

Abstract
Bisphosphonates are used in the treatment of hypercalcemia of malignancy, skeletal complications associated with metastastic bone disease, Paget’s disease, and osteoporosis. Osteonecrosis of the jaw (ONJ) is a recently described clinical condition that has been associated with the use of nitrogen-containing bisphosphonates. Reports describing this entity first appeared in the literature in 2003. While there have been significant numbers of case reports and a limited number of retrospective and prospective studies examining risk factors associated with ONJ, the pathophysiology of this condition remains elusive. In this review, we explore proposed mechanisms underlying ONJ development and identify potential areas for future investigation.
Keywords: bisphosphonates, pathophysiology, osteonecrosis, ONJ
Introduction
Bisphosphonates (BPs) are a widely used class of drugs indicated for the prevention and treatment of postmenopausal and steroid-induced osteoporosis; Paget’s disease of the bone; hypercalcemia of malignancy; multiple myeloma; and bone metastases associated with breast, prostate, lung, and other soft tissue tumors.1–4 In late 2003, a letter to the Journal of Oral and Maxillofacial Surgery described 36 patients who developed avascular necrosis of the jaw after receiving intravenous BPs.5 Twenty-four patients were on pamidronate (90 mg monthly) and 12 patients were receiving zoledronate (4 mg monthly). Patients were receiving BPs for multiple myeloma,18 metastatic breast cancer,17 and osteoporosis.1 All patients presented with painful, necrotic bone in the mandible (80%), maxilla (14%), or both (6%). In 78% of cases, the necrosis was associated with a prior history of tooth extraction. The remainder of cases appeared to arise spontaneously.5 Following this report, Migliorati et al.6 documented an additional five cases of suspected BP-associated osteonecrosis of the mandible. Three cases developed spontaneously and two cases occurred in patients with previous tooth extraction. No information regarding the patients’ diagnoses or duration of BP therapy was presented. Interestingly, prior to the initial report, Wang et al.7 reported on three females who developed jaw necrosis while receiving chemotherapy for metastatic breast cancer. Two of the patients had a previous history of dental manipulation. Histological evaluation of all three cases revealed necrotic bone without evidence of metastases. At the time of the initial report, the authors postulated that the osteonecrosis was a consequence of the cancer chemotherapy. In a follow-up communication, however, the authors noted that the patients had been treated with BPs.8
In late 2004, Ruggiero et al.9 reported on 63 cases of BP-associated osteonecrosis accrued over a 26-month period from two medical centers. Twenty-eight of the 63 patients had multiple myeloma, 20 patients had breast cancer, 3 patients had lung cancer, and 1 patient each had plasmacytoma and leukemia. All patients within the oncologic group were receiving chemotherapy and/or corticosteroids. Seven patients were diagnosed with osteoporosis and had no prior history of treatment with chemotherapeutic agents or corticosteroids. Thirty-nine patients presented with osteonecrosis of the mandible and 23 had maxillary involvement; one patient had necrotic lesions in both jaws. Fifty-four of 63 patients reported a recent extraction at the necrotic site while the remaining nine patients had apparent spontaneous bone exposure. Bacterial culture results were reported as normal oral flora in all cases. The reported length of BP therapy in this series ranged from 6 to 48 months although the treatment time for each individual patient was not specified. Treatment of the necrotic jaw lesions ranged from conservative management to resection with the majority of patients undergoing sequestrectomy. In a follow-up report to their 2003 letter, Marx et al. documented an additional 119 patients with osteonecrosis of the jaw (ONJ). Similar to other case series, the majority of patients were undergoing treatment for multiple myeloma (52.1%) and metastatic breast cancer (42%). The mandible was the most common site of bone exposure (68.1%) and ongoing periodontal disease was present in a significant number of patients (84%).10
ONJ is a clinical term that has been defined by a number of professional associations.11–17 Although there are slight differences in the various definitions, all involve a breach in mucosa leading to exposed bone that fails to heal in 6–8 weeks; furthermore, there should be no history of head and neck radiation in the affected patients. Over the past several years, there have been numerous case reports and series, a limited number of retrospective studies, and a handful of small prospective studies that strongly suggest a link between BP therapy and ONJ; however, a definitive cause-and-effect relationship between the two has yet to be established. Many questions regarding the incidence, pathoetiology, and natural history of this condition still need to be answered. In this paper, we review the potential mechanisms of ONJ development that have been proposed thus far. We will focus on the effect of BPs on epithelial cells, bone cells, and angiogenesis. Moreover, the potential role of immune dysfunction and other contributing factors in the pathogenesis of ONJ will be discussed. Finally, an instrument to risk stratify individual patients for the development of ONJ will be introduced.
Why the jaw?
Without a doubt, there are several features that make the oral cavity a unique environment. The anatomy is such that the alveolar bone in both the mandible and maxilla is covered by a thin layer of periosteum and epithelium with an attenuated layer of connective tissue. The oral structures are subjected to a wide variety of stresses, which may be physiologic (e.g., mastication), iatrogenic (e.g., dental procedures), or inflammatory (e.g., periodontal disease, caries) in nature. This combination of constant stress not only predisposes the thin mucosa to trauma, leading to exposure of bone, but likely demands an increase in metabolic compensation including bone remodeling. Furthermore, the oral cavity and teeth are colonized by a complex microbial flora, many of which are pathogenic organisms. The intimate relationship of teeth to the jaws allows a portal of entry for microbes and other inflammatory products to the underlying bone, a situation that is not found in any other part of the body.
The role of specific microbes in the development of ONJ is not completely resolved. Of interest, several authors have documented the presence of Actinomyces in their cases of ONJ. Lugassy et al.18 described two patients with multiple myeloma on long-term BP therapy. Histological examination of both specimens revealed necrotic bone surrounded by abundant microbial colonies with features of Actinomyces. Melo and Obeid19 reported a case of maxillary osteonecrosis with histological evidence of Actinomyces in patients with metastatic breast cancer on zoledronate. Hansen et al.20 examined histopathological specimens from 45 patients with Actinomycosis and confirmed the presence of Actinomyces colonies in 43/45 (93.5%) patients with either ONJ (58.7%) or infected osteomyelitis (35.6%). Three patients (6.7%) were not receiving chemotherapy of any kind. Histochemical analysis, standard gram stains, scanning electron microscopy, as well as polymerase chain reaction (PCR) testing was used to demonstrate Actinomyces species.
A recent retrospective study by Kos et al.21 examined 29 patients with ONJ. The authors performed a chart review and evaluated patients with delayed wound healing, bone sequestration, abscess formation, and recurrent wound dehiscence from January 2003 to December 2006. Cases of osteoradionecrosis (ORN) and open fractures were excluded. Patients were divided into two groups: those treated with BPs and those who had no documented history of BP use. The BP-treated group included 18 patients–12 with multiple myeloma, 3 with breast cancer, 2 with prostate carcinoma, and 1 with osteoporosis. The patients who had not been treated with BPs included two patients with breast cancer, two patients with lung cancer, and seven without cancer. Statistical analysis failed to show differences between the two groups in the recurrence rate of wound dehiscence. There were no effects from chemotherapy or steroid therapy on wound healing and there were no associations between wound dehiscence and the duration of BP treatment or the type of BP therapy used. Of interest was the observation of a statistically significant increase in Actinomyces colonization of exposed bone in the BP treated group (11/18; 61%) when compared to the nontreated group (2/11; 18%, P = 0.021).
Actinomyces are a normal inhabitant of the oral environment, and can be a component of dental plaque and calculus, gingival sulci, and periodontal pockets.22 The pathogenic potential of Actinomyces is well-recognized, and Actinomyces have been documented in various sites within the cervicofacial region including the jawbones (so-called “actinomycotic osteomyelitis”). The presence of Actinomyces species in significant numbers of ONJ histological specimens has led Naik and Russo23 to suggest a critical role of this organism in the development of ONJ. It remains unclear, however, whether actinomycotic colonization occurs as a primary event or as a secondary phenomenon due to its prevalence in the oral cavity. Furthermore, histological identification of Actinomyces can be challenging at times, as many bacterial organisms can exhibit actinomycotic-like morphology. Therefore, only a strong presumptive diagnosis of actinomycosis can be rendered in the absence of culture confirmation.24
Do BPs cause an oversuppression of bone turnover?
Several authors have proposed that BPs cause excessive reduction of bone turnover resulting in an increased risk of bone necrosis when the demand for osseous repair is present.25–27 This theory, however, fails to explain why exposed necrotic lesions are rarely seen in bones other than the jaws. In addition, a number of studies have failed to demonstrate that BPs reduce bone turnover to the extent that they cause adynamic bone. A study by Chapurlat et al.25 examined transiliac bone biopsies from postmenopausal women and found no significant difference from controls in the number of microcracks after at least three years’ treatment with a BP (3 subjects on intravenous pamidronate, 37 on oral alendronate, and 10 on oral risedronate), despite a marked reduction in bone turnover. Stepan et al.26 compared transiliac bone biopsies obtained from 38 postmenopausal women treated with daily or weekly alendronate for a mean duration greater than 5 years to biopsies from 28 treatment-naive women with postmenopausal osteoporosis. No differences in crack surface density or crack density between the two groups were found. There was, however, a correlation between low femoral neck density and increased microcrack accumulation following alendronate treatment after adjustment for potential confounders.26 In addition, several long-term clinical trials that evaluated the ability of BP treatment to reduce fracture risk in postmenopausal osteoporosis have showed that after the initiation of BP therapy, bone turnover rapidly returned to, and remained within the premenopausal range, with no additional fracture risk associated with long-term treatment.27 Furthermore, there have been no documented cases of ONJ due to an excessive reduction in bone turnover secondary to BP therapy.
