

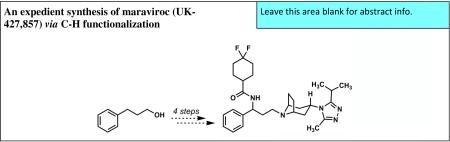
Abstract
A new, concise synthesis of the CCR-5 receptor antagonist maraviroc (UK-427,857) from 3-phenyl-1-propanol has been completed in four steps featuring a site-selective C–H functionalization.
Keywords: C-H functionalization, HIV/AIDS, CCR-5, Maraviroc, Amination
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
1. Introduction
As of 2013, there were 35.3 million people worldwide living with human immunodeficiency virus (HIV) and 2 million new infections acquired that year. At the same time, approximately 1.5 million individuals died from acquired immunodeficiency syndrome (AIDS), a rate of three every minute annually.1 Although the number of people currently infected with HIV has continued to grow over the last decade, the number of new infections has also dropped, indicating that HIV/AIDS has transitioned from a death sentence to what can be considered a manageable, chronic condition.2,3 The discovery and subsequent approval of the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine (1, AZT, Retrovir®) (Figure 1) in 1987 transformed the treatment of HIV infection,4 so much so that it has resided on the World Health Organization's Model Lists of Essential Medicines since 1998.5 Unfortunately, however, the use of monotherapy rapidly led to the emergence of drug resistant strains. The next leap forward for treatment came with the advent of highly active antiretroviral therapy (HAART) in 1996; this next revolution in HIV treatment was enabled by the synthesis, development, and approval of both new NRTIs, as well as new classes of drugs targeting other stages of the viral life cycle.2-4
Figure 1.

Selected FDA-approved HIV drugs
Pfizer's maraviroc (2, UK-427,857, Selzentry®) is a chemokine receptor antagonist that belongs to the fusion/entry inhibitor class. These compounds disrupt viral entry by inhibiting the binding of the HIV virus to CCR-5, a G protein-coupled receptor (GPCR) found primarily in cells of the immune system.6 Maraviroc acts as a CCR-5 antagonist by preventing this binding event, while enfuvirtide (3, Fuzeon®), an injectable polypeptide and the only other approved member of this class, binds a transmembrane protein of HIV (gp41) disrupting the final phase of viral fusion.7 Thus, maraviroc, which received FDA approval in 2007, is the only orally-dosed, small-molecule therapy targeting HIV viral entry.8
The molecular structure of maraviroc also induced diverse and creative efforts to construct its single, nitrogen-bearing benzylic stereocenter. The pioneering and highly convergent medicinal chemistry9 and process chemistry10,11 syntheses of maraviroc (2) by Pfizer (Scheme 1) utilized an intermolecular Mannich/enantiomer resolution process to produce β-amino ester 4, a key intermediate that was subsequently employed in coupling reactions with 4,4-difluorocyclohexane-1-carboxylic acid 5 and tropane triazole 612 to complete the synthesis. The subsequent syntheses of maraviroc by the laboratories of Schaus13 and Cordóva14 retained the logic of Pfizer's convergent synthesis design and featured chiral catalyst-controlled, asymmetric allylation and aza-Michael reactions, respectively, to establish the absolute configuration of the nitrogen-bearing stereocenter of maraviroc (2).
Scheme 1.

Pfizer's retrosynthetic analysis and key intermediates in prior syntheses of maraviroc (2).
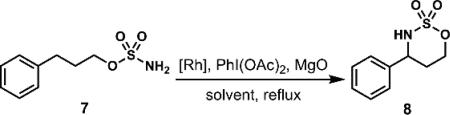
Through our ongoing collaborations with the Center for Selective C–H Functionalization (CCHF), we were drawn to the concept of constructing the benzylic C–N bond of maraviroc in the course of a Du Bois cyclization of acyclic sulfamate ester 7 (Scheme 2).15, 16-32 In the wake of the pivotal, ring-forming rhodium nitrene C–H insertion reaction (7 → 8), we would capitalize on the activation provided by the sulfonyl tethering element to achieve the final two C–N bond formations (8 → 9 → 2).
