

Abstract
Objective
An Alzheimer’s disease (AD) diagnosis is preceded by a long period of cognitive decline. We previously demonstrated increased risk of decline among individuals possessing one or more APOE ε4 alleles together with a family history of AD. The objective of this study is to investigate the possibility that such an increased risk might be due to AD risk genes with small effects in combination with APOE.
Methods
Participants in the Health and Retirement Study (HRS) over the age of 65, who contributed DNA, and had two or more evaluations with an abbreviated version of the modified Telephone Interview for Cognitive Status (TICS-m) were eligible for the study (n = 7451). A genetic risk score (g-score) was derived using AD risk genes’ meta-analyses data, assigning risk according to the number of risk alleles and summed over all the risk genes. Trajectories of cognitive function were modeled in four groups of Caucasian participants with and without one or more APOE ε4 alleles and either a high or low g-score: APOE ε4-/low g-score; APOE ε4-/high g-score; APOE ε4+/low g-score; and APOE ε4+/high g-score. Post hoc analyses evaluated interactions between individual genes and APOE.
Results
Individuals in the APOE ε4+/high g-score group exhibited the greatest cognitive decline over time (p<.0001). This risk appeared to be greater than the sum of the effects of either high g-score or APOE ε4 alone. When gene interactions were individually tested with APOE, a statistically significant interaction with CD33 was discovered (p = 0.04) although the interaction was no longer significant when adjusted for multiple comparisons.
Conclusions
Individuals with multiple AD risk genes in addition to having one or more APOE ε4 alleles are at greater risk of cognitive decline than individuals with either APOE ε4 or a high genetic risk score. Among those with one or more APOE ε4 alleles, having one or more copies of the CD33 C (risk) allele may further increase the risk of cognitive decline.
Introduction
An Alzheimer’s disease (AD) diagnosis is preceded by a long period of cognitive decline. During this time when cognitive changes are relatively mild, it may be possible to alter the trajectory of cognition. Researchers are seeking to identify and recruit into clinical trials, individuals who may be on a downward trajectory in order to test new therapeutic treatments and interventions. In some cases, those at highest genetic risk are recruited (e.g., individuals with PSEN1, PSEN2, or APP mutations), however this approach samples from a narrowly defined and relatively small group of individuals who are at the highest risk of early onset Alzheimer’s disease (EOAD). A number of genes with small effects have been identified as risk factors for late onset AD in a number of large, multi-cohort GWAS studies [1–4], so it stands to reason that individuals possessing multiple risk alleles have an even greater risk of AD and may be more likely to express an endophenotype of cognitive decline than those possessing fewer risk alleles. To date, little work has been done to demonstrate this potential association.
We previously reported the effects of family history of dementia and the APOE ε4 allele on cognitive performance over time with data from the Cache County Study of Memory Health and Aging (CCSMHA)[5]. In that study, family history was presumed to represent some combination of genetic predisposition or shared environment. Individuals possessing one or more APOE ε4 alleles declined faster than those without APOE ε4, and individuals who had a family history of dementia in addition to one or more APOE ε4 alleles had the worst performance over time on average. We now seek to test the hypothesis that other AD risk genes contribute to cognitive decline in a manner similar to the CCSMHA Study but now using genes instead of an endorsement of family history of AD. Specifically, we hypothesize that the combination of APOE ε4 and AD risk genes will be associated with a steeper downward trajectory of cognitive function.
Methods
Data for the study were drawn from the Health and Retirement Study (HRS) [6]. The HRS began data collection in 1992 and the Asset and Health Dynamics among the Oldest Old (AHEAD) study began in 1993. The two samples were merged in 1998 and are now referred to as HRS. Additional cohorts were added, including the War Baby study and the Children of the Depression study. Together, these studies provide comprehensive biennial data on a large sample that is representative of the U.S. population aged 50+. Interviews are conducted with all HRS respondents every two years by the Survey Research Center at the University of Michigan. All participants provided informed consent. Consent is inferred by the completion of mailed surveys; oral consent is obtained for telephone interviews; and written consent is obtained for biological samples. The study protocol, methods, and consent procedures were approved by the University of Michigan Institutional Review Board (IRB); the current secondary data analysis was approved by the Duke University Medical Center IRB. Interviews are conducted primarily by telephone, except for in-person interviews to cover specific sub-studies and with those aged 80 and older.