The effect of BPs on epithelial cells and wound healing
Numerous studies have explored the effect of BPs on a variety of epithelial cells, including gastrointestinal cells, cervical epithelial cells, renal cells, prostate epithelial cells, and oral mucosal cells. The development of gastric erosions and ulcers is a well-documented side effect associated with oral administration of nitrogen-containing BPs. Several investigators have demonstrated the adverse effects of exposing gastrointestinal cells to BPs. Twiss et al.28 confirmed the cytotoxic effects of pamidronate on Caco-2 intestinal cells by demonstrating increased cell permeability after BP exposure in vitro. In a subsequent investigation, this same group29 incubated Caco-2 epithelial cell cultures with various concentrations of two nitrogen-containing BPs, pamidronate and alendronate. They observed that in the presence of calcium, both BPs were toxic to the cells. However, only pamidronate exhibited toxicity when calcium was absent from the media. Furthermore, they demonstrated that pamidronate forms insoluble complexes with calcium that deposit on the cell surface, exposing them to locally increased and cytotoxic concentrations of the drug. Neither of the BPs induced apoptosis of the cells. Wallace et al.30 used an ex vivo gastric chamber model to show concentration-dependent epithelial injury and loss of integrity after exposure to pamidronate and alendronate. Exposure to the drug caused necrosis of the gastric epithelium, and the mechanism of damage was independent of microvascular injury, possibly due to disruption of surface-active phospholipids within the mucosal layer. Suri et al.31 exposed Caco-2 cells to various BPs in vitro and evaluated apoptosis by nuclear morphometry and TUNEL assays. In contrast to the results of Twiss et al.,29 they found a dose-dependent increase in apoptotic cells, reduction in cell viability, and inhibition of cell proliferation with pamidronate, among other BPs. Apoptosis and loss of viability was prevented when BP-induced inhibition of farnyldiphosphate (FPP) synthase—an enzyme in the mevalonate pathway—was bypassed by addition of geranylgeraniol, a downstream protein. They postulated that the toxic effects that BPs had on intestinal cells were likely related to disruption of the mevalonate pathway, which thereby reduces production of cholesterol precursors and interferes with cell membrane biosynthesis.
Giraudo et al.32 found that cervical epithelial cells treated with zoledronate in vitro exhibited increased apoptosis. Similarly, Perazella et al.33 demonstrated an increase in apoptosis in renal cells treated with pamidronate. Finally, Montague et al.34 examined prostate epithelial cells treated with zoledronate and confirmed a reduction in cell proliferation rate.
We have investigated the effects of BPs on oral epithelial cells. Mouse oral keratinocytes were isolated as previously described by Landesberg et al.35 and treated with pamidronate at various doses. Cell proliferation and apoptosis assays (Caspase-3, TUNEL assay, DAPI staining) were performed. In addition, the effect of pamidronate on in vitro wound healing was examined. Oral keratinocyte proliferation was assayed at 24, 48, 72, and 168 h with 0, 0.03, 0.1, and 0.3 mM pamidronate concentrations. The results, shown in Figure 1, demonstrate that pamidronate inhibits cellular proliferation at a concentration of 0.1 mM. When the cells were exposed to concentrations below 0.1 mM there was no significant difference in cell growth; however, exposure to pamidronate at higher doses caused the cells to be released from the plate within 2 h (data not shown). The relative number of viable cells in the 0.1 mM BP group did remain constant over the course of 7 days.
Figure 1.
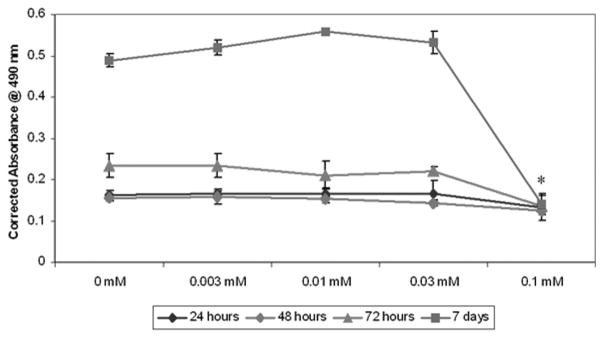
Cellular proliferation of oral keratinocytes with pamidronate. Low concentrations of pamidronate (0.003–0.03 mM) did not affect cell proliferation over the course of seven days. A higher dose (0.1 mM) significantly inhibited proliferation. *P < 0.05 when compared to controls.
The ability of pamidronate to induce apoptosis in oral keratinocytes was evaluated using three different methods: the upregulation of caspase-3, fragmentation of DNA by a TUNEL assay, and analysis of apoptotic nuclei visualized with DAPI staining. The results of the caspase-3 assays are shown in Figure 2. Enzyme levels in the wells treated with 0.03 and 0.1 mM pamidronate showed a significant increase at 24 and 48 h when compared to the 6-h sample; however, caspase-3 levels were not significantly different from the control levels. Cells that were treated with 0.3 mM of pamidronate showed a decrease in caspase-3 levels at 24 and 48 h coincident with the release of cells from the plate. The cells treated with staurosporine and cycloheximide, agents known to cause significant apoptosis, showed an upregulation of caspase-3 at all time points that was between four- and sixfold higher levels than that seen in any of the cells that received pamidronate.
Figure 2.
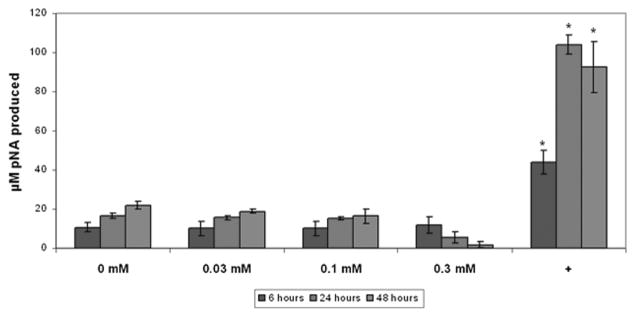
Caspase-3 activity in oral keratinocytes incubated with pamidronate. Pamidronate did not significantly increase caspase-3 activity in cells over a 48-h period when compared with controls. The positive control (+) was obtained by incubating cells with staurosporine and cycloheximide. * Significant increase compared with controls, P <0.05.
Analysis of the percentage of apoptotic cells was performed by counting the number of TUNEL positive cells in five high-power fields of approximately 100 cells. Approximately 2% of cells treated with 0.03, 0.1, and 0.3 mM pamidronate for 24 h were TUNEL positive, similar to untreated control cells. In contrast, the positive control, cells treated with 200 nM staurosporine and 10 um cycloheximide, had nearly 100% TUNEL-positive cells. At 48 h the findings were similar to that seen at 24 h; however, many of the cells in the 0.3 mM pamidronate samples had floated off of the slide. The 6-h time point showed minimal effects in the pamidronate-treated samples, with only about 50% of TUNEL-positive cells seen in the positive control (data not shown). These results are consistent with the caspase-3 assay results (discussed earlier) as well as quantitation of DAPI stained pyknotic nuclei in cells treated with 0, 0.03, 0.1, and 0.3 mM pamidronate, respectively (data not shown).
The in vitro model of wound healing was used to examine the effects of pamidronate treatment on the ability of oral keratinocytes to migrate and proliferate in a monolayer culture. The results are shown in Figure 3. In the preincubated samples, the control and 0.003, 0.01, and 0.06 mM pamidronate groups, cellular migration was appreciated beginning at 6 h and the wound was completely closed at 72 hours. The 0.1 mM preincubated group had a significantly delayed wound closure at all time points but did close when incubated for 5 days (data not shown). The plates that had BP added immediately after wounding (no preincubation) had a response that was identical to the control samples.
Figure 3.

Wound healing in oral keratinocytes with pamidronate. Cells were observed for 96 h after wounding in cell culture plates. Control wells exhibited significant migration into the wounded area from time of injury (A) to 96 h (B). Cells preincubated for 72 hours with 0.1 mM pamidronate prior to wounding had greatly reduced migration from 0 (C) to 96 h (D). Data shown are representative of three independent experiments.
Primary human oral epithelial cell cultures were established from discarded gingival tissue. The tissue was placed in dispase solution (Roche) and incubated at 4°C overnight. Epithelium was removed from the submucosal layer, minced, dissociated with TrypLE Express reagent (Invitrogen), pelleted, washed, and resuspended in low calcium DMEM with 15% chelated fetal bovine serum (FBS). The cells were cultured until colonies formed. Inhibition of proliferation by pamidronate was similar to that seen in mouse keratinocytes (Fig. 4).
