-domain. The H44 of 3′-minor domain (green cartoon) exhibits several CEPs with its neighboring central domain and 3′-major domain nucleotides.
Figure 5.
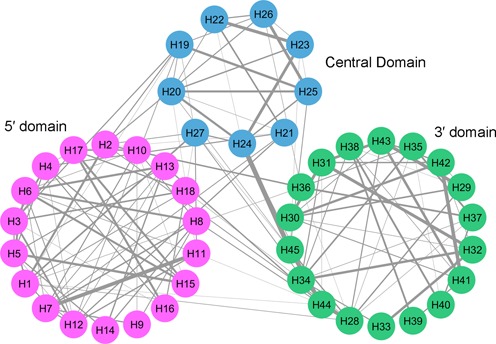
The inter-helix edge-weighted contact network of the 16S rRNA at its native assembled state. The network is constructed based on the number of van der Waals contacts between the atoms of the rRNA helices. A van der Waals contact is considered if two atoms are <6 Å separated in 3D space. In this image, each node represents a helix and lines connecting them represent the presence of van der Waals contacts between them. Edge thickness is proportional to the number of contacts. This network clearly demonstrates the intra-domain and inter-domain helix contacts. This network is prepared using the crystal structure having PDB identifier 2AVY (see Supplementary Dataset).
To understand the co-evolutionary pattern associated with helices involved in inter-domain packing, we transform the intra-rRNA CEPs into an edge-weighted network where each rRNA helix represents a node and PS values between helix pairs are the edge-weights. Module analyses of this network predominantly pick up a group of helices gathered around the APE sites as a dense-weighted module even with the highest computational stringency (Figure 4B). Since such initial modules do not significantly differ from the background network, stringency is further reduced. The growing module gradually incorporates 5WJ, 3WJ and the group of helices likely involved in 3′ domain kinetic trap (Figure 4B). Once the intra-module helices cover 90% of known 16S rRNA point mutation sites (65,66), the final module is projected onto the 16S rRNA crystal structure (Figure 4C).
APE-neighboring helices from all three domains co-assemble to construct the tRNA-movement path. Two coaxially stacked helix pairs from 5′ domain (H16 and H18) and 3′ domain (H31–H34) connect with one another to stabilize tRNA binding at the decoding center. The anticodon loop of the A-site tRNA interacts with the decoding center located at the conserved 530-loop of H18; and this conformation is further stabilized by few contacts between the anticodon arm and conserved H31–H34 pair. The P-site tRNA mostly connects helix H30 and the H42-pseudoknot of 3′ domain, while E-site tRNA connects the tip of helix H23 from central domain. We coin a term ‘APE-neighboring helices’ for those located within 20 Å from the tRNA-recognizing nucleotides (H16, H18, H23, H24, H27, H30, H31, H34, H43 and H44). Interestingly, these ten helices include those previously reconstituted at 5WJ (H16, H18) and 3WJ rearrangements (H23) and those experiencing 3′ domain kinetic trap (H30, H31, H34, H43 and H44). Gradual appearance of these previously reconstituted helices at the growing module (Figure 4B) illustrates local intra-domain constraints that can also act as global inter-domain constraints at different stages of assembly.
Structural constraints emerging from the inter-domain coupling likely drives strong preferable co-evolution among the APE-neighboring helices (Figure 4D). A majority of the intra-rRNA CEPs at this region (Figure 4E) connect helix H44 with neighboring 3'- and central domain rRNA segments. A majority of the CEPs connecting helix H44 (essential for translational fidelity (67)) are mediated by nucleotides flanking to conserved A1408, essential for 50S docking (67).
These results demonstrate how the local (intra-domain) and global (inter-domain) assembly constraints jointly determine the trajectories of sequence evolution. In the next step, we examine the association between co-evolution and the functional importance of individual nucleotides, validated by the point-mutation data.