Scheme 2.

A design for synthesizing maraviroc (2) featuring a Du Bois cyclization and the activation provided by a sulfonyl group.
In relation to the prior syntheses of maraviroc, this strategy would not require any oxidation state adjustments in the ascension to maraviroc (2).33 The simple –SO2– group that would be required for the key Du Bois cyclization would be expelled15 in the course of the final nucleophilic attack of the tropane building block on the terminal carbon of the activated, N-acylated cyclic sulfamate (9 → 2). The realization of this strategy featuring the use of a sulfonyl group as both a traceless directing and activating group in a synthesis of maraviroc (2) is described below.
Results and Discussion
The foundation for the strategy outlined in Scheme 2 is 3-phenyl-1-propanol, an inexpensive and readily starting material ($72/kg Sigma-Aldrich) comprising the phenyl-substituted propane backbone of the target, as well as the desired oxidation state at the terminal carbon atom. By a straightforward, known reaction,15,34 3-phenyl-1-propanol was converted to sulfamate ester 7, and, from that vantage point, we revisited Du Bois's attractive conversion of compound 7 to cyclic sulfamate 8.15 Our aim was to preserve the efficiency of this transformation while increasing the reaction concentration and reducing both the reaction time and catalyst loading. Using the original literature report for reference (entry 1, Table 1), we found that by replacing Rh2(oct)4 with Rh2(OAc)4 and increasing the reaction time from the originally reported 1-2 hours to 12 hours we could isolate the desired amination product in 93% yield (entry 5, Table 1) after column chromatography.
Table 1.
Rh-catalyzed C–H amination of 3-phenylpropylsulfamate 7.
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Cat. load (mol%) | Solvent | Concentration (M) | Yield (%) |
| 1 | Rh2(oct)4 | 2 | DCM | 0.16 | 84 |
| 2 | Rh2(OAc)4 | 0.5 | DCM | 0.10 | n.d. |
| 3 | Rh2(OAc)4 | 0.7 | DCM | 0.20 | 72 |
| 4 | Rh2(OAc)4 | 2 | DCM | 0.15 | 81a |
| 5 | Rh2(OAc)4 | 2 | DCM | 0.15 | 93 |
| 6 | Rh2(OAc)4 | 2 | DCM | 0.15 | 85b |
| 7 | Rh2(esp)2 | 0.25 | DCE | 0.10 | n.d. |
| 8 | Rh2(esp)2 | 0.5 | DCE | 0.10 | 66a,b |
| 9 | Rh2(esp)2 | 1 | DCE | 0.10 | 72a,b |
| 10 | Rh2(esp)2 | 1 | DCE | 0.25 | 77a,b |
| 11 | Rh2(esp)2 | 1 | DCE | 0.50 | 69a,b |
| 12 | Rh2(esp)2 | 1 | DCE | 0.20 | 81a,b |
DCM = dichloromethane; DCE = 1,2-dichloroethane.
Run open to air
Purified by recrystallization
The use of Rh2(esp)2, a catalyst with enhanced stability,35,36 in refluxing dichloroethane (DCE) permitted the desired aminations at concentrations as high as 0.5 M and catalyst loadings as low as 0.5 mol%; under these conditions, the desired cyclic sulfamate 8 was produced in moderate-to-good yields in less than 30 minutes. At lower catalyst loadings or concentrations above 0.2 M, small amounts of unidentified byproducts were observed, resulting in reduced yields of cyclic sulfamate 8 after purification. Nevertheless, these reactions conditions were reliable in cyclizations of 10 grams (50 mmol) of compound 7.