Figure 4.
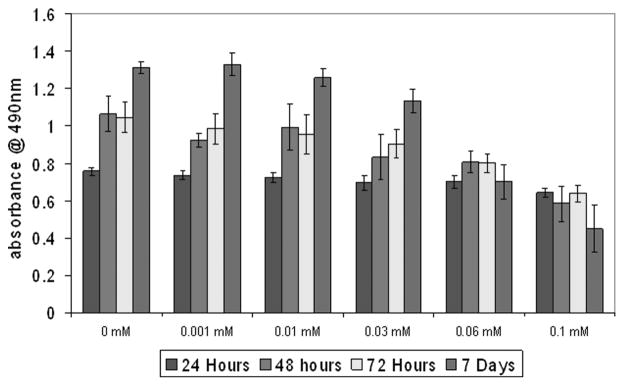
Cellular proliferation of human oral keratinocytes with pamidronate. Low concentrations of pamidronate (0.003–0.03 mM) did not affect cell proliferation over the course of seven days. A higher dose (0.06 mM and 0.1 mM) significantly inhibited proliferation. *P <0.05 when compared to controls.
Given these data, the question that invariably arises is, How do oral epithelial cells get exposed to the BPs in vivo? It is well known that following administration of BPs, both orally and intravenously, a significant amount of drug is sequestered in bone. In cancer patients with bone metastases and normal kidney function, this amount has been estimated to be around 65% of an intravenous dose.36 Data on actual concentrations of pamidronate as well as other BPs deposited in human bone are limited. Extrapolating pharmacokinetic data in humans on whole body retention after 4 years of treatment with 90 mg of intravenous pamidronate, an estimated bone concentration of around 475 ng/mg of bone tissue (which equals 1575 nmol/g) would be expected. Of note, such calculations, which also have been made by Reid for zoledronate,37 result in an estimated pamidronate bone concentration that is similar to those extrapolated from data on actual bone concentrations in animals such as dogs and mice assessed using 14C-labeled pamidronate. When extrapolated from the published canine data,38 90 mg of pamidronate every 4 weeks over a 4-year period would lead to a bone concentration in humans of 240 ng/mg, while data extrapolated from mice with a similar regimen would lead to a bone concentration of 155 ng/mg.39 It therefore seems reasonable to assume that the average bone concentration in humans after 4 years of 90 mg of pamidronate every 4 weeks ranges somewhere between 100 and 1000 ng/mg (or 0.33–3.32 nM assuming 1g bone = 1 mL). Little is known, however, about the concentration of BP in various parts of the skeleton, as well as concentrations in bone sites with increased bone turnover such as a Pagetic lesion or alveolar bone following a surgical procedure. Data gained by using 99mTc-labeled BPs have shown that there is preferential uptake of BP in these sites, but the exact concentrations have yet to be determined. Data collected in rodents clearly show a noneven distribution of BPs throughout the skeleton, and also throughout single bones, especially in case of metastatic bone disease.40–42 At the present time, the actual amount of BP that is deposited within the human maxillofacial bones following intravenous administration is not known. Furthermore, although it has been speculated that gingival crevicular concentrations of BP are similar to bone concentrations,37 there are no data to support this to our knowledge, and there are no data on BP concentrations in gingival crevicular fluid or saliva. Various factors may promote BP release from the bone reservoir, including but not limited to trauma from dentoalveolar manipulation and inflammation from active periodontal disease. It is reasonable to postulate, however, that such factors may stimulate the release of BPs from the underlying alveolar bone, resulting in local increases in BP concentrations within the oral environment. Should the BP concentrations achieve toxic levels, the oral epithelial changes described may ensue.
While the emphasis thus far in ONJ research has been focused on bone necrosis being the primary event—an “inside-out” theory—it is intriguing to speculate that the adverse effects of BPs on oral epithelium may play a critical role in the initiation of ONJ—an “outside-in” hypothesis. Furthermore, this theory is consistent with the currently accepted definitions of ONJ, all of which include a breach in mucosa as a necessary event in developing this condition.
Are osteoblasts from the craniofacial skeleton different from other osteoblasts?
Inherent differences between bone forming cells of the jaw and other parts of the skeleton may potentially influence the development of ONJ. During embryonic development, the skeleton arises from three distinct lineages: the craniofacial skeleton is formed by cranial neural crest cells, the axial skeleton is derived from paraxial mesoderm, and the appendicular skeleton is the product of lateral plate mesodermal cells.43 Additionally, in regions of the craniofacial skeleton and the clavicle, osteoblastic differentiation forms intramembranous bones directly, whereas in the remaining skeleton, bones are formed via endochondral ossification, which includes formation of cartilage models and their replacement by bone tissue.
Most of our knowledge about the skeleton is derived from experimental animal studies and investigations of human bone disorders,43,44 and there has been relatively little work comparing the nature of the cells in various parts of the skeleton. Declercq et al.45 isolated and cultured rat cells from fetal calvaria, fetal long bones, and adult long bone. They showed significant differences in cellular proliferation and in vitro mineralization between fetal and adult bone, as well as between different cell isolation procedures and passage number.46 A study of human cells derived from long bones of young men indicated that the sequence of in vitro osteogenesis is similar to that of rat calvarial cultures,47 and a study of human calvarial samples indicated age-related changes in bone formation, osteoblastic cell proliferation and differentiation.48 Interestingly, different expression patterns of bone morphogenic proteins were reported for human intramembraneous and endochondral bone and these were maintained in ex vivo osteoblastic cell cultures.49
Our preliminary results indicate differences in cell populations obtained from human alveolar and long bone samples (Fig. 5). Primary human osteoblasts were isolated from discarded alveolar and long bone surgical procedures. All protocols were approved by the IRB. The tissue was minced, washed, and cultured in DMEM/F12 medium supplemented with 20% FBS, gentamicin, and fungizone. Proliferation of the first and third passage cells was tested and compared between the two tissue sources. Alveolar bone cells exhibited sustained proliferation from days 1–7, reaching between 2–3 doublings, while long bone cells exhibited minimal proliferation, reaching up to one population doubling during 7 days of culture. Cell growth stopped in most long bone samples by passage two. Additionally, the morphology of the cultured cells was different between the two cell sources.
Figure 5.

Difference in cellular proliferation of human alveolar and long bone. Proliferation of long bone cells in culture was significantly less over the course of seven days compared with alveolar bone cells.
In a study by Stephanik et al.,50 pamidronate treatment of primary human mandibular and iliac crest bone marrow stem cells from the same donors showed differences in cell survival, osteogenesis, and osteoclast recruitment, suggesting possible dysregulation of mandible bone homeostasis after BPs exposure. Further studies detailing unique cellular characteristics of alveolar bone as well as the potential differences of alveolar bone cells in response to BPs when compared to long bone are clearly indicated in defining the potential pathophysiological mechanisms of ONJ.
Are osteoclasts from the craniofacial skeleton different from other osteoclasts?
Evidence exists that osteoclasts derived from site-specific locations may show significant differences in their functional activity. Several studies by Everts et al.51,52 have evaluated the proteolytic enzyme profiles of osteoclasts derived from calvarial bone (intramembranous bone) and long bone osteoclasts (endochondral bone). These authors performed morphometric analysis using specific inhibitors for cysteine proteinases (CP, cathepsins) and for Matrix MetalloProteinases (MMPs) in a bone explants cultures. Isolated calvarial and long bone osteoclasts were examined for tartrate resistant acid phosphatase (TRAP) activity, immunolocalization of αvβ3 integrin receptors, and the ability to form resorption pits on bone slices in the presence of the different CP and MMP inhibitors. While the TRAP and integrin receptors were comparable in the different osteoclasts, the morphometric results demonstrated that while osteoclastic activity in both the long bones and calvaria are dependent on CPs, MMPs are critical only in the osteoclasts derived from the calvarial bone.
BPs are known to be very effective in inhibiting osteoclasts; however, it is not known if these drugs exert differential effects on osteoclasts that are derived from craniofacial skeletal tissues. Recent reports of ONJ developing in cancer patients treated with denosamab (Prolia™), an anti-RANK (receptor activator of nuclear factor-κB) ligand inhibitor,53,54 suggest that the osteoclast derivation or the specific cell environment may influence the development of ONJ. Whether differences in the site-specific osteoclast response to BPs contribute to the pathophysiology and localization of ONJ is not known and bears further investigation.