Association of co-evolutionary patterns with point-mutation data
Interestingly, the reported 16S rRNA point-mutation sites (65,66) with significant phenotypic effects are located around the APE sites and early-protein recognition sites (Supplementary Materials section 3.5 and Table S3); we have classified these mutation sites into three categories (drastic, moderate and mild) according to severity of their phenotypic effects (66). The point-mutation sites are hubs/conserved positions and they are always identified in the neighborhood of other hubs and conserved nucleotides in the 3D structure. Performing a positional clustering using the Euclidean distances among the residues, we have identified 32 unique clusters of conserved/hub nucleotides (CH clusters) in the native 16S rRNA crystal structure (Supplementary Materials section 3.5.2). Interestingly, 21 of these 32 CH clusters include one or more documented point-mutation sites. Next, we have applied a random weighting method based on the number of mutational sites present at a CH cluster and the severity of their phenotypic effects (Supplementary Materials section 3.5.3). This method classifies the CH clusters into four groups according to gradually decreased number of mutations with decreased severity: highly sensitive (HS), moderately sensitive (MS), mildly sensitive (ms) and no-mutation (NM). HS and MS clusters are located mostly at the APE-neighboring and primary-binder recognition sites; these clusters are composed of few big hubs surrounded by many conserved residues and the contributing hubs co-evolve among themselves and with the proximal sites. Conversely, ms and NM clusters are composed of many small hubs and few conserved residues and they tend to co-evolve with distantly placed HS and MS hubs (Supplementary Figure S1 and S2). The ms and NM clusters are generally located at secondary and tertiary binder recognition sites, so their co-evolutions with distant HS and MS sites are likely driven by cooperative relationships.
Functionally essential sites exhibit stronger co-evolution with their neighborhood, compared to sites those are dispensable. Amino acid conservation is often used to predict functionally important sites; our results indicate that co-evolutionary information can also predict both functional sites and their interactions.
The putative important roles of the HS and MS-group residue clusters might be applied for antibiotic target site prediction. The known antibiotics target deleterious mutation sites of 16S rRNA; based on this observation, Yassin et al.(65) predicted 38 new deleterious mutation sites which might act as potential drug target sites. We pinpoint a subset of those sites that might be strong candidates of being targeted by antibiotics, based on their locations within HS and MS-group clusters (Supplementary Figure S3).
CONCLUSIONS
We have employed comparative sequence analysis and network theory on the self-assembly process of a macromolecular complex (which is central to cellular metabolism and phylogenetic analysis) to reveal statistical associations between co-evolutionary patterns and mechanistic of self-assembly process. We have developed a set of new metrics, which allow us to extract functionally relevant information from co-evolutionary signals. Protein–rRNA recognition, cooperativity phenomena and protein-induced helix rearrangements play crucial roles in driving inter-protein and protein–rRNA co-evolution. Molecular packing of rRNA helices plays a key role in driving intra-rRNA co-evolution. All three domains of the 16S rRNA fold independently and assemble at different rates. Folding rates of three domains are associated with the topological characteristics of their co-evolutionary networks. The co-evolutionary relationships provide biological bases for deleterious mutation sites and further allow prediction of putative antibiotic targeting sites. In summary, our results reveal that both molecular interactions in the native state as well as critical non-native interactions during the assembly drive co-evolutionary relationships.
These observations are a step toward understanding the relationship of assembly constraints of the ribosome and co-evolution among its protein–rRNA constituents. If such relationships hold for other macromolecular complexes, then co-evolutionary analyses, in combination with experimental approaches, might be helpful in illuminating allosteric mechanisms and multi-domain protein folding. For most large macromolecular complexes such as eukaryotic ribosomes, proteosomes and viral capsids, an understanding of the assembly process remains elusive. One can apply similar methods as described here to extract candidate sites involved in critical functional interactions during self-assembly of such complexes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors acknowledge the anonymous referees for many helpful suggestions during the review process.
FUNDING
Center of Excellence in Systems Biology and Biomedical Engineering (TEQIP Phase II), University of Calcutta, India; 2013 NIG Collaborative Research Grant A(82), National Institute of Genetics, Japan. Funding for open access charge: Center of Excellence in Systems Biology and Biomedical Engineering (TEQIP Phase II), University of Calcutta, India.
Conflict of interest statement. None declared.
