

Abstract
Transport of material between extensive neuronal processes and the cell body is crucial for neuronal function and survival. Growing evidence shows that deficits in axonal transport contribute to the pathogenesis of multiple neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). Here we review recent data indicating that defects in dynein-mediated retrograde axonal transport are involved in ALS etiology. We discuss how mutant copper-zinc superoxide dismutase (SOD1) and an aberrant interaction between mutant SOD1 and dynein could perturb retrograde transport of neurotrophic factors and mitochondria. A possible contribution of axonal transport to the aggregation and degradation processes of mutant SOD1 is also reviewed. We further consider how the interference with axonal transport and protein turnover by mutant SOD1 could influence the function and viability of motor neurons in ALS.
Keywords: amyotrophic lateral sclerosis, axonal transport, dynein, motor neuron, superoxide dismutase
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that leads to paralysis and death, typically within 5 years after onset because of loss of motor neurons in the spinal cord, brainstem, and motor cortex. Approximately 10% of ALS cases are inherited and of these approximately 20% are caused by dominantly inherited mutations in the Cu, Zn-superoxide dismutase 1 (SOD1) gene (Deng et al. 1993; Rosen et al. 1993). To date, more than 100 mutations scattered throughout the SOD1 protein have been identified and it has been established that SOD1 mutants acquire toxic properties (Gaudette et al. 2000). However, the nature of the toxicity and how the toxicity causes preferential motor neuron death is still debated. Several hypotheses of how mutant SOD1 could cause neurodegeneration including aberrant redox chemistry, mitochondrial damage, excitotoxicity, microglial activation, and inflammation as well as SOD1 aggregation have been proposed (Bruijn et al. 2004; Pasinelli and Brown 2006; Shaw and Valentine 2007).
Recent studies have shown that motor neurons are highly sensitive to defects in axonal transport, and reduced transport has been suggested to cause or contribute to the degeneration in ALS. Mutations in the retrograde motor complex dynein and in the dynein interacting complex dynactin cause motor neuron degeneration in humans and mice (Hafezparast et al. 2003; Puls et al. 2005). Decreased kinesin-mediated (anterograde) and dynein-mediated (retrograde) axonal transport have been observed both in ALS patients and in transgenic animal models (Breuer et al. 1987; Breuer and Atkinson 1988; Collard et al. 1995; Sasaki and Iwata 1996a; Williamson and Cleveland 1999; Ligon et al. 2005). In G93A SOD1 transgenic mice, a considerable inhibition of retrograde axonal transport was observed at a very early stage of disease before animals became symptomatic (Ligon et al. 2005). However, the mechanisms by which mutant SOD1 affects axonal transport have not been established. We recently showed that compared with wild-type (WT) SOD1, several SOD1 mutants interact with the dynein-dynactin complex much more stably (Zhang et al. 2007). Furthermore, we and others have shown that dynein co-localizes with protein inclusions formed by mutant SOD1 in ALS (Ligon et al. 2005; Zhang et al. 2007).
In this review, we will focus on recent data linking axonal transport defects to motor neuron degeneration and ALS etiology. We will discuss how mutant SOD1 and an aberrant interaction between mutant SOD1 and dynein-dynactin could influence neuronal survival by impairing retrograde transport of neurotrophic factors and mitochondria, as well as the potential role of this interaction in the aggregation process of mutant SOD1.
Intracellular transport in neurons
Motor neurons are highly specialized cells with extensive dendritic arbors and axonal processes that can extend up to 1 m from the cell body. The ability of the neuron to maintain this specialized morphology depends on cytoskeletal elements and continous transport of proteins and organelles to and from the cell body. The neuronal cytoskeleton comprises networks of microtubules, actin filaments, and neurofilaments (Fig. 1). Microtubules provide stability and polarity to the axonal compartment of the neuron as they are polarized with a slow-growing minus end directed toward the cell body and a fast growing plus end directed peripherally in the axon. Actin which is enriched at the cell membrane contributes mainly to the integrity of the cell periphery and plays a role in resistance to stress in mature neurons. Neurofilaments, which assemble from three subunits NF-L, NF-M, and NF-H primarily provide structural stabilization and regulate the axonal caliber, thereby controlling the speed of impulse conduction along the axon. Neurofilaments are particularly abundant in motor neurons with large caliber axons that depend on fast impulse conduction.
Fig. 1.