Participants
Participants for the current study are members of the HRS aged 65+, who provided genetic material for GWAS and completed HRS Telephone Interview for Cognitive Status-modified (TICS-m). Our sample is limited to participants aged 65+ because beginning in 1998, the HRS TICS-m was administered only to those age 65+ and participants not interviewed at the prior wave. If an individual was initially evaluated prior to age 65, only data from visits occurring after age 65 were used. To control for confounding due to population genetic substructure we focused on Whites only. Basic demographic information including sex and years of education were recorded as part of the baseline interview.
DNA Samples and Selection of SNPs
Saliva samples were collected from HRS participants beginning in 2006. GWA analyses were completed in 2013. The Illumina HumanOmi1-Quad and Illumina Human Omni-2.5 Quad bead chips were used as genotyping platforms. Genetic data are available through dbGaP. In order to facilitate comparison with other platforms, imputation strategies were used to increase the number of available markers. SNPs that best represent the genes of interest were identified in the GWA data.
Description of Variables
Genetic Risk Score
AD risk genes that were reported in a series of Nature Genetics papers from 2009–2011[1, 2],[3, 7] and represent the “top 10” AD risk genes in the Alzgene resource were selected [8]. Genes included: CLU, CR1, PICALM, MS4A6A/MS4A4E, CD33, CD2AP, ABCA7, CR1 and PICALM. APOE was considered separately.
A genetic risk score (g-score) was computed using the odds ratios for AD risk reported in published meta-analyses on the AlzGene website [8]. Odds ratios less than 1 were converted so that all the risks were reported in the same direction. The number of alleles possessed by each participant was multiplied by the odds ratio for each AD risk gene. The g-score is the sum of all the risks weighted by the strength of the reported odds ratio. The resulting continuous risk score was then dichotomized at the median into high and low scores. A separate variable was created based on quartiles of risk.
Genetic risk groups
Four mutually exclusive risk groups were determined based on APOE ε4 and g-score status. APOE was modeled separately because the strength of its association with cognitive function may mask other genetic effects. The reference group had no APOE ε4 and a low g-score (APOE-/low g-score), other groups included those with no APOE ε4 and a high g-score alleles(APOE-/high g-score), APOE ε+ and a low g-score(APOE+/low g-score), and the highest risk group with APOE ε4+ and a high g-score(APOE+/high g-score).
HRS TICS-m for identification of Cognitive Decline
The Telephone Interview for Cognitive Status (TICS) has been used extensively. The original version of the TICS [9], was modeled after the Mini-Mental State Examination (MMSE)[10] and has good discriminability between normal and demented elderly[11]. The HRS TICS-m, an abbreviated version of the modified version of the TICS, has a total of 35 points: 10 points each for immediate and delayed recall; 10 points for counting backwards, naming, and orientation; and 5 points for serial 7s. The HRS TICS-m has been administered serially since 1993 in the AHEAD sample and since 1996 in the HRS sample. The measure has been cross-validated for administration by telephone or in person[12]. Age-related changes in longitudinal performance on the HRS TICS-m have been reported[13] and are similar to other cognitive measures. If respondents were unable to complete the HRS TICS-m, a proxy informant was administered the IQCODE[14] to obtain an assessment of cognitive and functional decline of the respondent in the preceding two years. Only individuals with HRS TICS-m scores were included in the analysis.