Effect of BPs on bone vascularity and angiogenesis
Several authors have postulated that the initiating event in ONJ is a compromise in bone-blood supply caused by exposure to BPs. As such, similarities have been drawn between ONJ and ORN, where patients develop necrotic bone as a consequence of decreased bone vascularity in the setting of radiation-induced fibrosis. In support of this theory, Kapitola et al.55 found that rats treated with pamidronate alone experienced an overall reduction in bone-blood flow and bone resorption rates (as measured by the uptake of 85Sr-microspheres, density and ash weight of bone, and urinary excretion of the resorption markers pyridinoline and deoxypyridinoline) compared to controls being treated with growth hormone alone or growth hormone plus pamidronate. However, there appear to be distinct differences between ONJ and ORN, both clinically and microscopically. While ORN almost never occurs in the maxilla, the ratio of maxillary to mandibular lesions in ONJ is less dramatic (2:1),11 with a significant number of cases described in the maxillary bone. Anatomically, the overwhelming mandibular predilection of ORN—a condition characterized by hypovascularity, hypoxia, and hypocellularity56—makes sense as the mandible is supplied by one major vessel. Moreover, ONJ may also differ from ORN histologically. Hansen et al.20 found an increase in vessel hyalinization and decrease in cellularity in ORN specimens compared to ONJ specimens.
The significant incidence of ONJ in the maxilla suggests that alteration in bone blood flow at the tissue level may not be solely responsible for induction of bone necrosis. A number of studies have shown that BPs may inhibit angiogenesis at the level of the endothelial cell. Wood et al.57 demonstrated in vitro that zoledronate inhibited proliferation of human endothelial cells stimulated with fetal calf serum, basic fibroblast growth factor, and vascular endothelial growth factor (VEGF). This group also found that endothelial cell adhesion, migration, and vessel sprouting were also significantly reduced by zoledronate. Bezzi et al.58 investigated the effects of zoledronate and clodronate on human umbilical vein endothelial cell (HUVEC) in vitro and confirmed that zoledronate but not clodronate inhibited integrin-mediated adhesion and migration. Furthermore, they found that zoledronate disrupted focal adhesions and actin stress fibers and enhanced tumor necrosis factor-induced cell death of HU-VECs that was independent of nuclear fragmentation or capase activity.58 Fournier et al.59 found that endothelial cells treated with a variety of BPs, including zoledronate, reduced proliferation, induced apoptosis, and decreased capillary-like tube formation in vitro. To investigate the effects of BPs in vivo, these authors examined revascularization in the prostate gland of rats induced by testosterone. They found a 50% reduction in revascularization in rats treated with zoledronate. In a mouse model of human cervical carcinoma (estrogen treated K14-HPV16 female transgenic mice or HPV/E2 mice), Giraudo et al.32 found that treatment with zoledronate inhibited angiogenesis around the tumor, as well as progression of premalignant lesions and tumor growth. The authors suggested that BPs may be efficacious as an adjunct to treatment for cervical carcinoma.32 Hamma-Kourbali et al.60 used a non-nitrogen-containing BP (BP7033) to treat a human epidermoid carcinoma cell line and were able to inhibit Ras processing as well as decrease VEGF and matrix metalloproteinase-2 (MMP-2) in vitro. When these tumor cells were introduced into nude mice, the BP was able to inhibit tumor growth and angiogenesis. An in vitro study by Okamoto et al.61 showed that incadronate inhibited advanced glycation endproduct (AGE)-induced angiogenesis, a process that is accelerated in diabetics, suggesting that BPs may prove valuable in the inhibition of vascular endothelial proliferation in diabetic retinopathy. Finally, BPs have been shown in vitro to inhibit MMPs 1, 2, 3, 7, 8, 9, 12, 13, and 14. MMPs are up-regulated in a number of inflammatory conditions and appear to play a role in tumor metastases.62
In human studies, Santini et al. have shown in several series that patients with advanced solid cancers and associated bone metastases exhibit significantly reduced serum VEGF when treated with zoledronate or pamidronate.63–65 Vincenzi et al.66 confirmed these findings and further observed that the reductions in VEGF levels were maintained throughout their study period, and that the greatest decrease was noted at 21 days. This group also found that cancer patients receiving a single infusion of zoledronate showed significant decreases in platelet-derived growth factor (PDGF) levels, although this reduction was only sustained for 2 days.64
A recent meta-analysis of three large clinical trials compared the incidence of ONJ in breast cancer patients receiving bevacizumab (avastin, anti-VEGF-1 monoclonal antibody) versus placebo and in patients with and without BP therapy. Although there were significant limitations to the study, the analysis failed to show an increased incidence in ONJ secondary to bevacizumab therapy in patients with locally recurrent or metastatic breast cancer treated with chemotherapy, with or without BPs.67
Many questions remain as to whether the antiangiogenic properties of BPs play a major role in the pathogenesis of ONJ. The notable incidence of ONJ within the maxilla appears to refute BP-induced inhibition of angiogenesis as the sole cause of ONJ. Furthermore, the antiangiogenic theory fails to explain why bone necrosis associated with BPs is not commonly seen in bones other than the jaws. It is likely, therefore, that the antiangiogenic properties of BPs contribute to the development of ONJ but that other factors, including intrinsic anatomic and physiologic differences in the gnathic bones, may play roles as well.
ONJ, altered wound healing, and immune dysfunction
Diabetes
It is well known that patients with diabetes have impaired wound healing that often results in chronic skin ulcerations, nonunion of fractures, and severe periodontal disease.68,69 Wound healing is a complex, multistep process that results in the restoration of an intact epidermal barrier. Appropriate wound healing relies on three overlapping functions of keratinocytes and fibroblasts: migration, proliferation, and differentiation. Directed migration is critical to wound healing, and defects in this process are associated with the clinical phenotype of chronic nonhealing wounds. Endothelial cells, macrophages, and platelets as well as numerous growth factors, surface receptors, and extracellular matrix proteins are also critical in the wound healing process. While it seems plausible that the impaired wound healing seen in diabetic patients could play a role in the development of ONJ, evidence is limited to a single brief report from Khamaisi et al.70 This group evaluated the frequency of diabetes mellitus in 31 patients with ONJ compared with a control group of 33 oncological patients without ONJ. The diagnosis of diabetes mellitus was based on the presence of two consecutive fasting blood glucose levels greater than 7mmol/L. Impaired fasting glucose levels were defined as levels of 100 or greater and 7mmol/L or less. Of the 31 patients with ONJ, 58% were found to either have diabetes or impaired fasting glucose (including one patient with gestational diabetes). Twelve percent of patients in the control group had diabetes. Statistical analysis was significant between the two groups when all diabetic-related diagnoses (diabetes, impaired fasting glucose, and gestational diabetes) were grouped together and compared to controls. Clearly, further investigations to define the role that impaired healing in diabetic patients plays in the development of ONJ are critical.
Chemotherapy and corticosteroids
Osteonecrosis (aseptic necrosis, avascular necrosis) of bones other than the craniofacial skeletal has been associated with sickle cell anemia, impairment of local bone circulation, Gaucher’s disease, and collagen vascular diseases. Although it is rare, several cases of osteonecrosis have been documented in patients receiving chemotherapy without concomitant corticosteroids.71–73 Interestingly, one of these cases was a patient with acute myelogenous leukemia who developed a large area of necrotic bone associated with several periodontally involved teeth. Chemotherapeutic agents have multiple effects on cells and are known to inhibit proliferation of oral mucosal cells often leading to mucositis and stomatitis. It would therefore seem likely that many of these agents could potentially play a role in the initiation or perpetuation of an ONJ lesion. The paucity of large well-controlled prospective studies and the fact that most patients reported in ONJ case series are being treated for metastatic bone disease with BPs and concomitant chemotherapy makes it difficult to assess the exact role of chemotherapeutic agents in the pathophysiology of this condition.
The inhibition of wound healing by corticosteroids is a well-recognized side effect of these medications. In addition, osteonecrosis, secondary to both intravenous and oral corticosteroids, has been well documented, most often seen in patients treated for malignancies or rheumatic diseases. The most common site of steroid-induced osteonecrosis is the femoral head; however, involvement of the knees, wrists, humerus, patella, and scapula has also been described.73 Similar to ONJ, the etiology and natural history of steroid-induced osteonecrosis is not well understood; however, there is some evidence that suggests a genetic predisposition to this condition.74 Several retrospective, uncontrolled studies have noted a high percentage of patients with ONJ that are receiving corticosteroids.10,14,74–77
Recently several animal models for ONJ have been developed. Bi et al.78 were the first to report on a murine model that showed exposed bone in the jaws following a 3-week intraperitoneal administration of zoledronate and dexamethasone, tooth extraction, and an additional 3- to 12-week course of drugs. While ONJ-like lesions developed in the animals treated with zoledronate and dexamethasone, the addition of doxetaxel resulted in significantly larger areas of ONJ-like lesions in these mice. Clearly, this murine model, as well as other newly described animal models, will aid in defining the specific role that steroids and chemotherapy play in the pathogenesis of ONJ.