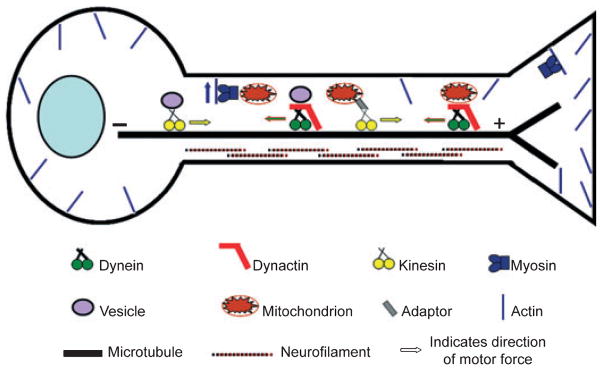
Cytoskeleton, motor proteins, and intracellular transport in neurons. Neurons depend on the cytoskeleton and molecular motor proteins to maintain cell homeostasis and communication between the cell body and the periphery. The neuronal cytoskeleton comprises microtubules, actin, and neurofilaments. Kinesin motor proteins move cargos along microtubules away from the cell body (anterograde transport), whereas dynein together with the dynactin complex move cargos in the retrograde direction toward the cell body. Myosin motors use actin filaments and are responsible for short-ranged dispersive distribution of cargos. Myosin motors have for instance been implicated in pulling mitochondria away from microtubules and facilitate their anchorage to F-actin. The interaction between a motor and its cargo may be direct or may be mediated by adaptor proteins.
In addition to their structural roles, both microtubules and actin filaments provide conduits for intracellular transport. The microtubules provide long-range pathways for fast anterograde movement (away from cell body) of kinesin motor proteins and the retrograde movement (toward cell body) of the dynein motor complex (Fig. 1). Actin filaments are used by myosin motor proteins for short-range, dispersive distribution of vesicles, and/or organelles to the cell periphery. Transport on microtubules and actin is coordinated to distribute organelles such as mitochondria throughout the cell body and neurites.
The first microtubule-based anterograde transport motor protein identified was kinesin 1, also called conventional kinesin. By hydrolyzing ATP, kinesin 1 moves cargo toward the plus end of microtubules (anterograde transport). To date, more than 45 members of the kinesin family have been identified in humans. Although a majority of the kinesins transport cargo in the anterograde direction, a few have also been shown to function in the retrograde direction, whereas others play a role in regulating microtubule dynamics rather than transport (for review see Miki et al. (2005, 2001).
Cytoplasmic dynein is the major motor driving retrograde transport in cells. Dynein is a large complex (approximately two million daltons) and consists of two dynein heavy chains (DHC), two dynein intermediate chains (DIC), four dynein light intermediate chains (DLIC), and various dynein light chains (DLC). The dimeric DHC forms the core of the dynein complex. Each DHC subunit folds to form a globular head containing the motor domain and a flexible stalk that is involved in dimerization of the two heavy chains as well as interaction with other dynein subunits [reviewed in Pfister et al. (2006)]. In vivo, dynein requires the co-complex dynactin for most functions. Dynactin functions both to increase the motor efficiency of dynein and to serve as an adaptor between dynein and various cargos [reviewed in Schroer (2004)]. The dynactin assembly compises multiple subunits that form a distinct structure: a filament base and a projecting sidearm linked to the base by a shoulder domain. The base of dynactin is formed by a small filament of the actin-related protein, Arp1, capped by other subunits such as CapZ, Arp11, p25, and p27. The projecting sidearm of dynactin is formed by two dimeric p150Glued subunits and is involved in the interaction with dynein via the DIC, but it also interacts with microtubules. A tetramer of the dynamitin-p50 subunit forms the shoulder of dynactin that links the sidearm to the filament base. Over-expression of p50 competitively dissociates the p150Glued sidearm from the filament base (Melkonian et al. 2007).
Recent studies suggest that dynein and kinesin can function in an interdependent manner, such that disruption of movement in one direction will affect movement in the opposite direction (Brady et al. 1990; Waterman-Storer et al. 1997; Martin et al. 1999). Moreover, many cargos can move bi-directionally along microtubules [reviewed in Welte (2004)]. In some cases the overall direction of motility is regulated, but in other cases the movement appears to be stochastic with multiple apparently random changes in direction. Several models have been proposed to explain the bi-directionality and interdependency of cargo transport [reviewed in Gross et al. (2002) and Holzbaur (2004)]. In the ‘tug-of-war’ model, kinesins and dynein are suggested to bind to the cargo simultaneously and the direction of movement is determined by the dominant motor at any given time. In another model, either kinesin or dynein is active at a certain time. The slow transport of neurofilaments has been suggested to occur in the ‘tug-of-war’ manner whereas cargos such as peroxisomes and vesicles have been suggested to be transported in a coordinated manner (Gross et al. 2002; Holzbaur 2004). The coordination of the motor activities could be regulated by direct interaction between motor proteins, by adaptor proteins linking different motors, or by other regulatory pathways. Recent studies have supplied evidence for all of the proposed mechanisms. A direct interaction between kinesin-1 and dynein has been observed (Ligon et al. 2004). In addition, dynactin has been reported to interact with both dynein and several kinesins (Blangy et al. 1997; Deacon et al. 2003). Furthermore, microtubule associated proteins such as tau might also differentially influence the motility of kinesin and dynein motors (Dixit et al. 2008).