Analytic Approach
Demographic characteristics of the sample based on the four mutually exclusive genetic risk groups described above were compared with chi-square tests or generalized linear models (SAS PROC GLM). Repeated measures fixed effects models are used to estimate cognitive trajectories[15] using the SAS PROC MIXED procedure [16]. The dependent variable for the main analysis was a continuous measure of decline on the TICS-m. With this approach, individual differences in cognitive performance over time are captured while accounting for the correlation in repeated measures. Models were adjusted for baseline age, sex, and years of education. Age was used as the time scale and a quadratic term for age was tested to allow for non-linear progression of cognitive decline. Post hoc analyses evaluated interactions individually between each gene and APOE using similar mutually exclusive four-level factorial variable models and interaction terms.
Results
A total of 8,709 individuals who contributed DNA and cognitive assessments after age 65 were identified from a total of 12,507 participants who provided DNA for genetic analysis (Fig 1). 7,451 of these self-identified as White and had complete data for this analysis. Demographic characteristics of the sample by genetic risk group are shown in Table 1.
Fig 1. Sample Selection Flowchart.
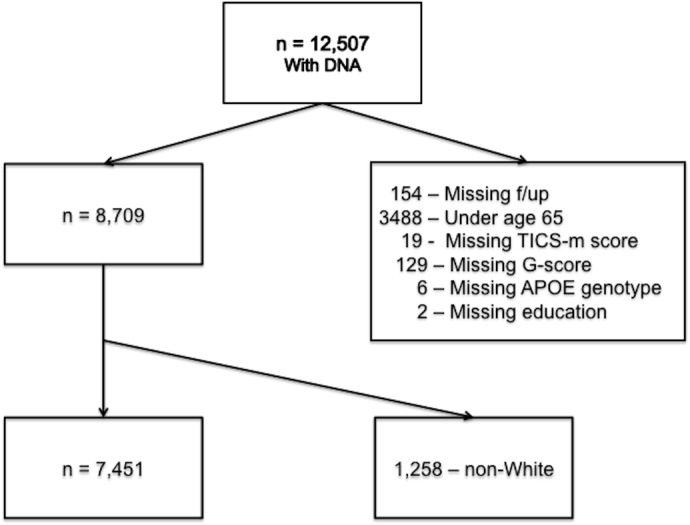
Abbreviations: f/up: follow-up; TICS-m: Telephone Interview for Cognitive Status-modified; G-score: genetic risk score.
Table 1. Demographic Characteristics of HRS Sample.
| Characteristic | Group1 APOE ε4+ G-Score high | Group2 APOE ε4-G-Score high | Group3 APOE ε4+ G-Score low | Group4 APOE ε4-G-Score low | Total |
|---|---|---|---|---|---|
| N (%) | 723 (9.7) | 2783 (37.4) | 840 (11.3) | 3105 (41.7) | 7451 |
| Mean # Observations(SD) * | 4.9 (2.4) | 5.1 (2.4) | 5.0 (2.4) | 5.2 (2.4) | 5.0 (2.4) |
| Mean Baseline Age (SD) | 67.4 (3.3) | 67.7 (3.6) | 67.7 (3.8) | 67.8 (3.7) | 67.7 (3.7) |
| Sex: Female (%) | 424 (58.6) | 1561 (56.1) | 475 (56.6) | 1790 (57.7) | 4250 (57.0) |
| Male (%) | 299 (41.4) | 1222 (43.9) | 365 (43.5) | 1315 (42.4) | 3201 (43.0) |
| Mean Yrs Education (SD) | 12.7 (3.2) | 12.5 (3.1) | 12.6 (3.1) | 12.6 (3.0) | 12.6 (3.1) |
| Mean Baseline TICS-m (SD) | 23.7 (4.4) | 23.8 (4.3) | 23.7 (4.3) | 24.0 (4.2) | 23.8 (4.3) |
Abbreviations: G-score: genetic risk score HRS: Health and Retirement Study; N: number; SD: standard deviation; TICS-m: Telephone Interview for Cognitive Status-modified; Yrs: years.
*There were statistically significant differences in the mean number of visits between the reference Group (Group 4) and Groups 3 and 1. There were also significant differences between Groups 1 and 2.