REFERENCES
- 1.Saiz L., Vilar J.M. Stochastic dynamics of macromolecular-assembly networks. Mol. Syst. Biol. 2006;2:0024. doi: 10.1038/msb4100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollard T.D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 3.Williamson J.R. Cooperativity in macromolecular assembly. Nat. Chem. Biol. 2008;4:458–465. doi: 10.1038/nchembio.102. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T., Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 5.Moisant P., Neeman H., Zlotnick A. Exploring the paths of (virus) assembly. Biophys. J. 2010;99:1350–1357. doi: 10.1016/j.bpj.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marsh J.A., Hernández H., Hall Z., Ahnert S.E., Perica T., Robinson C.V., Teichmann S.A. Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell. 2013;153:461–470. doi: 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy E.D., Boeri Erba E., Robinson C.V., Teichmann S.A. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woese C.R., Fox G.E. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl. Acad. Sci. U.S.A. 1977;74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mears J.A., Cannone J.J., Stagg S.M., Gutell R.R., Agrawal R.K., Harvey S.C. Modeling a minimal ribosome based on comparative sequence analysis. J. Mol. Biol. 2002;321:215–234. doi: 10.1016/s0022-2836(02)00568-5. [DOI] [PubMed] [Google Scholar]
- 10.Roberts E., Sethi A., Montoya J., Woese C.R., Luthey-Schulten Z. Molecular signatures of ribosomal evolution. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13953–13958. doi: 10.1073/pnas.0804861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramakrishnan V., White S.W. Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends Biochem. Sci. 1998;23:208–212. doi: 10.1016/s0968-0004(98)01214-6. [DOI] [PubMed] [Google Scholar]
- 12.de Juan D., Pazos F., Valencia A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 13.Lovell S.C., Robertson D.L. An integrated view of molecular coevolution in protein–protein interactions. Mol. Biol. Evol. 2010;27:2567–2575. doi: 10.1093/molbev/msq144. [DOI] [PubMed] [Google Scholar]
- 14.Fares M.A., Travers S.A. A novel method for detecting intramolecular coevolution: adding a further dimension to selective constraints analyses. Genetics. 2006;173:9–23. doi: 10.1534/genetics.105.053249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madaoui H., Guerois R. Coevolution at protein complex interfaces can be detected by the complementarity trace with important impact for predictive docking. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7708–7713. doi: 10.1073/pnas.0707032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flock T., Venkatakrishnan A., Vinothkumar K., Babu M.M. Deciphering membrane protein structures from protein sequences. Genome Biol. 2012;13:160. doi: 10.1186/gb-2012-13-6-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunstan M.S., Guhathakurta D., Draper D.E., Conn G.L. Coevolution of protein and RNA structures within a highly conserved ribosomal domain. Chem. Biol. 2005;12:201–206. doi: 10.1016/j.chembiol.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 18.Brandman R., Brandman Y., Pande V.S. Sequence coevolution between RNA and protein characterized by mutual information between residue triplets. PLoS ONE. 2012;7:e30022. doi: 10.1371/journal.pone.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traub P., Nomura M. Structure and function of E. coli ribosomes v. reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc. Natl. Acad. Sci. U.S.A. 1968;59:777–784. doi: 10.1073/pnas.59.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samaha R.R., O'Brien B., O'Brien T.W., Noller H.F. Independent in vitro assembly of a ribonucleoprotein particle containing the 3' domain of 16S rRNA. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7884–7888. doi: 10.1073/pnas.91.17.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Recht M.I., Williamson J.R. RNA tertiary structure and cooperative assembly of a large ribonucleoprotein complex. J. Mol. Biol. 2004;344:395–407. doi: 10.1016/j.jmb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Adilakshmi T., Bellur D.L., Woodson S.A. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talkington M.W., Siuzdak G., Williamson J.R. An assembly landscape for the 30S ribosomal subunit. Nature. 2005;438:628–632. doi: 10.1038/nature04261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Held W.A., Ballou B., Mizushima S., Nomura M. Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J. Biol. Chem. 1974;249:3103–3111. [PubMed] [Google Scholar]