Gamma/delta T cells
Nitrogen-containing BPs are well-known to cause a flu-like acute phase reaction in about one third of patients, predominantly after a first intravenous administration.79 This acute phase reaction has been linked to the inhibition of the enzyme farnesyl pyrophosphate (FPP)-synthase by the N-containing BPs. As a result of this enzyme inhibition, isopentenyl pyrophosphate (IPP) accumulates intracellularly, and is presented to Vγ9Vδ2+ T cells by an as yet unidentified mechanism.79 The result is the activation and proliferation of γ, δ T cells,80–82 cytokine release, and flu-like symptoms.83,84 This mechanism is now also being explored for immunotherapy purposes to treat tumors, as N-containing BPs have been shown to increase intracellular IPP concentrations in tumor cells in vitro.79 Inflammatory cells have been shown in bone biopsies from ONJ patients, hence, ONJ may also be linked to gamma-delta T cell activation and proliferation. To our knowledge, however, there are no data available on a potential relationship between developing ONJ and flu-like symptoms induced by N-containing BPs. Although interesting, the involvement of gamma-delta T cells in ONJ presently remains a hypothesis.
Other contributing factors
Hypercoagulable states
In the mid-1990s, a series of reports from one group of investigators surfaced implicating hypercoagulopathies with idiopathic osteonecrosis of the femoral head. Idiopathic osteonecrosis—as opposed to secondary osteonecrosis that occurs in the setting of predisposing factors like alcoholism, corticosteroid use, and systemic diseases—is characterized histologically by venous hypertension of the bone.85–88 This group theorized that patients suffering from a hypercoagulopathy—such as an increased tendency to develop thrombosis (thrombophilia) or a reduced ability to lyse thrombi (hypofibrinolysis)89—are predisposed to persistent thrombosis, which may lead to impaired venous drainage, increased medullary pressure, and subsequent anoxic bone necrosis.90–95 The existence of a hypercoagulable state can be assessed by a number of specialized serologic tests, including measurement of heritable thrombophilic Factor VIII levels, resistance to activated protein C (RAPC), protein C deficiency levels, anticardiolipin antibodies (ACLA) levels, tissue plasminogen activator activity (tPA-Fx), plasminogen activator inhibitor activity (PAI-Fx), and lipoprotein A levels (LP(a)). In one of their largest series,96 these authors evaluated 71 patients with idiopathic osteonecrosis of the femoral head for coagulopathies. They found that 19/71 (27%) of patients with idiopathic osteonecrosis had serologic measure of thrombophilia compared to 3/66 (5%) of race- and sex-matched controls. Similarly, 25/69 (36%) of the osteonecrosis patients had a serologic measure of hypofibrinolysis compared to 12/67 (18%) of controls. Similar results were found in other studies by the same group of investigators.91,94,97 Of particular interest, these authors have recently suggested that such hypercoagulable states may contribute to the development of necrotic lesions of the jaws.98 The role of hypercoagulopathies in the development of ONJ warrants further investigation.
Calcium and parathyroid hormone levels
It has been previously shown that patients taking BP can develop hypocalcemia and secondary hyperparathyroidism.99,100 The role of parathyroid hormone (PTH) in promoting cell proliferation and cancer progression is of interest in the oncologic population. One study showed that metastatic breast and prostate patients treated with zoledronate who demonstrated an elevation in PTH levels had an overall increased risk of mortality.89 Similarly, PTH increase in metastatic prostate patients receiving zoledronate had an increased risk of disease progression.101 Intriguingly, it has been shown that BP-induced secondary hyperparathyroidism and consequent renal osteodystrophy may predispose to spontaneous osteonecrosis of the femoral head.102
In a short letter to the Annals of Oncology, Ardine et al.103 examined a small group of breast cancer patients who developed ONJ while being treated with zoledronate. The median time from initiation of therapy to ONJ development was 14 months. The control population consisted of breast cancer patients also receiving zoledronate but who did not develop ONJ over 18 months of treatment. All patients were assessed for corrected calcium and PTH levels at baseline and the following time points: each of the 4 months prior to ONJ onset in the ONJ group and between the 12th and 14th months of zoledronate treatment in the control group. These authors found that the ONJ patients exhibited lower calcium and higher PTH values compared to controls at all evaluated time points and that the differences often achieved statistical significance. Interestingly, both patient populations exhibited an initial increase in PTH levels upon administration of the BP, although the increase was transient in the control group. These findings suggest that ONJ patients may be predisposed to persistent hypocalcemia and hyperparathyroidism compared to patients who do not develop ONJ, suggesting a role for calcium and PTH in ONJ. It is difficult to draw definitive conclusions from this study given the small size of the ONJ subset and that little information was provided regarding the control population. However, it appears that this is a potential avenue of future investigations in the study of ONJ pathogenesis.
From the bench to the bedside: which patients are at risk of ONJ?
A number of mechanisms have been described above that are likely involved in the pathophysiology of ONJ. In addition, we have outlined some important clinical risk factors that have been implicated in the development of ONJ lesions. The challenge for the clinician is to stratify risk for the development of ONJ in the clinical setting. Recently, a new clinical scale, the UCONNS (University of Connecticut OsteoNecrosis Numerical Scale), an instrument that weighs the various risk factors described herein, has been developed in order to address this issue (Appendix A). Future validation of this scale in our patient populations will be carried out with the goal that clinicians will be able to appropriately monitor and prevent ONJ.
Conclusion
The pathophysiology of ONJ is not well defined and likely is multifactorial in nature. While this condition is most likely associated with BP use, a causal effect has yet to be demonstrated. The unique anatomy, biomechanical forces, and microbial environment of the oral cavity may explain why ONJ lesions show preference for the craniofacial region. Furthermore, it appears likely that immunosuppression and altered wound healing play a role in the development of ONJ. We have presented data supporting the theory that ONJ lesions are initiated by trauma to the oral mucosa and BPs sequestered in the jaw bones inhibit wound closure—an “outside-in theory.” It is evident that much work remains before the pathophysiology of ONJ is definitively elucidated.
Acknowledgments
This work was supported by NIH R21-DEO17164 (RL and SC), NIH P41-EB002520 (GVN), and the New York Stem Cell Foundation (D.M. is a NYSCF-Stanley and Fional Druckenmiller fellow).
Appendix
The University of Connecticut OsteoNecrosis Numerical Scale (UCONNS): A Proposed Predictive Index for ONJ Risk Assessment
Regina Landesberg,1 Pam Taxel,2 Susan Tannebaum,3 David Shafer,4 David Pendrys,5 and Khalid Almas6
1Associate Professor, Division of Oral and Maxillofacial Surgery, School of Dental Medicine. 2Associate Professor, Department of Medicine, Division of Endocrinology and Metabolism, School of Medicine. 3Associate Professor, Department of Medicine, Division of Oncology, School of Medicine. 4Associate Professor, Division of Oral and Maxillofacial Surgery, School of Dental Medicine. 5Associate Professor, Division of Behavioral Sciences and Community Health, School of Dental Medicine. 6Assistant Professor, Division of Periodontology, School of Dental Medicine, University of Connecticut Health Center, Farmington, Connecticut
Osteonecrosis of the jaws (ONJ) was first reported in 2003 and, despite numerous case reports and a limited number of retrospective and prospective clinical studies, the pathophysiology, comorbidities, and potential risk factors for the development of the condition remain ill-defined. While ONJ appears to be associated with the use of nitrogen-containing BPs, numerous other factors potentially contribute to the occurrence of this disease. Furthermore, while a number of medical and dental societies have put forth clinical definitions and recommendations for prevention and treatment, determination of the importance of specific risk factors is often hampered by incomplete or nonspecific patient data. Consequently, we have developed an instrument, the University of Connecticut OsteoNecrosis Numerical Scale (UCONNS), to assess some of the suspected risk factors for the development of ONJ (Appendix Table A1). It appears that in order for our knowledge of ONJ and its associated risks to move forward, there needs to be a standardized reporting system and assessment of patients between institutions. The great majority of suspected risk factors for ONJ have not been scientifically validated; thus, the values assigned within are based on our best assessment of the presently available literature. The scale is meant to be a starting point and will require modification as more data becomes available. We plan to test the validity of this instrument by applying it to our patient population data in both prospective and retrospective analyses.
Appendix Table A1.