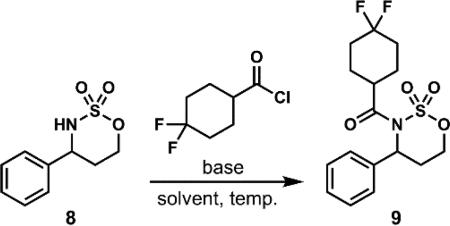
After optimization of the intramolecular C–H amination, attention was focused on improving the acylation of the cyclic sulfamate 8 with the acid chloride derived from 4,4-difluorocyclohexane-1-carboxylic acid 5. Treatment of compound 8 with NaOt-Bu as reported by Du Bois15 resulted in the formation of the acylated sulfamate product 9 in 27% yield (entry 1, Table 2). Initial attempts at optimization began by employing NaH in 1,2-dimethoxyethane (DME) at room temperature and proved to be effective (entry 2, Table 2). However, on scales greater than 1 mmol of substrate 8, competing decomposition of 8 was observed. By changing the base to KOt-Bu in DME (entry 3, Table 2), the reaction became more consistent and reliably afforded greater than 60% yield of the desired acylated sulfamate 9 on scales up to approximately 1.5 grams.
Table 2.
Optimization of acylation conditions.
| |||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | temp (°C) | conc. (M) | Yield (%) |
| 1 | NaOt-Bu | DME | rt | 0.1 | 27 |
| 2 | NaH | DME | rt | 0.1 | 74 |
| 3 | KOt-Bu | DME | rt | 0.1 | 63 |
| 4 | n-BuLi | THF | –78 | 0.1 | n.d. |
| 5 | n-BuLi | THF | 0 | 0.1 | n.d. |
| 6 | i-PrMgCl | THF | 0 | 0.1 | 90a |
| 7 | i-PrMgCl | THF | 0 | 0.2 | 74 |
DME = 1,2-dimethoxyethane
Additional reactions were also conducted in THF as solvent with stronger bases. Use of n-butyllithium (n-BuLi) at –78 °C (entry 4) led to incomplete conversion of compound 8 to acylated sulfamate 9. Disappointingly, the same reaction, when performed at 0 °C, resulted in decomposition of the starting material with no desired product observed. Gratifyingly, the use of ipropylmagnesium chloride (i-PrMgCl) at 0 °C (entry 6) led to another significant improvement in yield. These conditions were found to not only be scalable (up to 9.5 grams/45 mmol, albeit with a decrease in yield), but, by employing i-PrMgCl, we also found that we were able to double the concentration of the acylation reaction with no detriment to the conversion, yield, and purity of 9 (entry 7, see Supporting Information).
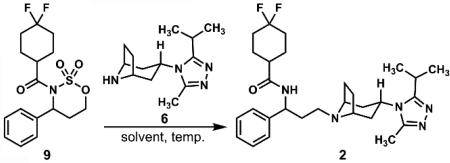
Having formed two of the three key bonds targeted in our retrosynthesis, we could address the final union of N-acylated cyclic sulfamate 9 and the tropane triazole 6. The pioneering synthesis of maraviroc (2) by Pfizer9-11 had established the feasibility of forming the same C–N bond by reductive amination after attempts at displacing a primary mesylate by the secondary amine of the tropane fragment had failed to provide greater than 20 percent yield of 2 under a variety of conditions. While the difficulties that the Pfizer group had encountered in alkylations of the tropane building block with primary methanesulfonate esters were a concern, we reasoned that the conformationally-constrained nature and the electron-withdrawing acyl group of cyclic sulfamate 9 would heighten the intrinsic electrophilic reactivity of the terminal carbon that would be attacked by the secondary amine of compound 6.
Combining 9 and 6 in a variety of solvents produced the results outlined in Table 3. Polar aprotic solvents, (except for DMSO) were best for this transformation, with the reaction in acetonitrile providing the N-alkylated product in 62% yield. We also found that it was also possible to intercept the Pfizer -amino alcohol 1011 in four steps from 3-phenyl-1-propanol by reacting 9 with water in hot acetonitrile. These results constitute a four step and formal, six-step synthesis of maraviroc (2) from 3-phenyl-1-propanol in approximately 35% and 40% overall yield, respectively.
Table 3.