Dynein defects and motor neuron disease
Dynein and dynactin have multiple cellular house-keeping roles, including participation in mitosis and endoplasmic reticulum to Golgi vesicular trafficking. However, the dynein-dynactin machinery is also required for multiple neuron-specific processes, such as neuronal migration (Sasaki et al. 2000), neurite outgrowth, and synapse formation (Barakat-Walter and Riederer 1996; Cheng et al. 2006) as well as retrograde axonal transport of proteins and organelles (Schnapp and Reese 1989; He et al. 2005). Disruption of dynein or dynactin is therefore expected to compromise severely the function and health of neurons with long axons, including motor neurons.
Two independent N-ethyl-N-nitrosourea-induced missense mutations in the stalk domain of the DHC1 gene, Loa and Cra1, decreased retrograde transport in motor neurons and produced late-onset motor neuron degeneration in heterozygous mice (Hafezparast et al. 2003). Furthermore, a mutation (G59S) in the p150Glued dynactin subunit was recently linked to a slowly progressive form of motor neuron disease in a North American family and heterozygous knock-in mice as well as transgenic mice carrying this mutation also developed motor neuron degeneration (Puls et al. 2003, 2005; Lai et al. 2007; Laird et al. 2008). Moreover, impaired dynein-dynactin complexes upon over-expression of the dynactin p50 subunit in mice also cause motor neuron disease (LaMonte et al. 2002).
Axonal transport defects have been suggested to produce motor neuron degeneration in ALS. Studies both in ALS patients and in transgenic animals have revealed decreased axonal transport in both anterograde and retrograde directions (Breuer et al. 1987; Breuer and Atkinson 1988; Collard et al. 1995; Sasaki and Iwata 1996a; Williamson and Cleveland 1999; Ligon et al. 2005). In motor nerve specimens from ALS patients, decreased axonal transport of organelles such as mitochondria was observed (Breuer et al. 1987; Breuer and Atkinson 1988). In ALS transgenic mice, slowed anterograde transport of cargos such as neurofilaments appeared prior to disease onset (Warita et al. 1999; Williamson and Cleveland 1999). Interestingly, inhibition of retrograde transport has been suggested to be one of the earliest events in the G93A SOD1 mouse model of ALS. In these mice, decreased retrograde transport was evident long before mice showed visible symptoms. However, this reduction in retrograde transport coincided with the onset of neuromuscular junction destabilization and onset of muscle weakness (Ligon et al. 2005). Furthermore, defects in retrograde transport have been demonstrated even in embryonic motor neurons isolated from G93A embryos (Kieran et al. 2005).
Surprisingly, crossing heterozygous Loa and Cra1 mutant mice with G93A SOD1 mice ameliorated the transport defect, delayed disease onset, and slowed disease progression in G93A mice (Kieran et al. 2005; Teuchert et al. 2006). Though the precise mechanism(s) leading to this effect remain unclear, several explanations have been suggested. One hypothesis states that the dynein mutations alter intracellular transport and thereby change the subcellular localization of SOD1 or the interaction of SOD1 with other proteins or organelles (Kieran et al. 2005; El-Kadi et al. 2007). For example, it is possible that decreased interaction of mutant SOD1 with mitochondria could improve cell survival by reducing apoptosis or other downstream consequences (Wong et al. 1995; Menzies et al. 2002; Liu et al. 2004; Pasinelli et al. 2004). Interestingly, SOD1-positive aggregates have been observed in homozogous Loa mice suggesting that this dynein mutation can affect WT SOD1 distribution in the cell (Hafezparast et al. 2003). A second hypothesis is that the decreased retrograde transport rates caused by the DHC Loa or Cra1 mutations might counterbalance an inhibition of anterograde transport caused by G93A mutant SOD1, thereby restoring the balance between anterograde and retrograde transport (Kieran et al. 2005; El-Kadi et al. 2007).
Other studies, however, have reported that disrupting dynein-dynactin by means other than Loa and Cra1 mutations did not influence survival or disease progression in G93A mice. Lai et al. (2007) reported that mice with the G59S mutation in the dynactin subunit p150Glued showed signs of reduced motor neuron axonal transport and developed motor neuron disease which were similar to Loa and Cra1 mice. However, when heterozygous G59S knock-in mice were crossed with G93A mice, no improvement of the G93A phenotype could be observed. Furthermore, a recent study showed that a novel DHC mutation, Swl, caused proprioceptive sensory neuropathy without causing motor neuron deficits (Chen et al. 2007). Crossing the Swl mice with G93A SOD1 mice did not affect ALS disease onset or progression either. The same study also reported that Loa and Cra1 mice not only developed late-onset motor neuron disease, but also suffered from sensory neuropathy that occurred prior to the onset of motor symptoms (Chen et al. 2007).
The studies crossing different dynein and dynactin mutant mice with G93A mice are summarized in Table 1. Two hypotheses could be envisioned to explain the various effects on motor neuron viability and G93A SOD1 mice phenotype by the different dynein and dynactin mutants. First, the various dynein and dynactin mutations could inhibit dynein transport of all cargos but to different degrees. The Swl mutation could affect dynein-mediated transport to a less extent, thus causing harm only to very sensitive sensory proprioceptive neurons. Loa and Cra1 on the other hand could cause a higher level of dynein inhibition, thereby causing both sensory and motor neuron degeneration. It is possible that only reduction of dynein transport within a certain range could have beneficial effect on G93A SOD1 mice. To address this hypothesis, the rate of axonal transport in motor and sensory neurons in the Swl and G59S p150Glued mice needs to be determined.