A compound symmetry correlation structure best fit the repeated measures models. Models were constructed using the four risk groups described above and adjusted for baseline age, age*age, sex, sex*age, education in years, and education*age. The four-level factorial variable was also multiplied by age. The results for the four-level variable and age interaction are presented in Table 2. Individuals in the highest risk group (APOE ε4+/high g-score) had statistically significantly greater decline than individuals with neither risk factor (APOE ε4-/low g-score -0.11, 95% CI -0.13 to -0.08) (Table 2). The high-risk group also had greater decline than the sum of either genetic risk factor alone (Fig 2). Results were similar when we restricted the sample to just the highest and lowest quartiles of genetic risk score.
Table 2. Mixed Effects Models for Cognitive Decline by APOE/G-score group.
| Group | Group*Age | |||
|---|---|---|---|---|
| B(se) | p-value | B(se) | p-value | |
| APOE ε4+/G-Score high | -0.075 (0.15) | 0.6245 | -0.106 (0.012) | <.0001 |
| APOE ε4+/G-Score low | -0.088 (0.15) | 0.5447 | -0.046 (0.011) | <.0001 |
| APOE ε4-/G-Score high | -0.128 (0.10) | 0.1889 | 0.0008 (0.007) | 0.9129 |
| APOE ε4-/G-Score low | ref | ref | ref | ref |
Abbreviations: se: standard error; G-score: genetic risk score; ref: reference group.
Model adjusted for age, age2, sex, sex*age, education, and education*age.
Fig 2. Mixed effects models for cognitive trajectories by APOE ε4/G-score risk group.

Abbreviations: TICS-m: Telephone interview for Cognitive Status-modified; high G-score: high genetic risk score; low G-score: low genetic risk score. Model included age, age2, sex, sex*age, education, education*age, and group*age. Corresponding model results for the four-level variable and interaction terms are presented in Table 2.
To further evaluate this phenomenon, we ran post-hoc mixed effects models to evaluate each gene in combination with APOE (controlling for age, sex, and education) and found that the increased decline was driven by a statistically significant interaction with CD33 (-0.68, 95% CI -1.33 to -0.03; p = 0.04). When a Bonferroni correction[17] for multiple comparisons was applied, findings were no longer statistically significant. Separate factorial models were constructed to evaluate APOE/CD33 combinations in four mutually exclusive groups; APOE ε4-/CD33-, APOE ε4-/CD33+, APOE ε4+/CD33-, and APOE ε4+/CD33+ (Fig 3). This model was adjusted for baseline age, age*age, sex, sex*age, education, education*age, and interaction terms for the four-level variable*age. The group with both APOE ε4+ and CD33+ was significantly different than the group with only APOE ε4+ (-0.638, SE 0.24; Tukey-Kramer adjusted p = 0.045). To rule out effects of the APOE ε2 allele, we ran models stratified by APOE ε2 status (yes/no) and the association was relatively unchanged in the sample without APOE ε2.
Fig 3. Mixed effects models for cognitive trajectories by APOE ε4/CD33 risk group.

Abbreviations: TICS-m: Telephone interview for Cognitive Status-modified. Covariates included age, age2, sex, sex*age, education, education*age, group, and group*age.
Discussion
In this study of 7,451 participants from the HRS, individuals with one or more APOE ε4 alleles and a high g-score appear to experience greater cognitive decline over time than individuals with either APOE ε4 or a high g-score alone. Post hoc analyses restricting the sample to individuals in the highest or lowest quartiles of g-score did not produce any meaningful change in the results. However, further investigation revealed that the increased decline in individuals with both APOE and other AD risk genes was driven primarily by an interaction between APOE and CD33. This is the first study we know of to suggest the possibility of an interaction between APOE ε4 and CD33 affecting cognitive function over time in the HRS data, although we acknowledge that the interaction was no longer significant once a correction for multiple comparisons was applied.