Exploration of solvents for tropane alkylation.
| ||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yield (%) |
| 1 | DMSO | rt | 24 | n.d. |
| 2 | DMF | rt | 12 | 56 |
| 3 | NMP | rt | 12 | 44 |
| 4 | Acetonitrile | rt | 12 | 58 |
| 5 | Acetonitrile | 75 | 6 | 62 |
| 6 | Acetonitrile | 75 | 12 | 56 |
DMSO = dimethylsulfoxide; DMF = N,N-dimethylformamide; NMP = N-methyl-2-pyrolidinone
Overall, this synthesis produces racemic maraviroc in only four operations from commercially available 3-phenyl-1-propanol and two of the three building blocks of the pioneering synthesis by Pfizer. Since the pivotal Du Bois cyclization of sulfamate ester 7 has been achieved in an enantioselective fashion,32 a path to either enantiomer of maraviroc featuring the concept of C–H amination is now at hand. This achievement takes its place beside a rapidly expanding number of C–H functionalizations that require substrate directivity and yet it is distinguished by its complete utilization of the potentialities of a simple sulfonyl group; this simple tethering element directs the pivotal rhodium nitrene C–H insertion, exerts a favorable influence over the two subsequent C–N bond formations, and disappears in the course of the final coupling step. In light of the previously reported difficulties in the alkylation of Pfizer's tropane building block 6 with simple sulfonate esters, the successful pairing of N-acylated cyclic sulfamate 9 with compound 6 to directly give maraviroc (2) is noteworthy. The particular combination of concepts on which this synthesis is founded permitted a direct transformation of an inexpensive and abundant starting material to an important HIV drug without the need for protecting groups or oxidation state adjustments.33 Our efforts to probe the capabilities of the expanding menu of C–H functionalization methods in syntheses of structurally intricate natural products and pharmaceutical agents are continuing.
Supplementary Material
Scheme 3.

Synthesis of maraviroc (2) from 3-phenyl-1-propanol
Acknowledgements
The authors would like to thank Pfizer for the generous gift of the tosylate salt of compound 6. This work was supported by the National Institute of General Medical Sciences (GM065483), CCI Center for Selective C–H Functionalization National Science Foundation (CHE-1205646) and Princeton University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO . WHO; 2014. pp. 1–168. [Google Scholar]
- 2.Fauci AS, Folkers GK. J. Am. Med. Assoc. 2012;308:343–344. [Google Scholar]
- 3.Deeks SG, Lewin SR, Havlir DV. Lancet. 2013;382:1525–1533. doi: 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dube K, Henderson GE, Margolis DM. Trends Microbiol. 2014;22:547–549. doi: 10.1016/j.tim.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO EMLsChanges 1977_2011.xls Ed [Google Scholar]
- 6.Lederman MM, Penn-Nicholson A, Cho M, Mosier DJ. Am. Med. Assoc. 2006;296:815–826. doi: 10.1001/jama.296.7.815. [DOI] [PubMed] [Google Scholar]
- 7.LaBonte J, Lebbos J, Kirkpatrick P. Nat. Rev. Drug Discov. 2003;2:345–346. doi: 10.1038/nrd1091. [DOI] [PubMed] [Google Scholar]
- 8.Abel S, Davis JD, Ridgway CE, Hamlin JC, Vourvahis M. Antivir. Ther. 2009;14:831–837. doi: 10.3851/IMP1297. [DOI] [PubMed] [Google Scholar]
- 9.Price DA, Gayton S, Selby MD, Ahman J, Haycock-Lewandowski S, Stammen BL, Warren A. Tetrahedron Lett. 2005;46:5005–5007. [Google Scholar]