Table 1.
The effect of different dynein and dynactin mutations on axonal transport and protein aggregation in the G93A ALS mouse model
| Mice | Retrograde transport in motor neurons | SOD1 aggregates in motor neurons | Development of motor neuron disease | Phenotypic outcome of crossing | References |
|---|---|---|---|---|---|
| G93A | Reduced | Yes | Yes | NA | (Gurney et al. 1994; Kieran et al. 2005) |
| Loa +/+ | Reduced | Yes, WT SOD1 positive inclusions seen | Yes, severe phenotype, mice die at P0–P1 | NA | (Hafezparast et al. 2003; Kieran et al. 2005) |
| Loa −/+ | No change | ND | Yes, but normal lifespan | NA | (Hafezparast et al. 2003; Kieran et al. 2005) |
| Loa −/+ x G93A | No change | ND | Yes | Delayed onset and extended survival of G93A | (Kieran et al. 2005) |
| Cra1+/+ | Reduced | No | Yes, severe phenotype, mice die at P0–P1 | NA | (Hafezparast et al. 2003) |
| Cra1−/+ | ND | ND | Yes, but normal lifespan | NA | (Hafezparast et al. 2003) |
| Cra1 −/+ x G93A | ND | ND | Yes | Delayed onset and extended survival of G93A | (Teuchert et al. 2006) |
| Swl +/+ | ND | ND | Embryonic lethal | NA | (Chen et al. 2007) |
| Swl −/+ | ND | ND | No | NA | (Chen et al. 2007) |
| Swl −/+ x G93A | ND | ND | Yes | No effect on G93A phenotype | (Chen et al. 2007) |
| G59S p150Glued +/+ | ND | ND | Embryonic lethal | NA | (Lai et al. 2007) |
| G59S p150Glued −/+ | Reduced (accumulation of cargos, but transport rate not measured) | ND | Yes | NA | (Lai et al. 2007) |
| G59S p150Glued x G93A | ND | ND | Yes | No effect on G93A phenotype | (Lai et al. 2007) |
ND, not determined; NA, not applicable; ALS, amyotrophic lateral sclerosis; WT SOD, wild-type superoxide dismutase.
A second hypothesis where different dynein and dynactin mutations could affect transport of different dynein-dynactin cargos of varied relevance to different neuronal populations could also be envisioned. Cargos bind to dynein in multiple ways [reviewed in Chevalier-Larsen and Holzbaur (2006); Karcher et al. (2002)]. Most known cargos including vesicles and organelles bind to dynein via the DIC interacting complex dynactin. Other cargos such as neurofilaments can bind directly to the DIC, while a third set of cargos including the Fyn tyrosine kinase and rhodopsin bind via DLCs. A fourth set of proteins including pericentrin bind through DLIC. The G59S p150Glued dynactin mutation could clearly affect the subset of dynein cargos using dynactin as adaptor. Although the Loa, Cra1, and Swl mutations are all predicted to affect homodimerization of DHC, Loa is also predicted to effect interaction with DIC, whereas Swl is predicted to affect a region important for cargo interaction (Chen et al. 2007). It is therefore plausible that the different dynein and dynactin mutations could affect transport of different subsets of cargos. The positive effect of Loa and Cra1 on G93A SOD1 mice could be because of reduction of certain cargos which are not affected by the Swl and G59S-p150Glued mutations.
Mutant SOD1, dynein and axonal transport
Although the underlying mechanism is not well understood, it is clear that decreased retrograde axonal transport is a component of familial ALS caused by mutations in SOD1 (Breuer et al. 1987; Breuer and Atkinson 1988; Collard et al. 1995; Sasaki and Iwata 1996b; Williamson and Cleveland 1999; Ligon et al. 2005). Several different mechanisms to explain how mutant SOD1 could cause this can be envisioned and are illustrated in Fig. 2. These mechanisms include: (i) physical blockade of dynein by mutant SOD1 aggregates, (ii) disruption of microtubule formation or stability, (iii) disturbance of dynein motor activity, (iv) disruption of the dynein-dynactin complex integrity, (v) disruption of dynein-dynactin microtubule interaction, or (vi) masking of cargo-binding sites.
Fig. 2.

Possible mechanisms by which mutant superoxide dismutase 1 could interfere with dynein-dynactin motor activity.
Axonal inclusions, including spheroids as large as 20 μm in diameter, have been reported in ALS patients (Kato et al. 2003). In ALS transgenic mice, we and others have shown that dynein co-localizes with mutant SOD1 aggregates in motor neuron axons (Ligon et al. 2005; Zhang et al. 2007). It is possible that mutant SOD1 aggregates could directly hinder dynein transport on microtubules in axons. A similar mechanism was proposed in Huntington’s disease after huntingtin aggregates were observed with diameters exceeding those of axons; consistent with this axonal swelling around aggregates and loss of axonal transport was reported in Drosophila models (Gunawardena et al. 2003; Lee et al. 2004).