Both the APOE and CD33 genes are located on chromosome 19, however these two genes are not in linkage disequilibrium as they are 6Mb apart [3]. The C allele of CD33 (rs3865444) has been associated with an increased risk of AD in genome-wide association studies [3, 4], has been tied to more severe cognitive decline in AD [18, 19], and correlated with lower MMSE scores [20]. Using data from the Religious Orders Study, Memory and Aging Project, and the Chicago Health and Aging Project, Bradshaw et al found that the C allele accounts for a significant portion of the variance in CD33 gene expression (>70.0%), is associated with PiB positivity, neuritic plaque burden, and reduced phagocytic activity [21]. The relationship between genotype and gene expression appears to be consistent over the life course, suggesting that CD33 may be a marker of susceptibility [21]. CD33 is involved in clearance of Aβ from the brain [22] so one might speculate that the cognitive effects of CD33 become apparent later in the disease process. This notion aligns with a model of the disease that specifies cognitive deficits as occurring later, after the accumulation of Aβ [23]. Others have speculated that the association between APOE and AD decreases with increasing age [24, 25]. These theories both correspond with our model where the effects of CD33 seem indistinguishable from APOE early on but appear to strengthen later. When combined with the cumulative effects of APOE later in life, the underlying function of this gene may become more important. Although the actual mechanism is unknown, an interaction between these two genes resulting in potentially increased accumulation of β-amyloid is plausible.
The level of cognitive decline among those with a high genetic risk score in the absence of APOE ε4 was not statistically significantly different than the level of decline in individuals with a low genetic risk score. A number of investigators have reported similar results using genetic risk scores calculated in much the same manner[26–28] including those using the HRS cohort [29, 30]. Most of these studies have failed to find significant differences in cognition (including cross sectional performance[26, 27], progression to mild cognitive impairment [26], or dementia outcomes [27, 30]). The current study differs in several important ways. First, the cohort was restricted to ages 65 and older where cognitive decline is more likely to be of a detectable magnitude. Other studies included younger individuals, which may influence the ability to detect cognitive decline [27, 29, 30]. Second, we looked at cognitive decline over time in a cohort with evaluations that took place every two years on average using a global measure of cognition that is heavily weighted to episodic memory. Third, our sample size was larger than most prior studies. And finally, we controlled for education in our models as this is an important covariate in studies of cognitive function at baseline and over time.
Limitations
The HRS TICS-m has been criticized as a blunt cognitive measure; however for the purposes of this study, it appears to be an adequate measure of global cognitive function over time. In this study, we selected only those who self-reported White race to determine the sample used for genetic analysis, which may result in some misclassification. It is likely that the percentage of Northern European ancestry is relatively high among individuals who self-report as White. In addition, a paper by Reitz et al, suggested that Whites and African Americans share common AD risk genes although the frequencies of some risk alleles may differ [31]. Indeed the frequencies of CD33 alleles are different in African Americans (0.02) than in Whites (0.34). Given this difference and the fact that the current study is not a GWAS study where the genetic association can be confounded by admixture, it seems appropriate to focus only on White race only. Further, the non-White sample is likely too small to yield significant results. When we tested the full sample including individuals with self-reported African American heritage, results were similar, although we note that the proportion of non-White participants was relatively small. Findings were not significant when run in subsample of non-White participants. This suggests that if there were misclassification, its effects were not meaningful. We note the SNPs selected for this study are based on genes identified in the Alzgene top ten list [8]. While this list is continually updated with information from GWA studies, there are likely genes associated with AD that have yet to be identified. For the current study, we chose to focus on the genes that have the strongest associations in the existing literature.
Conclusions
The ε4 allele of the APOE gene is a potent risk factor for cognitive decline over time. Here we have shown in a sample of White participants from the HRS, that cognitive decline among individuals with one or more APOE ε4 alleles in addition to AD risk genes of small effect are greater than the risk associated with APOE ε4 alone. This risk appears to be pronounced in individuals who also possess one or more copies of the CD33 C allele although further study and replication of findings is needed.