- 10.Haycock-Lewandowski SJ, Wilder A, Ahman J. Org. Proc. Res. Dev. 2008;12:1094–1103. [Google Scholar]
- 11.Åhman J, Birch M, Haycock-Lewandowski SJ, Long J, Wilder A. Org. Proc. Res. Dev. 2008;12:1104–1113. [Google Scholar]
- 12.Price DA, Gayton S, Selby MD, Ahman J, Haycock-Lewandowski S. Synlett. 2005;2005:1133–1134. [Google Scholar]
- 13.Lou S, Moquist PN, Schaus SE. J. Am. Chem. Soc. 2007;129:15398–15404. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao GL, Lin S, Korotvicka A, Deiana L, Kullberg M, Cordova A. Adv. Synth. Catal. 2010;352:2291–2298. [Google Scholar]
- 15.Espino CG, Wehn PM, Chow J, Du Bois J. J. Am. Chem. Soc. 2001;123:6935–6936. [Google Scholar]
- 16.Liang JL, Yuan SX, Huang JS, Yu WY, Che CM. Angew. Chem. Int. Ed. 2002;41:3465–3468. doi: 10.1002/1521-3773(20020916)41:18<3465::AID-ANIE3465>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 17.Liang JL, Yuan SX, Huang JS, Che CM. J. Org. Chem. 2004;69:3610–3619. doi: 10.1021/jo0358877. [DOI] [PubMed] [Google Scholar]
- 18.Cui Y, He C. Angew. Chem. Int. Ed. 2004;43:4210–4212. doi: 10.1002/anie.200454243. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Chan PWH, Che CM. Tetrahedron Lett. 2005;46:5403–5408. [Google Scholar]
- 20.Zhang JL, Huang JS, Che CM. Chem.-Eur. J. 2006;12:3020–3031. doi: 10.1002/chem.200501510. [DOI] [PubMed] [Google Scholar]
- 21.Brodsky BH, Bois JD. Chem. Commun. 2006:4715–4717. doi: 10.1039/b611280c. [DOI] [PubMed] [Google Scholar]
- 22.Liu P, Wong EL-M, Yuen AW-H, Che C-M. Org. Lett. 2008;10:3275–3278. doi: 10.1021/ol801157m. [DOI] [PubMed] [Google Scholar]
- 23.Zalatan DN, Du Bois J. Synlett. 2009;2009:143–146. doi: 10.1055/s-0028-1087392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barman DN, Nicholas KM. Eur. J. Org. Chem. 2011;2011:908–911. [Google Scholar]
- 25.Kornecki KP, Berry JF. Eur. J. Inorg. Chem. 2012;2012:562–568. [Google Scholar]
- 26.Paradine SM, White MC. J. Am. Chem. Soc. 2012;134:2036–2039. doi: 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]
- 27.Kornecki KP, Berry JF. Chem. Commun. 2012;48:12097–12099. doi: 10.1039/c2cc36614b. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Guan X, Wong EL-M, Liu P, Huang J-S, Che C-M. J. Am. Chem. Soc. 2013;135:7194–7204. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]
- 29.Harvey ME, Musaev DG, Du Bois J. J. Am. Chem. Soc. 2011;133:17207–17216. doi: 10.1021/ja203576p. [DOI] [PubMed] [Google Scholar]
- 30.Huang G-H, Li J-M, Huang J-J, Lin J-D, Chuang GJ. Chem.-Eur. J. 2014;20:5240–5243. doi: 10.1002/chem.201304633. [DOI] [PubMed] [Google Scholar]
- 31.Milczek E, Boudet N, Blakey S. Angew. Chem. Int. Ed. 2008;47:6825–6828. doi: 10.1002/anie.200801445. [DOI] [PubMed] [Google Scholar]
- 32.Zalatan DN, Du Bois J. J. Am. Chem. Soc. 2008;130:9220–9221. doi: 10.1021/ja8031955. [For other enantioselective aminations of 3-phenylpropylsulfamate, see references 19 and 31.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns NZ, Baran PS, Hoffmann RW. Angew. Chem. Int. Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 34.Guthikonda K, Du Bois J. J. Am. Chem. Soc. 2002;124:13672–13673. doi: 10.1021/ja028253a. [DOI] [PubMed] [Google Scholar]
- 35.Espino CG, Fiori KW, Kim M, Du Bois J. J. Am. Chem. Soc. 2004;126:15378–15379. doi: 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]
- 36.Du Bois J. Org. Process Res. Dev. 2011;15:758–762. doi: 10.1021/op200046v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.