In a recent study, we showed that mutant SOD1 interacted with dynein more stably than did WT SOD1 in both ALS cell culture and animal models (Zhang et al. 2007). Though co-immunoprecipitation of SOD1 and dynein-dynactin showed no evidence suggesting that mutant SOD1 altered the dynein-dynactin subunit interactions, we did observe an increase in the amount of mutant SOD1 that interacted with dynein as the disease progressed (Zhang et al. 2007). It is still unclear which subunit(s) of dynein and/or dynactin interacts with mutant SOD1. If mutant SOD1 is a bona fide cargo of dynein, the interaction could be mediated by either DLC or dynactin, which are known to bind cargos. However, it is also possible that this aberrant interaction involves novel components distinct from DLC or dynactin. Given the beneficial effects of the Loa and Cra1 DHC mutations on ALS disease (Kieran et al. 2005; Teuchert et al. 2006), it would be interesting to test whether these dynein mutations affect the interaction of mutant SOD1 with the dynein complex.
On the basis of the data showing increased association of mutant SOD1 with dynein as the ALS progressed in mice (Zhang et al. 2007), we hypothesize that the transport capacity of dynein might be overwhelmed by handling mutant SOD1 over time. Interestingly, a recent study in monkeys showed that the dynein-dynactin complex undergoes age-related changes including increased amounts of dynein in nerve endings and a decrease in the dynein-dynactin interaction (Kimura et al. 2007). These changes suggest that less functional dynein-dynactin complexes may be available during aging. Both factors, i.e. the increase in dynein-mutant SOD1 association and the decrease in functional dynein-dynactin while aging, could contribute to reduced retrograde transport to levels that are no longer able to sustain neuronal survival. This could thereby produce a phenotype of adult onset motor neuron degeneration.
Reduced retrograde transport of cargos such as neurotrophic factors, mitochondria, and membrane vesicles could damage motor neurons by multiple mechanisms. First, reduced retrograde axonal transport of neurotrophic factors could have dire consequences for motor neurons as these factors stimulate neuronal survival. Neurotrophic factors including nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 are secreted by target tissues (e.g. muscles) and then bind to Trk receptors on the surface of neurons. The Trk receptor-neurotrophin complex is then internalized and initiates signaling cascades that regulate neuronal cell growth, survival, and repair pathways (Campenot and MacInnis 2004; Bronfman et al. 2007). For the neurotrophins to act effectively and promote long-term survival, transport of the neurotrophin-receptor complex to the cell body by the dynein complex may be required (Yano et al. 2001; Delcroix et al. 2003; Ye et al. 2003; Heerssen et al. 2004). Some ALS studies have demonstrated a benefit upon administration of exogenous neurotrophic factors, but others have shown little or no effect (Wang et al. 2002; Azari et al. 2003; Feeney et al. 2003; Kaspar et al. 2003; Azzouz et al. 2004; Zheng et al. 2004; Dobrowolny et al. 2005; Storkebaum et al. 2005; Pun et al. 2006; Li et al. 2007). Conclusions from such studies may be confounded by issues related to exactly which neurotrophic factors motor neurons requires the timing and the effectiveness of neurotrophic factor delivery.
Second, mitochondrial distribution and dynamics can be affected by reduced axonal transport. Mitochondria are the energy factories of cells and are particularly abundant in areas of neurons with high metabolic demands such as the axon hillock, the nodes of Ranvier, and the synaptic regions. These organelles generate reactive oxygen species (ROS) as a byproduct from energy production. Mitochondria cannot be synthesized de novo in cells and relay on constant transport, fission, and fusion to maintain normal morphology and function [see Frazier et al. (2006) for review]. Dynein is important not only for axonal transport of mitochondria but also for mitochondrial fission (Frazier et al. 2006). Furthermore, damaged mitochondria with reduced membrane potential are degraded by autophagy, an intracellular turnover process in which dynein has been suggested to play a role (Twig et al. 2008). Inhibition of dynein function not only could impair energy homeostasis, but also could cause accumulation of damaged mitochondria leading to increased ROS production at nerve terminal regions. Increased levels of ROS could damage synaptic structures and contribute both to the distal neuropathy observed early in ALS and to impaired re-innervation. Many studies have implicated mitochondria in the pathophysiology of ALS. Although WT SOD1 is traditionally believed to be a cytoplasmic protein, mutant SOD1s also localize on and in mitochondria (Vande Velde et al. 2008) and have been suggested to induce mitochondria-mediated apoptosis (Wong et al. 1995; Menzies et al. 2002; Zhu et al. 2002; Liu et al. 2004; Pasinelli et al. 2004). As both mitochondria and mutant SOD1 interact with dynein, it is possible that locally high concentrations of mutant SOD1 in the proximity of mitochondria may contribute to aggregation of mutant SOD1 in or on mitochondria or to direct impairment of mitochondrial function.