University of Connecticut OsteoNecrosis Numerical Scale
| 1 Bisphosphonate Use (Maximum 15) | |
| Oral | |
|
| |
| Points | Criteria |
|
| |
| 1 | Initiation of therapy - <3 Years |
| 2 | 3 – 5 Years |
| 3 | > 5 Years |
|
| |
| Reclast (IV Once a Year) | |
|
| |
| Points | Criteria |
|
| |
| 1 | < 3 Doses 5 – 10 mg |
| 2 | 3 – 5 Doses 15 – 20 mg |
| 3 | 6 or greater Doses 25 mg or greater |
|
| |
| Zoledronate (Zometa) | |
|
| |
| Points | Criteria |
|
| |
| 3 | 1–6 Doses 4 –24 mg |
| 4 | 7–12 Doses 28 – 48 mg |
| 5 | 13–18 Doses 52 –72 mg |
| 6 | 19 – 24 Doses 76 –96 mg |
| 7 | 25–36 Doses 100 – 144 mg |
| 8 | > 36 Doses > 144 mg |
|
| |
| Pamidronate (Aredia) | |
|
| |
| Points | Criteria |
|
| |
| 2 | 1 – 12 Doses |
| 3 | 13 – 24 Doses |
| 4 | > 25 Doses |
|
| |
| 2 Proposed Oral/Maxillofacial/Dental/Surgical Procedures (Maximum 5) | |
|
| |
| Points | Criteria |
|
| |
| 0 | Prophylaxis |
| 0 | Restorative procedure to tooth structure (Amalgam, crown & bridge) |
| 1 | Nonsurgical endodontic treatment |
| 1 | Denture Fabrication/New Denture insertion (prior 12 months) |
| 2 | Scaling and Root Planning, soft tissue surgery, non-immediate implant placement (prior 12 months) |
| 3 | Periodontal osseous surgery, surgical endodontic procedure, immediate implant placement (prior 12 months) |
| 4 | Routine extraction (prior 12 months) |
| 5 | Surgical extraction, surgical endodontic procedure, bone augmentation (prior 12 months) |
| 6 | Surgical resection of mandible/maxilla (prior 12 months) |
|
| |
| 3 Oral Health (Maximum 7) | |
|
| |
| Points | Criteria |
|
| |
| 0 | Good Oral Hygiene, minimal or no plaque (zone 1) no tooth mobility, no spontaneous bleeding, no visible inflammation |
| 1 | Fair Oral Hygiene, visible plaque (zone 2), 1+ tooth mobility visible inflammation, papillary inflammation |
| 2 | Poor oral hygiene, visible plaque (zone 3), 2+ tooth mobility, spontaneous bleeding, 3+ inflammation |
| 4 | Active infection/suppuration |
| 5 | Osteomyelitis |
|
| |
| 4 Medical Condition (Maximum 10) | |
|
| |
| Points | Criteria |
|
| |
| 0 | Health |
| 1 | HIV |
| 2 | Diabetes, Rheumatic Disorders |
| 2 | Soft tissue cancers other than breast/prostate |
| 3 | Breast, Prostate Cancer |
| 5 | Multiple myeloma |
|
| |
| 5 Comorbidities (Maximum 10) | |
|
| |
| Points | Criteria |
|
| |
| 0 | Nonsmoker |
| 1 | Prior smoker, Quit > 6 mos ago Inhaled steroids (within 12 months) |
| 2 | Smoker, current or last month, >10 cigs/day Steroids PO (within 12 months) |
| 3 | Steroids IV, IM (within 12 months) |
| 5 | Immunosuppression, Chemotherapy, immune modulators (for rheumatic disorders, transplant patients), (within 12 months) |
|
| |
| Risk Assessment | |
| < 10 | Minimal Risk |
| 10 to 15 | Moderate Risk |
| 16 or above | Significant Risk |
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Major P. The use of zoledronic acid, a novel, highly potent bisphosphonate, for the treatment of hypercalcemia of malignancy. Oncologist. 2002;7:481–491. doi: 10.1634/theoncologist.7-6-481. [DOI] [PubMed] [Google Scholar]
- 2.Rosen LS, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001;7:377–387. [PubMed] [Google Scholar]
- 3.Saad F, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458–1468. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 4.Van Poznak CH. The use of bisphosphonates in patients with breast cancer. Cancer Control. 2002;9:480–489. doi: 10.1177/107327480200900605. [DOI] [PubMed] [Google Scholar]
- 5.Marx RE. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 2003;61:1115–1117. doi: 10.1016/s0278-2391(03)00720-1. [DOI] [PubMed] [Google Scholar]
- 6.Migliorati CA. Bisphosphanates and oral cavity avascular bone necrosis. J Clin Oncol. 2003;21:4253–4254. doi: 10.1200/JCO.2003.99.132. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Goodger NM, Pogrel MA. Osteonecrosis of the jaws associated with cancer chemotherapy. J Oral Maxillofac Surg. 2003;61:1104–1107. doi: 10.1016/s0278-2391(03)00328-8. [DOI] [PubMed] [Google Scholar]
- 8.Pogrel MA. Bisphosphonates and bone necrosis. J Oral Maxillofac Surg. 2004;62:391–392. doi: 10.1016/j.joms.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Ruggiero SL, et al. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62:527–534. doi: 10.1016/j.joms.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Marx RE, et al. Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis) of the jaws: risk factors, recognition, prevention, and treatment. J Oral Maxillofac Surg. 2005;63:1567–1575. doi: 10.1016/j.joms.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Ruggiero SL, et al. American Association of Oral and Maxillofacial Surgeons. Position paper on bisphosphonate-related osteonecrosis of the jaws. J Oral Maxillofac Surg. 2009;67(Suppl 5):2–12. doi: 10.1016/j.joms.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Khosla S, et al. Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2007;22:1479–1491. doi: 10.1359/jbmr.0707onj. [DOI] [PubMed] [Google Scholar]
- 13.Kyle RA, et al. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25:2464–2472. doi: 10.1200/JCO.2007.12.1269. [DOI] [PubMed] [Google Scholar]
- 14.Ruggiero SL, Fantasia J, Carlson E. Bisphosphonate-related osteonecrosis of the jaw: background and guidelines for diagnosis, staging and management. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:433–441. doi: 10.1016/j.tripleo.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Toshiyki T, Hiroshi H, Toshitsugu T, et al. Bisphosphonate-related osteonecrosis of the jaw: position paper from the Allied Task Force Committee of Japanese Society for Bone and Mineral Research, Japan Osteoporosis Society, Japanese Society of Periodontology, Japanese Society for Oral and Maxillofacial Radiology, and Japanese Society of Oral and Maxillofacial Surgeons. J Bone Miner Metab. 2010;28:385–383. doi: 10.1007/s00774-010-0162-7. [DOI] [PubMed] [Google Scholar]
- 16.Patel V, McLeod NMH, Rogers SM, Brennan PA. Bisphosphonate osteonecrosis of the jaw-a literature review of UK policies versus international policies on bisphosphonates, risk factors and prevention. Br J Oral Maxillofac Surg. 2010 doi: 10.1016/j.bjoms.2010.05.007. In press. [DOI] [PubMed] [Google Scholar]
- 17.Ruggiero S, Dodson TB, Assael LA, et al. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaw. Australian Endodontic J. 2009;35:119–130. doi: 10.1111/j.1747-4477.2009.00213.x. [DOI] [PubMed] [Google Scholar]
- 18.Lugassy G, et al. Severe osteomyelitis of the jaw in long-term survivors of multiple myeloma: a new clinical entity. Am J Med. 2004;117:440–441. doi: 10.1016/j.amjmed.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Melo MD, Obeid G. Osteonecrosis of the maxilla in a patient with a history of bisphosphonate therapy. J Can Dent Assoc. 2005;71:111–113. [PubMed] [Google Scholar]
- 20.Hansen T, et al. Actinomycosis of the jaws-histopathological study of 45 patients shows significant involvement in bisphosphonate-associated osteonecrosis and infected osteoradionecrosis. Virchows Arch. 2007;451:1009–1017. doi: 10.1007/s00428-007-0516-2. [DOI] [PubMed] [Google Scholar]
- 21.Kos M, Brusco D, Kuebler J, Engelke W. Clinical comparison of patients with osteonecrosis of the jaws, with and without a history of bisphosphonates administration. Int J Oral Maxillofac Surg. 2010;39:1097–1102. doi: 10.1016/j.ijom.2010.04.054. [DOI] [PubMed] [Google Scholar]
- 22.Hall V. Actinomyces: gathering evidence of human colonization and infection. Anaerobe. 2008;14:1–7. doi: 10.1016/j.anaerobe.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Naik NH, Russo TA. Bisphosphonate-related osteonecrosis of the jaw: the role of Actinomyces. CID. 2009;49:1729–1732. doi: 10.1086/648075. [DOI] [PubMed] [Google Scholar]
- 24.Kolenbrander PE. Oral microbial communities: biofilms, interactions, and genetic systems. Annu Rev Microbiol. 2000;54:413–437. doi: 10.1146/annurev.micro.54.1.413. [DOI] [PubMed] [Google Scholar]
- 25.Chapurlat RD, et al. Microcrack frequency and bone remodeling in postmenopausal osteoporotic women on long-term bisphosphonates: a bone biopsy study. J Bone Miner Res. 2007;22:1502–1509. doi: 10.1359/jbmr.070609. [DOI] [PubMed] [Google Scholar]