- 25.Bunner A.E., Beck A.H., Williamson J.R. Kinetic cooperativity in Escherichia coli 30S ribosomal subunit reconstitution reveals additional complexity in the assembly landscape. Proc. Natl. Acad. Sci. U.S.A. 2010;107:5417–5422. doi: 10.1073/pnas.0912007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaczanowska M., Rydén-Aulin M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. R. 2007;71:477–494. doi: 10.1128/MMBR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartman H., Favaretto P., Smith T.F. The archaeal origins of the eukaryotic translational system. Archaea. 2006;2:1–9. doi: 10.1155/2006/431618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D'Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M., et al. The comparative RNA web (CRW) site: an online database of comparative sequence & structure information for ribosomal, intron, & other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fodor A.A., Aldrich R.W. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins. 2004;56:211–221. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 30.Fares M.A., Travers S.A.A. A novel method for detecting intramolecular coevolution: adding a further dimension to selective constraints analyses. Genetics. 2006;173:9–23. doi: 10.1534/genetics.105.053249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleishman S.J., Yifrach O., Ben-Tal N. An evolutionarily conserved network of amino acids mediates gating in voltage-dependent potassium channels. J. Mol. Biol. 2004;340:307–318. doi: 10.1016/j.jmb.2004.04.064. [DOI] [PubMed] [Google Scholar]
- 32.Dutheil J., Pupko T., Jean-Marie A., Galtier N. A model-based approach for detecting coevolving positions in a molecule. Mol. Biol. Evol. 2005;22:1919–1928. doi: 10.1093/molbev/msi183. [DOI] [PubMed] [Google Scholar]
- 33.Wollenberg K.R., Atchley W.R. Separation of phylogenetic & functional associations in biological sequences by using the parametric bootstrap. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3288–3291. doi: 10.1073/pnas.070154797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gouveia-Oliveira R., Pedersen A.G. Finding coevolving amino acid residues using row and column weighting of mutual information and multi-dimensional amino acid representation. Algorithms Mol. Biol. 2007;2:12. doi: 10.1186/1748-7188-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn S.D., Wahl L.M., Gloor G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- 36.Buslje C.M., Santos J., Delfino J.M., Nielsen M. Correction for phylogeny, small number of observations & data redundancy improves the identification of coevolving amino acid pairs using mutual information. Bioinformatics. 2009;25:1125–1131. doi: 10.1093/bioinformatics/btp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin L.C., Gloor G.B., Dunn S.D., Wahl L.M. Using information theory to search for co-evolving residues in proteins. Bioinformatics. 2005;21:4116–4124. doi: 10.1093/bioinformatics/bti671. [DOI] [PubMed] [Google Scholar]
- 38.Yeang C.-H., Haussler D. Detecting coevolution in and among protein domains. PLoS Comp. Biol. 2007;3:e211. doi: 10.1371/journal.pcbi.0030211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayashida M., Kamada M., Song J., Akutsu T. Prediction of protein-RNA residue-base contacts using two-dimensional conditional random field with the lasso. BMC Syst. Biol. 2013;7(Suppl. 2):S15. doi: 10.1186/1752-0509-7-S2-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahony S., Auron P.E., Benos P.V. Inferring protein-DNA dependencies using motif alignments and mutual information. Bioinformatics. 2007;23:i297–i304. doi: 10.1093/bioinformatics/btm215. [DOI] [PubMed] [Google Scholar]
- 41.Tillier E.R., Lui T.W. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics. 2003;19:750–755. doi: 10.1093/bioinformatics/btg072. [DOI] [PubMed] [Google Scholar]
- 42.Fodor A.A., Aldrich R.W. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins. 2004;56:211–221. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 43.Newman M.E.J. Mixing patterns in networks. Phys. Rev. E. 2003;67:026126. doi: 10.1103/PhysRevE.67.026126. [DOI] [PubMed] [Google Scholar]
- 44.Dinner A.R., Karplus M. The roles of stability and contact order in determining protein folding rates. Nat. Struct. Biol. 2001;8:21–22. doi: 10.1038/83003. [DOI] [PubMed] [Google Scholar]
- 45.Nepusz T., Yu H., Paccanaro A. Detecting overlapping protein complexes from protein-protein interaction networks. Nat. Methods. 2012;9:471–472. doi: 10.1038/nmeth.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones S., Thornton J.M. Protein-protein interactions: a review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 47.Tsodikov O.V., Record M.T. Jr, Sergeev Y.V. A novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J. Comput. Chem. 2002;23:600–609. doi: 10.1002/jcc.10061. [DOI] [PubMed] [Google Scholar]