Third, the structural and functional integrity of the endoplasmic reticulum-golgi network is dependent on microtubule-mediated vesicular membrane trafficking. Impairment of dynein-dynactin function by p50 dynamitin over-expression has been shown to interfere with endosome trafficking and cause golgi fragmentation (Burkhardt et al. 1997; Valetti et al. 1999). Fragmentation of the golgi has been observed in both sporadic and SOD1-mediated familial ALS (Gonatas et al. 2006). In ALS mouse models, golgi fragmentation can also be observed before onset of paralysis (Gonatas et al. 2006).
Dyneins role in SOD1 protein aggregation and degradation
Dynein-mediated transport has been suggested to play a role in protein degradation and accumulation of misfolded proteins in cells. The observed interaction between mutant SOD1 and dynein could therefore also influence SOD1 aggregation and degradation as discussed below.
The removal of misfolded and/or aggregated proteins may pose a vulnerability for neurons, as suggested by the aberrant accumulation of protein aggregates in many neurodegenerative diseases. Proteins are mainly degraded by two pathways, the ubiquitin-proteasome system (UPS) and macroautophagy (Fig. 3). Proteins destined to be degraded by the UPS are marked for elimination by the covalent attachment of ubiquitin and then shuttled to the proteasome for proteolytic cleavage (Goldberg 2003). As most UPS activity is believed to occur in the cell body, it is possible that dynein-mediated retrograde transport could be involved in shuttling targeted proteins to the proteasome. The proteasome comprises multiple subunits forming a barrel-like chamber within which proteolysis occurs. Because of the restricted opening of the proteasome, larger molecular complexes and organelles cannot enter for degradation by the proteasome (Goldberg 2003).
Fig. 3.

Potential role(s) of dynein-dynactin in aggregation and degradation of mutant superoxide dismutase 1 (SOD1). Proteins are mainly degraded by two pathways, the ubiquitin-proteasome system (UPS) and autophagy. Proteins degraded by UPS are ubiquitinated and then shuttled to the proteasome. Dynein could have a potential role in transporting targeted proteins from the cell periphery to the proteasome, as most UPS degradation is believed to occur in the cell body. Autophagy could not only degrade monomeric SOD1, but could also clear SOD1 aggregates. During autophagy, proteins/aggregates are enveloped by membrane and form autophagosomes which are then fused to lysosomes containing hydrolytic enzymes. Dynein is believed to be involved in the fusion of the autophagosome and the lysosome. Dynein transport has also been implicated in the formation of large perinuclear protein aggregates called aggresomes that may be degraded by autophagy. Mutant SOD1 has also been suggested to aggregate on or in mitochondria and cause mitochondrial damage. Dynein transport and dynein-dependent autophagy is used to degrade damaged mitochondria.
Macroautophagy (or autophagy) is an intracellular process in which proteins, organelles, or protein aggregates are degraded. During autophagy, the substrate is engulfed by membranous structures to form autophagosomes, which then fuse with lysosomes containing hydrolytic enzymes (Mizushima 2007). Autophagy is an important degradation pathway for neurons as mice lacking the autophagy gene Atg7 develops axonal dystrophy characterized by distal accumulation of membrane structures and swelling of axonal terminals (Komatsu et al. 2007b). Dynein has been linked to autophagic clearance of aggregated proteins by several studies (Fig. 3). First, dynein-mediated transport has been shown to collect misfolded proteins from the cell periphery and transport them to the perinuclear area, where they form inclusions called aggresomes; it is suggested that these structures may become membrane-bound and associate with lysosomes (Johnston et al. 2002; Taylor et al. 2003). Dynein has also been suggested to play an important role in lysosomal transport and to mediate the fusion of the autophagosome and the lysosome (Burkhardt et al. 1997; Ravikumar et al. 2005).
Mutant SOD1 is believed to be degraded by both the UPS and autophagy as shown in Fig. 3 (Kabuta et al. 2006). We recently demonstrated that while mutant SOD1 strongly interacts with dynein, WT SOD1 does not or to very little extent (Zhang et al. 2007). Furthermore, we have shown that mutant, but not WT SOD1 interacts with p62/sequestosome 1 (Gal et al. 2007), a protein that has been linked to autophagy (Komatsu et al. 2007a; Pankiv et al. 2007). Moreover, we showed that over-expression of p62/sequestosome 1 increased the formation of large mutant SOD1 inclusions resembling aggresomes (Gal et al. 2007). These data suggest that cells might utilize dynein-mediated transport to collect mutant SOD1 and form aggresomes that are targeted for autophagic degradation. For this purpose, the interaction between mutant SOD1 and dynein might be of beneficial effect to the cell. Supporting the importance of autophagic clearance of aggregates in preventing neuronal toxicity, a recent study showed that blocking of dynein by the Loa mutation caused decreased autophagic clearance of mutant Huntingtin protein resulting in increased inclusion/aggre-some burden and toxicity in Huntington’s disease model mice (Ravikumar et al. 2005). However, this mechanism may not be relevant in SOD1-mediated ALS as this negative effect of Loa was not seen in G93A ALS mice (Kieran et al. 2005). In fact, as discussed earlier, both Loa and Cra1 had a beneficial effect in the G93A mice and speculations that impairment of aggresome formation/autophagy might even have a positive effect in ALS have been raised (Teuchert et al. 2006).