- 26.Stepan JJ, et al. Low bone mineral density is associated with bone microdamage accumulation in post-menopausal women with osteoporosis. Bone. 2007;41:378–385. doi: 10.1016/j.bone.2007.04.198. [DOI] [PubMed] [Google Scholar]
- 27.Woo SB, Hellstein JW, Kalmar JR. [Corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med. 2006;144:753–761. doi: 10.7326/0003-4819-144-10-200605160-00009. Narrative. [DOI] [PubMed] [Google Scholar]
- 28.Twiss IM, et al. Cytotoxic effects of pamidronate on monolayers of human intestinal epithelial (Caco-2) cells and its epithelial transport. J Pharm Sci. 1994;83:699–703. doi: 10.1002/jps.2600830521. [DOI] [PubMed] [Google Scholar]
- 29.Twiss IM, et al. The effects of nitrogen-containing bisphosphonates on human epithelial (Caco-2) cells, an in vitro model for intestinal epithelium. J Bone Miner Res. 1999;14:784–791. doi: 10.1359/jbmr.1999.14.5.784. [DOI] [PubMed] [Google Scholar]
- 30.Wallace JL, et al. N-bisphosphonates cause gastric epithelial injury independent of effects on the microcirculation. Aliment Pharmacol Ther. 1999;13:1675–1682. doi: 10.1046/j.1365-2036.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- 31.Suri S, et al. Nitrogen-containing bisphosphonates induce apoptosis of Caco-2 cells in vitro by inhibiting the mevalonate pathway: a model of bisphosphonate-induced gastrointestinal toxicity. Bone. 2001;29:336–343. doi: 10.1016/s8756-3282(01)00589-0. [DOI] [PubMed] [Google Scholar]
- 32.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perazella MA. Drug-induced renal failure: update on new medications and unique mechanisms of nephro-toxicity. Am J Med Sci. 2003;325:349–362. doi: 10.1097/00000441-200306000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Montague R, et al. Differential inhibition of invasion and proliferation by bisphosphonates: anti-metastatic potential of Zoledronic acid in prostate cancer. Eur Urol. 2004;46:389–401. doi: 10.1016/j.eururo.2004.04.022. discussion 401–402. [DOI] [PubMed] [Google Scholar]
- 35.Landesberg R, et al. Inhibition of oral mucosal cell wound healing by bisphosphonates. J Oral Maxillofac Surg. 2008;66:839–847. doi: 10.1016/j.joms.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leyvraz S, et al. Pharmacokinetics of pamidronate in patients with bone metastases. J Natl Cancer Inst. 1992;84:788–792. doi: 10.1093/jnci/84.10.788. [DOI] [PubMed] [Google Scholar]
- 37.Reid IR, Bolland MJ, Grey AB. Is bisphosphonate-associated osteonecrosis of the jaw caused by soft tissue toxicity? Bone. 2007;41:318–320. doi: 10.1016/j.bone.2007.04.196. [DOI] [PubMed] [Google Scholar]
- 38.King LE, Vieth R. Extraction and measurement of pamidronate from bone samples using automated pre-column derivatization, high-performance liquid chromatography and fluorescence detection. J Chromatogr B Biomed Appl. 1996;678:325–330. doi: 10.1016/0378-4347(95)00531-5. [DOI] [PubMed] [Google Scholar]
- 39.Hoggarth CR, Bennett R, Daley-Yates PT. The pharmacokinetics and distribution of pamidronate for a range of doses in the mouse. Calcif Tissue Int. 1991;49:416–420. doi: 10.1007/BF02555853. [DOI] [PubMed] [Google Scholar]
- 40.Bauss F, Russell RG. Ibandronate in osteoporosis: preclinical data and rationale for intermittent dosing. Osteoporos Int. 2004;15:423–433. doi: 10.1007/s00198-004-1612-7. [DOI] [PubMed] [Google Scholar]
- 41.Monkkonen J, Koponen HM, Ylitalo P. Comparison of the distribution of three bisphosphonates in mice. Pharmacol Toxicol. 1990;66:294–298. doi: 10.1111/j.1600-0773.1990.tb00750.x. [DOI] [PubMed] [Google Scholar]
- 42.Usui T, et al. Pharmacokinetics of incadronate, a new bisphosphonate, in healthy volunteers and patients with malignancy-associated hypercalcemia. Int J Clin Pharmacol Ther. 1997;35:239–244. [PubMed] [Google Scholar]
- 43.Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 44.Karsenty G. The complexities of skeletal biology. Nature. 2003;423:316–318. doi: 10.1038/nature01654. [DOI] [PubMed] [Google Scholar]
- 45.Declercq H, et al. Isolation, proliferation and differentiation of osteoblastic cells to study cell/biomaterial interactions: comparison of different isolation techniques and source. Biomaterials. 2004;25:757–768. doi: 10.1016/s0142-9612(03)00580-5. [DOI] [PubMed] [Google Scholar]
- 46.Declercq HA, et al. Calcification as an indicator of osteoinductive capacity of biomaterials in osteoblastic cell cultures. Biomaterials. 2005;26:4964–4974. doi: 10.1016/j.biomaterials.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 47.Siggelkow H, et al. Development of the osteoblast phenotype in primary human osteoblasts in culture: comparison with rat calvarial cells in osteoblast differentiation. J Cell Biochem. 1999;75:22–35. [PubMed] [Google Scholar]
- 48.de Pollak C, et al. Age-related changes in bone formation, osteoblastic cell proliferation, and differentiation during postnatal osteogenesis in human calvaria. J Cell Biochem. 1997;64:128–139. [PubMed] [Google Scholar]
- 49.Suttapreyasri S, et al. Expression of bone morphogenetic proteins in normal human intramembranous and endochondral bones. Int J Oral Maxillofac Surg. 2006;35:444–452. doi: 10.1016/j.ijom.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 50.Stefanik D, Sarin J, Lam T, et al. Disparate osteogenic response of mandible and iliac crest bone marrow stromal cells to pamidronate. Oral Dis. 2008;14:465–471. doi: 10.1111/j.1601-0825.2007.01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Everts V, Korper W, Jansen DC, et al. Functional heterogeneity of osteoclasts: matrix metalloproteinases participate in osteoclastic resorption of calvarial bone but not in resorption of long bone. FASEB J. 1999;13:1219–1230. doi: 10.1096/fasebj.13.10.1219. [DOI] [PubMed] [Google Scholar]
- 52.Everts V, Korper W, Docherty AJ, Beertsen W. Matrix metalloproteinase inhibitors block osteoclastic resorption of calvarial bone but not the resorption of long bone. Ann NY Acad Sci. 1999;878:603–608. doi: 10.1111/j.1749-6632.1999.tb07739.x. [DOI] [PubMed] [Google Scholar]
- 53.Aghaloo TL, Felsenfeld AL, Tetradis S. Osteonecrosis of the jaw in a patient on Denosumab. J Oral Maxillofac Surg. 2010;68:959–963. doi: 10.1016/j.joms.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor KH, Middlefell LS, Mizen KD. Osteonecrosis of the jaws induced by anti-RANK ligand therapy. Br J Oral Surg. 2010;48:221–223. doi: 10.1016/j.bjoms.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 55.Kapitola J, Zak J. Effect of pamidronate on bone blood flow in oophorectomized rats. Physiol Res. 1998;47:237–240. [PubMed] [Google Scholar]
- 56.Marx RE. Osteoradionecrosis: a new concept of its pathophysiology. J Oral Maxillofac Surg. 1983;41:283–288. doi: 10.1016/0278-2391(83)90294-x. [DOI] [PubMed] [Google Scholar]
- 57.Wood J, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055–1061. doi: 10.1124/jpet.102.035295. [DOI] [PubMed] [Google Scholar]
- 58.Bezzi M, et al. Zoledronate sensitizes endothelial cells to tumor necrosis factor-induced programmed cell death: evidence for the suppression of sustained activation of focal adhesion kinase and protein kinase B/Akt. J Biol Chem. 2003;278:43603–43614. doi: 10.1074/jbc.M308114200. [DOI] [PubMed] [Google Scholar]
- 59.Fournier P, et al. Bisphosphonates inhibit angiogenesis in vitro and testosterone-stimulated vascular regrowth in the ventral prostate in castrated rats. Cancer Res. 2002;62:6538–6544. [PubMed] [Google Scholar]
- 60.Hamma-Kourbali Y, et al. A novel non-containing-nitrogen bisphosphonate inhibits both in vitro and in vivo angiogenesis. Biochem Biophys Res Comm. 2003;310:816–823. doi: 10.1016/j.bbrc.2003.09.083. [DOI] [PubMed] [Google Scholar]
- 61.Okamoto T, et al. Incadronate disodium inhibits advanced glycation end products-induced angiogenesis in vitro. Biochem Biophys Res Comm. 2002;297:419–424. doi: 10.1016/s0006-291x(02)02218-0. [DOI] [PubMed] [Google Scholar]
- 62.Teronen O, et al. MMP inhibition and downregulation by bisphosphonates. Ann N Y Acad Sci. 1999;878:453–465. doi: 10.1111/j.1749-6632.1999.tb07702.x. [DOI] [PubMed] [Google Scholar]
- 63.Santini D, et al. Pamidronate induces modifications of circulating angiogenetic factors in cancer patients. Clin Cancer Res. 2002;8:1080–1084. [PubMed] [Google Scholar]