- 48.Hammer Ø., Harper D.A.T., Ryan P.D. PAST: Paleontological Statistics software package for education and data analysis. Paleontol. Elect. 2001;4:9. [Google Scholar]
- 49.Kim H., Abeysirigunawarden S.C., Chen K., Mayerle M., Ragunathan K., Luthey-Schulten Z., Ha T., Woodson S.A. Protein-guided RNA dynamics during early ribosome assembly. Nature. 2014;506:334–338. doi: 10.1038/nature13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freire E., Murphy K.P. Molecular basis of co-operativity in protein folding. J. Mol. Biol. 1991;222:687–698. doi: 10.1016/0022-2836(91)90505-z. [DOI] [PubMed] [Google Scholar]
- 51.Cui Q., Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agalarov S.C., Sridhar Prasad G., Funke P.M., Stout C.D., Williamson J.R. Structure of the S15, S6, S18-rRNA complex: assembly of the 30S ribosome central domain. Science. 2000;288:107–113. doi: 10.1126/science.288.5463.107. [DOI] [PubMed] [Google Scholar]
- 53.Brodersen D.E., Clemons W.M. Jr, Carter A.P., Wimberly B.T., Ramakrishnan V. Crystal structure of the 30 s ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16 S RNA. J. Mol. Biol. 2002;316:725–768. doi: 10.1006/jmbi.2001.5359. [DOI] [PubMed] [Google Scholar]
- 54.Mayerle M., Bellur D.L., Woodson S.A. Slow formation of stable complexes during coincubation of minimal rRNA and ribosomal protein S4. J. Mol. Biol. 2011;412:453–465. doi: 10.1016/j.jmb.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davies C., Gerstner R.B., Draper D.E., Ramakrishnan V., White S.W. The crystal structure of ribosomal protein S4 reveals a two-domain molecule with an extensive RNA-binding surface: one domain shows structural homology to the ETS DNA-binding motif. EMBO J. 1998;17:4545–4558. doi: 10.1093/emboj/17.16.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sayers E.W., Gerstner R.B., Draper D.E., Torchia D.A. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry. 2000;39:13602–13613. doi: 10.1021/bi0013391. [DOI] [PubMed] [Google Scholar]
- 57.Winker S., Woese C.R. A definition of the domains Archaea, Bacteria and Eucarya in terms of small subunit ribosomal RNA characteristics. Syst. Appl. Microbiol. 1991;14:305–310. doi: 10.1016/S0723-2020(11)80303-6. [DOI] [PubMed] [Google Scholar]
- 58.Ramaswamy P., Woodson S.A. Global stabilization of rRNA structure by ribosomal proteins S4, S17, and S20. J. Mol. Biol. 2009;392:666–677. doi: 10.1016/j.jmb.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Batey R.T., Williamson J.R. Effects of polyvalent cations on the folding of an rRNA three-way junction and binding of ribosomal protein S15. RNA. 1998;4:984–997. doi: 10.1017/s1355838298980426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer N., Konevega A.L., Wintermeyer W., Rodnina M.V., Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 61.Bagler G., Sinha S. Assortative mixing in Protein Contact Networks and protein folding kinetics. Bioinformatics. 2007;23:1760–1767. doi: 10.1093/bioinformatics/btm257. [DOI] [PubMed] [Google Scholar]
- 62.Batey R.T., Williamson J.R. Effects of polyvalent cations on the folding of an rRNA three-way junction & binding of ribosomal protein S15. RNA. 1998;4:984–997. doi: 10.1017/s1355838298980426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serganov A.A., Masquida B., Westhof E., Cachia C., Portier C., Garber M., Ehresmann B., Ehresmann C. The 16S rRNA binding site of Thermus thermophilus ribosomal protein S15: comparison with Escherichia coli S15, minimum site & structure. RNA. 1996;2:1124–1138. [PMC free article] [PubMed] [Google Scholar]
- 64.Randles L.G., Batey S., Steward A., Clarke J. Distinguishing specific and nonspecific interdomain interactions in multidomain proteins. Biophys. J. 2008;94:622–628. doi: 10.1529/biophysj.107.119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yassin A., Fredrick K., Mankin A.S. Deleterious mutations in small subunit ribosomal RNA identify functional sites and potential targets for antibiotics. Proc. Natl. Acad. Sci. U.S.A. 2005;102:16620–16625. doi: 10.1073/pnas.0508444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Triman K.L., Peister A., Goel R.A. Expanded versions of the 16S and 23S ribosomal RNA mutation databases (16SMDBexp and 23SMDBexp) Nucleic Acids Res. 1998;26:280–284. doi: 10.1093/nar/26.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin D., Liu Q., Devaraj A., Fredrick K. Role of helix 44 of 16S rRNA in the fidelity of translation initiation. RNA. 2012;18:485–495. doi: 10.1261/rna.031203.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.