Whether aggregates, aggresomes, and/or inclusions are toxic is still debated. However, multiple mechanisms by which mutant SOD1 aggregates/inclusions could be toxic to cells have been proposed. For instance, inclusions have been suggested to disrupt organelles like mitochondria, cause depletion of essential proteins via co-aggregation or physically block intracellular transport as discussed above.
Conclusions and future investigations
It is clear that dynein-mediated retrograde axonal transport is affected in motor neurons in ALS, however, the underlying mechanism(s) are still unclear. Mutations that perturb the dynein-dynactin machinery cause phenotypes that may influence multiple processes including neurotrophic factor delivery, transport, and homeostasis of mitochondria and protein aggregation or degradation. Disruption of dynein function could therefore link several proposed pathways relevant to ALS pathophysiology, including axonal transport defects, mitochondrial dysfunction, and mutant SOD1 aggregation. Genetic manipulation of in vivo models of motor neuron disease continues to provide important mechanistic clues. Crossing Loa and Cra1 mice with G93A SOD1 mice has shown, surprisingly, that disruptions of dynein by some mutations can ameliorate the ALS disease process for reasons that remain to be clarified. A better understanding is needed of how the transport of specific cargos is regulated and how perturbations of both retrograde and anterograde transport are integrated to produce a given phenotype.
Acknowledgments
This study was in part supported by NIH grants R01-NS49126 (to H. Z.), R01-NS045087 (to E. J. K), R01-NS44170 (to L. J. H.), and a Muscular Dystrophy Association grant (to L. J. H.).
Abbreviations used
- ALS
amyotrophic lateral sclerosis
- DHC
dynein heavy chains
- DIC
dynein intermediate chains
- DLC
dynein light chains
- DLIC
dynein light intermediate chains
- NF
neurofilament
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- Trk
tyrosine kinase
- UPS
ubiquitin-proteasome system
- WT
wild-type
References
- Azari MF, Lopes EC, Stubna C, Turner BJ, Zang D, Nicola NA, Kurek JB, Cheema SS. Behavioural and anatomical effects of systemically administered leukemia inhibitory factor in the SOD1 (G93A G1H) mouse model of familial amyotrophic lateral sclerosis. Brain Res. 2003;982:92–97. doi: 10.1016/s0006-8993(03)02989-5. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Barakat-Walter I, Riederer BM. Triiodothyronine and nerve growth factor are required to induce cytoplasmic dynein expression in rat dorsal root ganglion cultures. Brain Res Dev Brain Res. 1996;96:109–119. [PubMed] [Google Scholar]
- Blangy A, Arnaud L, Nigg EA. Phosphorylation by p34cdc2 protein kinase regulates binding of the kinesin-related motor HsEg5 to the dynactin subunit p150. J Biol Chem. 1997;272:19418–19424. doi: 10.1074/jbc.272.31.19418. [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS. A monoclonal anti-body against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer AC, Atkinson MB. Fast axonal transport alterations in amyotrophic lateral sclerosis (ALS) and in parathyroid hormone (PTH)-treated axons. Cell Motil Cytoskeleton. 1988;10:321–330. doi: 10.1002/cm.970100136. [DOI] [PubMed] [Google Scholar]
- Breuer AC, Lynn MP, Atkinson MB, Chou SM, Wilbourn AJ, Marks KE, Culver JE, Fleegler EJ. Fast axonal transport in amyotrophic lateral sclerosis: an intra-axonal organelle traffic analysis. Neurology. 1987;37:738–748. doi: 10.1212/wnl.37.5.738. [DOI] [PubMed] [Google Scholar]
- Bronfman FC, Escudero CA, Weis J, Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev Neurobiol. 2007;67:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB, MacInnis BL. Retrograde transport of neurotrophins: fact and function. J Neurobiol. 2004;58:217–229. doi: 10.1002/neu.10322. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HH, Liu SH, Lee HC, Lin YS, Huang ZH, Hsu CI, Chen YC, Chang YC. Heavy chain of cytoplasmic dynein is a major component of the postsynaptic density fraction. J Neurosci Res. 2006;84:244–254. doi: 10.1002/jnr.20898. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Deacon SW, Serpinskaya AS, Vaughan PS, Lopez Fanarraga M, Vernos I, Vaughan KT, Gelfand VI. Dynactin is required for bidirectional organelle transport. J Cell Biol. 2003;160:297–301. doi: 10.1083/jcb.200210066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musaro A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kadi AM, Soura V, Hafezparast M. Defective axonal transport in motor neuron disease. J Neurosci Res. 2007;85:2557–2566. doi: 10.1002/jnr.21188. [DOI] [PubMed] [Google Scholar]
- Feeney SJ, Austin L, Bennett TM, Kurek JB, Jean-Francois MJ, Muldoon C, Byrne E. The effect of leukaemia inhibitory factor on SOD1 G93A murine amyotrophic lateral sclerosis. Cytokine. 2003;23:108–118. doi: 10.1016/s1043-4666(03)00217-5. [DOI] [PubMed] [Google Scholar]
- Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT. Mitochondrial morphology and distribution in mammalian cells. Biol Chem. 2006;387:1551–1558. doi: 10.1515/BC.2006.193. [DOI] [PubMed] [Google Scholar]
- Gal J, Strom AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:11068–11077. doi: 10.1074/jbc.M608787200. [DOI] [PubMed] [Google Scholar]
- Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against mis-folded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci. 2006;246:21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Gross SP, Welte MA, Block SM, Wieschaus EF. Coordination of opposite-polarity microtubule motors. J Cell Biol. 2002;156:715–724. doi: 10.1083/jcb.200109047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7:596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL. Motor neurons rely on motor proteins. Trends Cell Biol. 2004;14:233–240. doi: 10.1016/j.tcb.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu, Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem. 2006;281:30524–30533. doi: 10.1074/jbc.M603337200. [DOI] [PubMed] [Google Scholar]
- Karcher RL, Deacon SW, Gelfand VI. Motor-cargo interactions: the key to transport specificity. Trends Cell Biol. 2002;12:21–27. doi: 10.1016/s0962-8924(01)02184-5. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kato S, Shaw P, Wood-Allum C, Leigh PN, Shaw C. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. ISN Neuropath Press; Basel: 2003. Amyotrophic lateral sclerosis. [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Imamura O, Ono F, Terao K. Aging attenuates dynactin-dynein interaction: down-regulation of dynein causes accumulation of endogenous tau and amyloid precursor protein in human neuroblastoma cells. J Neurosci Res. 2007;85:2909–2916. doi: 10.1002/jnr.21408. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007a;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007b;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Lin X, Chandran J, Shim H, Yang WJ, Cai H. The G59S mutation in p150 (glued) causes dysfunction of dynactin in mice. J Neurosci. 2007;27:13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Farah MH, Ackerley S, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci USA. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Brakefield D, Pan Y, Hunter D, Myckatyn TM, Parsadanian A. Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp Neurol. 2007;203:457–471. doi: 10.1016/j.expneurol.2006.08.028. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- Ligon LA, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur EL. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport. 2005;16:533–536. doi: 10.1097/00001756-200504250-00002. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkonian KA, Maier KC, Godfrey JE, Rodgers M, Schroer TA. Mechanism of dynamitin-mediated disruption of dynactin. J Biol Chem. 2007;282:19355–19364. doi: 10.1074/jbc.M700003200. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZMA, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA. 2001;98:7004–7011. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–476. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Shah PR, Hummerich H, Russ A, Cotton J, Annuar AA, King SM, Fisher EM. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1. doi: 10.1371/journal.pgen.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996a;47:535–540. doi: 10.1212/wnl.47.2.535. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Synaptic loss in anterior horn neurons in lower motor neuron disease. Acta Neuropathol (Berl) 1996b;91:416–421. doi: 10.1007/s004010050444. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA. 1989;86:1548–1552. doi: 10.1073/pnas.86.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Shaw BF, Valentine JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- Teuchert M, Fischer D, Schwalenstoecker B, Habisch HJ, Bockers TM, Ludolph AC. A dynein mutation attenuates motor neuron degeneration in SOD1G93A mice. Exp Neurol. 2006;198:271–274. doi: 10.1016/j.expneurol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valetti C, Wetzel DM, Schrader M, Hasbani MJ, Gill SR, Kreis TE, Schroer TA. Role of dynactin in endocytic traffic: effects of dynamitin overexpression and colocalization with CLIP-170. Mol Biol Cell. 1999;10:4107–4120. doi: 10.1091/mbc.10.12.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci USA. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LJ, Lu YY, Muramatsu S, et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci. 2002;22:6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki SB, Kuznetsov SA, Tabb JS, Weiss DG, Langford GM, Holzbaur EL. The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc Natl Acad Sci USA. 1997;94:12180–12185. doi: 10.1073/pnas.94.22.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14:R525–R537. doi: 10.1016/j.cub.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Yano H, Lee FS, Kong H, Chuang J, Arevalo J, Perez P, Sung C, Chao MV. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci. 2001;21:RC125. doi: 10.1523/JNEUROSCI.21-03-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Kuruvilla R, Zweifel LS, Ginty DD. Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron. 2003;39:57–68. doi: 10.1016/s0896-6273(03)00266-6. [DOI] [PubMed] [Google Scholar]
- Zhang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial ALS-linked SOD1 mutants and the dynein complex: implications of retrograde axonal transport in ALS. J Biol Chem. 2007;282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]
- Zheng C, Nennesmo I, Fadeel B, Henter JI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004;56:564–567. doi: 10.1002/ana.20223. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]