- 64.Santini D, et al. Zoledronic acid induces significant and long-lasting modifications of circulating angiogenic factors in cancer patients. Clin Cancer Res. 2003;9:2893–2897. [PubMed] [Google Scholar]
- 65.Santini D, et al. Repeated intermittent low-dose therapy with zoledronic acid induces an early, sustained, and long-lasting decrease of peripheral vascular endothelial growth factor levels in cancer patients. Clin Cancer Res. 2007;13(15 Pt 1):4482–4486. doi: 10.1158/1078-0432.CCR-07-0551. [DOI] [PubMed] [Google Scholar]
- 66.Vincenzi B, et al. Zoledronic acid-related angiogenesis modifications and survival in advanced breast cancer patients. J Interferon Cytokine Res. 2005;25:144–151. doi: 10.1089/jir.2005.25.144. [DOI] [PubMed] [Google Scholar]
- 67.Van Poznack C. Osteonecrosis of the jaw and bevacizumab therapy. Breast Cancer Res Treat. 2010;122:189–191. doi: 10.1007/s10549-010-0933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes.[comment] J Clin Inv. 2007;117:1219–1222. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 70.Khamaisi M, et al. Possible association between diabetes and bisphosphonate-related jaw osteonecrosis. [see comment] J Clin Endocrinol Metabol. 2007;92:1172–1175. doi: 10.1210/jc.2006-2036. [DOI] [PubMed] [Google Scholar]
- 71.Harper PG, Trask C, Souhami RL. Avascular necrosis of bone caused by combination chemotherapy without corticosteroids. Br Med J Clin Res Ed. 1984;288:267–268. doi: 10.1136/bmj.288.6413.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marymont JV, Kaufman EE. Osteonecrosis of bone associated with combination chemotherapy without corticosteroids. Clin Orthopead Rel Res. 1986;204:150–153. [PubMed] [Google Scholar]
- 73.Sung EC, et al. Osteonecrosis of the maxilla as a complication to chemotherapy: a case report. Spec Care Dentist. 2002;22:142–146. doi: 10.1111/j.1754-4505.2002.tb01178.x. [DOI] [PubMed] [Google Scholar]
- 74.Jones LC, Hungerford DS. Osteonecrosis: etiology, diagnosis, and treatment. Cur Opin Rheumatol. 2004;16:443–9. doi: 10.1097/01.moo.0000127829.34643.fd. [DOI] [PubMed] [Google Scholar]
- 75.Ortega C, et al. Osteonecrosis of the jaw in prostate cancer patients with bone metastases treated with zoledronate: a retrospective analysis. Acta Oncologica. 2007;46:664–668. doi: 10.1080/02841860601185917. [DOI] [PubMed] [Google Scholar]
- 76.Tosi P, et al. Osteonecrosis of the jaws in newly diagnosed multiple myeloma patients treated with zoledronic acid and thalidomide-dexamethasone. Blood. 2006;108:3951–3952. doi: 10.1182/blood-2006-07-033571. [DOI] [PubMed] [Google Scholar]
- 77.Van Den Wyngaert T, Huizing MT, Vermorken JB. Bisphosphonates and osteonecrosis of the jaw: cause and effect or a post hoc fallacy? Ann Oncol. 2006;17:1197–1204. doi: 10.1093/annonc/mdl294. [DOI] [PubMed] [Google Scholar]
- 78.Bi Y, Gao Y, Ehirchiou D, et al. Bisphosphonates cause osteonecrosis of the jaw-like disease in mice. Am J Pathol. 2010;177:280–290. doi: 10.2353/ajpath.2010.090592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roelofs AJ, et al. Molecular mechanisms of action of bisphosphonates: current status. Clin Cancer Res. 2006;12(20 Pt 2):6222s–6230s. doi: 10.1158/1078-0432.CCR-06-0843. [DOI] [PubMed] [Google Scholar]
- 80.Gober HJ, et al. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. 2003;197:163–168. doi: 10.1084/jem.20021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hewitt RE, et al. The bisphosphonate acute phase response: rapid and copious production of proinflammatory cytokines by peripheral blood gd T cells in response to aminobisphosphonates is inhibited by statins. Clin Exp Immunol. 2005;139:101–111. doi: 10.1111/j.1365-2249.2005.02665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thompson K, Rogers MJ. Statins prevent bisphosphonate-induced gamma, delta-T-cell proliferation and activation in vitro. J Bone Miner Res. 2004;19:278–288. doi: 10.1359/JBMR.0301230. [DOI] [PubMed] [Google Scholar]
- 83.Kunzmann V, et al. Stimulation of gamma delta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 2000;96:384–392. [PubMed] [Google Scholar]
- 84.Kunzmann V, Bauer E, Wilhelm M. Gamma/delta T-cell stimulation by pamidronate. N Engl J Med. 1999;340:737–738. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 85.Atsumi T, Kuroki Y, Yamano K. A microangiographic study of idiopathic osteonecrosis of the femoral head. Clin Orthop Relat Res. 1989;246:186–194. [PubMed] [Google Scholar]
- 86.Jones JP., Jr Intravascular coagulation and osteonecrosis. Clin Orthop Relat Res. 1992;277:41–53. [PubMed] [Google Scholar]
- 87.Mankin HJ. Nontraumatic necrosis of bone (osteonecrosis) N Engl J Med. 1992;326:1473–1479. doi: 10.1056/NEJM199205283262206. [DOI] [PubMed] [Google Scholar]
- 88.Jones JP. Arthritis and Allied Conditions: A Textbook of Rheumatology. 14. Lippincot Williams and Wilkins; Philadelphia, PA: 2001. Osteonecrosis. [Google Scholar]
- 89.Glueck CJ, et al. Heterozygosity for the Leiden mutation of the factor V gene, a common pathoetiology for osteonecrosis of the jaw, with thrombophilia augmented by exogenous estrogens. J Lab Clin Med. 1997;130:540–543. doi: 10.1016/s0022-2143(97)90132-3. [DOI] [PubMed] [Google Scholar]
- 90.Glueck CJ, et al. Hypofibrinolysis: a common, major cause of osteonecrosis. Am J Hematol. 1994;45:156–166. doi: 10.1002/ajh.2830450212. [DOI] [PubMed] [Google Scholar]
- 91.Glueck CJ, et al. Idiopathic osteonecrosis, hypofibrinolysis, high plasminogen activator inhibitor, high lipoprotein(a), and therapy with Stanozolol. Am J Hematol. 1995;48:213–220. doi: 10.1002/ajh.2830480402. [DOI] [PubMed] [Google Scholar]
- 92.Glueck CJ, et al. Thrombophilia and hypofibrinolysis: pathophysiologies of osteonecrosis. Clin Orthop Relat Res. 1997;334:43–56. [PubMed] [Google Scholar]
- 93.Glueck CJ, et al. Familial high plasminogen activator inhibitor with hypofibrinolysis, a new pathophysiologic cause of osteonecrosis? Thromb Haemost. 1993;69:460–465. [PubMed] [Google Scholar]
- 94.Glueck CJ, et al. Familial idiopathic osteonecrosis mediated by familial hypofibrinolysis with high levels of plasminogen activator inhibitor. Thromb Haemost. 1994;71:195–198. [PubMed] [Google Scholar]
- 95.Glueck CJ, et al. Thrombophilia, hypofibrinolysis, and alveolar osteonecrosis of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:557–566. doi: 10.1016/s1079-2104(96)80047-3. [DOI] [PubMed] [Google Scholar]
- 96.Glueck CJ, Freiberg RA, Wang P. Heritable thrombophilia-hypofibrinolysis and osteonecrosis of the femoral head. Clin Orthop Relat Res. 2008;466:1034–1040. doi: 10.1007/s11999-008-0148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Glueck CJFR, Gruppo R, Crawford A, et al. Thrombophilia and hypofibrinolysis: reversible pathogenetic etiologies of osteonecrosis. In: Urbaniak James R, Jones John Paul., Jr, editors. Osteonecrosis Etiology, Diagnosis and Treatment. Chapter 14. Publication of the American Association of Orthopedic Surgeons; Rosemont, IL: 1997. [Google Scholar]
- 98.McMahon RE, et al. Osteonecrosis: a multifactorial etiology. J Oral Maxillofac Surg. 2004;62:904–905. doi: 10.1016/j.joms.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 99.Tanvetyanon TSP. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol. 2006;17:897–907. doi: 10.1093/annonc/mdj105. [DOI] [PubMed] [Google Scholar]
- 100.Berruti A, et al. Metabolic effects of single-dose pamidronate administration in prostate cancer patients with bone metastases. Int J Biol Markers. 2002;17:244–252. doi: 10.5301/jbm.2008.2194. [DOI] [PubMed] [Google Scholar]
- 101.Berruti ADL, Tucci M. Metabolic bone disease induced by prostate cancer. Rationale for the use of bisphosphonates. J Urol. 2001;166:2023–2031. [PubMed] [Google Scholar]
- 102.Elder G. Pathophysiology and recent advances in the management of renal osteodystrophy. J Bone Miner Res. 2002;17:2094–2105. doi: 10.1359/jbmr.2002.17.12.2094. [DOI] [PubMed] [Google Scholar]
- 103.Ardine M, et al. Could the long-term persistence of low serum calcium levels and high serum parathyroid hormone levels during bisphosphonate treatment predispose metastatic breast cancer patients to undergo osteonecrosis of the jaw? Ann Oncol. 2006;17:1336–1337. doi: 10.1093/annonc/mdl045. [DOI] [PubMed] [Google Scholar]