Data Availability
Data cannot be shared with the manuscript because data were obtained from a third party, the University of Michigan Health and Retirement Study. The University of Michigan makes the data available to qualified researchers upon request: http://hrsonline.isr.umich.edu/index.php?p=reg. Per HRS, “Co-investigators and persons not under the direct supervision of another researcher are required to submit separate applications in order to obtain access to HRS public files and datasets.” We used RAND Version M fat file data. The RAND versions of the data are similarly available from the HRS website and the HRS rules for access and distribution apply to the RAND data. Genetic data for the HRS were obtained from dbGaP. These data are available through an application process whereby assurances for data security must be put in place in order to qualify: https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login. Other researchers can access the data used for this analysis by registering with HRS and accessing the data from the HRS website. Researchers will need to apply to dbGaP and gain approval for access to the genetic data.
Funding Statement
This work was supported funded by the National Institute on Aging, grant # R01AG042633 (http://www.nia.nih.gov/) to KMH, MWL, MK, CG, and BLP. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41(10):1088–93. Epub 2009/09/08. doi: ng.440 [pii] 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094–9. Epub 2009/09/08. doi: ng.439 [pii] 10.1038/ng.439 . [DOI] [PubMed] [Google Scholar]
- 3. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nature genetics. 2011;43(5):436–41. Epub 2011/04/05. 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genetics. 2011;43(5):429–+. 10.1038/Ng.803 PubMed Central PMCID: PMC3084173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayden KM, Zandi PP, West NA, Tschanz JT, Norton MC, Corcoran C, et al. Effects of family history and apolipoprotein E epsilon4 status on cognitive decline in the absence of Alzheimer dementia: the Cache County Study. Arch Neurol. 2009;66(11):1378–83. Epub 2009/11/11. doi: 66/11/1378 [pii] 10.1001/archneurol.2009.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juster FT, Suzman R. An overview of the health and retirement study. J Hum Resour. 1995;30:S7–S56. 10.2307/146277 . [DOI] [Google Scholar]
- 7. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43(5):429–35. doi: http://www.nature.com/ng/journal/v43/n5/abs/ng.803.html#supplementary-information PubMed Central PMCID: PMC3084173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23. Epub 2006/12/29. 10.1038/ng1934 . [DOI] [PubMed] [Google Scholar]
- 9. Brandt J, Spencer M, Folstein M. The Telephone Interview for Cognitive Status. Neuropsychiatry, Neuropsychology, and Behavioral Neurology. 1988;1:111–7. [Google Scholar]
- 10. Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–98. Epub 1975/11/01. doi: 0022-3956(75)90026-6 [pii]. . [DOI] [PubMed] [Google Scholar]
- 11. Manly JJ, Schupf N, Stern Y, Brickman AM, Tang M-X, Mayeux R. Telephone-Based Identification of Mild Cognitive Impairment and Dementia in a Multicultural Cohort. Arch Neurol. 2011;68(5):607–14. 10.1001/archneurol.2011.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ofstedal MB, Fisher G, Herzog AR. Documentation of cognitive functioning measures in the Health and Retirement Study Ann Arbor: University of Michigan; 2005. [December 14, 2007]. Available from: http://hrsonline.isr.umich.edu/docs/userg/dr-006.pdf. [Google Scholar]
- 13. McArdle JJ, Fisher GG, Kadlec KM. Latent variable analyses of age trends of cognition in the Health and Retirement Study, 1992–2004. Psychol Aging. 2007;22(3):525–45. Epub 2007/09/19. 10.1037/0882-7974.22.3.525 . [DOI] [PubMed] [Google Scholar]
- 14. Jorm AF, Jacomb PA. The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): socio-demographic correlates, reliability, validity and some norms. Psychol Med. 1989;19(4):1015–22. Epub 1989/11/01. . [DOI] [PubMed] [Google Scholar]
- 15. Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–74. Epub 1982/12/01. . [PubMed] [Google Scholar]
- 16. The Mixed Procedure 1992. In: SAS/STAT Software: Changes and Enhancements release 6 07 [Internet]. SAS Institute Inc. [Google Scholar]
- 17. Dunn OJ. Multiple Comparisons Among Means. Journal of the American Statistical Association. 1961;56(293):52–64. [Google Scholar]
- 18. Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PloS one. 2012;7(11):e50976 10.1371/journal.pone.0050976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–43. 10.1016/j.neuron.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu N, Tan MS, Sun L, Jiang T, Wang YL, Tan L, et al. Decreased expression of CD33 in peripheral mononuclear cells of Alzheimer's disease patients. Neuroscience letters. 2014;563:51–4. 10.1016/j.neulet.2014.01.004 . [DOI] [PubMed] [Google Scholar]
- 21. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nature neuroscience. 2013;16(7):848–50. 10.1038/nn.3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang T, Yu JT, Hu N, Tan MS, Zhu XC, Tan L. CD33 in Alzheimer's disease. Molecular neurobiology. 2014;49(1):529–35. 10.1007/s12035-013-8536-1 . [DOI] [PubMed] [Google Scholar]
- 23. Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119–28. 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA: the journal of the American Medical Association. 1997;278(16):1349–56. . [PubMed] [Google Scholar]
- 25. Qiu C, Winblad B, Fastbom J, Fratiglioni L. Combined effects of APOE genotype, blood pressure, and antihypertensive drug use on incident AD. Neurology. 2003;61(5):655–60. . [DOI] [PubMed] [Google Scholar]
- 26. Carrasquillo MM, Crook JE, Pedraza O, Thomas CS, Pankratz VS, Allen M, et al. Late-onset Alzheimer's risk variants in memory decline, incident mild cognitive impairment, and Alzheimer's disease. Neurobiology of aging. 2015;36(1):60–7. 10.1016/j.neurobiolaging.2014.07.042 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verhaaren BF, Vernooij MW, Koudstaal PJ, Uitterlinden AG, van Duijn CM, Hofman A, et al. Alzheimer's disease genes and cognition in the nondemented general population. Biological psychiatry. 2013;73(5):429–34. 10.1016/j.biopsych.2012.04.009 . [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Rodriguez E, Sanchez-Juan P, Vazquez-Higuera JL, Mateo I, Pozueta A, Berciano J, et al. Genetic risk score predicting accelerated progression from mild cognitive impairment to Alzheimer's disease. J Neural Transm. 2012. Epub 2012/11/28. 10.1007/s00702-012-0920-x . [DOI] [PubMed]
- 29. Zhang C, Pierce BL. Genetic susceptibility to accelerated cognitive decline in the US Health and Retirement Study. Neurobiology of aging. 2014;35(6):1512 e11-8. 10.1016/j.neurobiolaging.2013.12.021 . [DOI] [PubMed] [Google Scholar]
- 30. Marden JR, Walter S, Tchetgen Tchetgen EJ, Kawachi I, Glymour MM. Validation of a polygenic risk score for dementia in black and white individuals. Brain and behavior. 2014;4(5):687–97. 10.1002/brb3.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA: the journal of the American Medical Association. 2013;309(14):1483–92. Epub 2013/04/11. 10.1001/jama.2013.2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data cannot be shared with the manuscript because data were obtained from a third party, the University of Michigan Health and Retirement Study. The University of Michigan makes the data available to qualified researchers upon request: http://hrsonline.isr.umich.edu/index.php?p=reg. Per HRS, “Co-investigators and persons not under the direct supervision of another researcher are required to submit separate applications in order to obtain access to HRS public files and datasets.” We used RAND Version M fat file data. The RAND versions of the data are similarly available from the HRS website and the HRS rules for access and distribution apply to the RAND data. Genetic data for the HRS were obtained from dbGaP. These data are available through an application process whereby assurances for data security must be put in place in order to qualify: https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login. Other researchers can access the data used for this analysis by registering with HRS and accessing the data from the HRS website. Researchers will need to apply to dbGaP and gain approval for access to the genetic data.