

Abstract
Objective:
We report the clinical features, comorbidities, and outcome of 22 newly identified patients with antibodies to the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR).
Methods:
This was a retrospective review of patients diagnosed between May 2009 and March 2014. Immunologic techniques have been reported previously.
Results:
Patients' median age was 62 years (range 23–81; 14 female). Four syndromes were identified: 12 (55%) patients presented with distinctive limbic encephalitis (LE), 8 (36%) with limbic dysfunction along with multifocal/diffuse encephalopathy, one with LE preceded by motor deficits, and one with psychosis with bipolar features. Fourteen patients (64%) had a tumor demonstrated pathologically (5 lung, 4 thymoma, 2 breast, 2 ovarian teratoma) or radiologically (1 lung). Additional antibodies occurred in 7 patients (3 onconeuronal, 1 tumor-related, 2 cell surface, and 1 tumor-related and cell surface), all with neurologic symptoms or tumor reflecting the concurrent autoimmunity. Treatment and outcome were available from 21 patients (median follow-up 72 weeks, range 5–266): 5 had good response to immunotherapy and tumor therapy, 10 partial response, and 6 did not improve. Eventually 5 patients died; all had a tumor or additional paraneoplastic symptoms related to onconeuronal antibodies. Coexistence of onconeuronal antibodies predicted a poor outcome (p = 0.009).
Conclusion:
Anti-AMPAR encephalitis usually manifests as LE, can present with other symptoms or psychosis, and is paraneoplastic in 64% of cases. Complete and impressive neurologic improvement can occur, but most patients have partial recovery. Screening for a tumor and onconeuronal antibodies is important because their detection influences outcome.
The recent characterization of autoimmune synaptic disorders has led to the identification of subtypes of limbic, multifocal, or generalized encephalitis that often respond to immunotherapy. One of the antibodies targets the GluA1 or GluA2 (previously called GluR1 or GluR2) subunits of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), an ionotropic receptor that belongs to the family of glutamate receptors. AMPAR mediates most of the fast excitatory synaptic transmission in the brain, and is important for synaptic plasticity, memory, and learning.1 The initial description of the encephalitis associated with these antibodies was published in 2009 and included 10 patients, all with limbic encephalitis (LE) who had CSF and serum antibodies that reacted with the neuropil of rat brain and the cell surface of cultures of rat hippocampal neurons, leading to precipitate and characterize the target antigens as the GluA1 or GluA2 subunits of the AMPAR.2 Recent studies have shown that these antibodies cause a selective decrease in the total surface amount and synaptic localization of GluA1 and GluA2-containing AMPAR through increased internalization and degradation,3 resulting also in a decrease of AMPAR-mediated currents.3,4 Since the initial description of this disorder, few cases with similar antibodies have been reported and therefore the clinical manifestations are largely unknown.5–8 We report 22 additional patients and describe the clinical presentation, cancer association, response to treatment, comorbidities, prognostic factors, and outcome.
METHODS
Patients.
Sera or CSF of 10,573 patients with suspected autoimmune encephalitis or paraneoplastic neurologic syndromes (including LE, nonfocal encephalitis, encephalomyelitis, psychiatric disorders, dementia, Morvan syndrome, and cerebellar dysfunction) were included in the studies of antibody screening. The samples were received between May 2009 and March 2014 in the Department of Neurology, University of Pennsylvania; Department of Neurology, Erasmus Medical Center Rotterdam; and the Center of Neuroimmunology at Institut d´Investigació Biomèdica August Pi i Sunyer (IDIBAPS), Hospital Clinic, University of Barcelona. Clinical information was obtained by the investigators or from questionnaires completed by the referring physicians and telephone interviews. One patient was previously published as an isolated case report.9
Standard protocol approvals, registrations, and patient consents.
Informed consent for antibody studies was obtained in all patients. The study was approved by the Institutional Review Boards of the Hospitals of the University of Pennsylvania, Erasmus Medical Center Rotterdam, and University of Barcelona.
Screening for antineuronal antibodies.
Serum and CSF samples were tested for antibodies to intracellular and cell surface antigens using immunohistochemistry on rat brain as previously reported.2,10 Samples showing specific tissue staining were further examined with in-house or immunoblot assays for antibodies to onconeuronal antigens (Hu, Yo, Ri, amphiphysin, Ma1/2, Tr), tumor-associated antigens (SOX1, ZIC4), and non-tumor-associated antigens (GAD65, AK5, Homer3).11,12 The identity of the target cell surface or synaptic autoantigens was determined with HEK293 cells expressing LGI1, CASPR2, NMDAR, AMPAR, and GABA(B)R, as reported.2,13–16 The type of AMPAR subunit identified by patients' antibodies was investigated using HEK293 cells expressing only the GluA1 or GluA2 subunits of the receptor.2 We did not include in these investigations the determination of antibodies using immunoblot of recombinant, denatured AMPAR subunits,4 given that these antibodies do not produce the typical neuropil immunostaining of the antibodies studied here and their syndrome association remains to be established.
Review of previously reported cases of tumor-associated anti-AMPAR encephalitis.
In order to assess the significance of the presence of an underlying tumor or additional paraneoplastic antibodies in the clinical outcome (survival and relapse frequency), we reviewed the current data along with all previously reported oncologic or paraneoplastic cases of anti-AMPAR encephalitis.2,5,6,8
Statistical analysis.
Age between groups was compared by Mann-Whitney U test, time until diagnosis by Student t test, and comparison of serum and CSF by McNemar test. For survival, Kaplan-Meier curves were created, using log-rank tests. The association of relapses with type of therapy (aggressive, nonaggressive) was compared with Fisher exact test. Statistical significance was defined as p value less than 0.05.
RESULTS
Patients.
We identified 22 patients with AMPAR antibodies. A summary of the clinical information is shown in tables 1 and 2. The median age was 62 years (interquartile range [IQR] 37.3–70.3 years; range 23–81 years) with no differences between patients with or without tumor (60.5 years, IQR 38.8–70.3, and 64 years, IQR 35–72, respectively; p = 0.80). The female:male ratio was 14:8. Fourteen patients (64%) had an underlying tumor; in 13, the tumor was demonstrated pathologically, including 5 small cell lung cancer (SCLC), 4 thymoma, 2 breast cancer, and 2 ovarian teratoma, and in another patient the tumor was demonstrated radiologically (lung cancer). One patient had onconeuronal antibodies (CRMP5), but no tumor has been identified until now (30 weeks follow-up).
Table 1.
Clinical presentation in patients with AMPAR autoimmune encephalitis
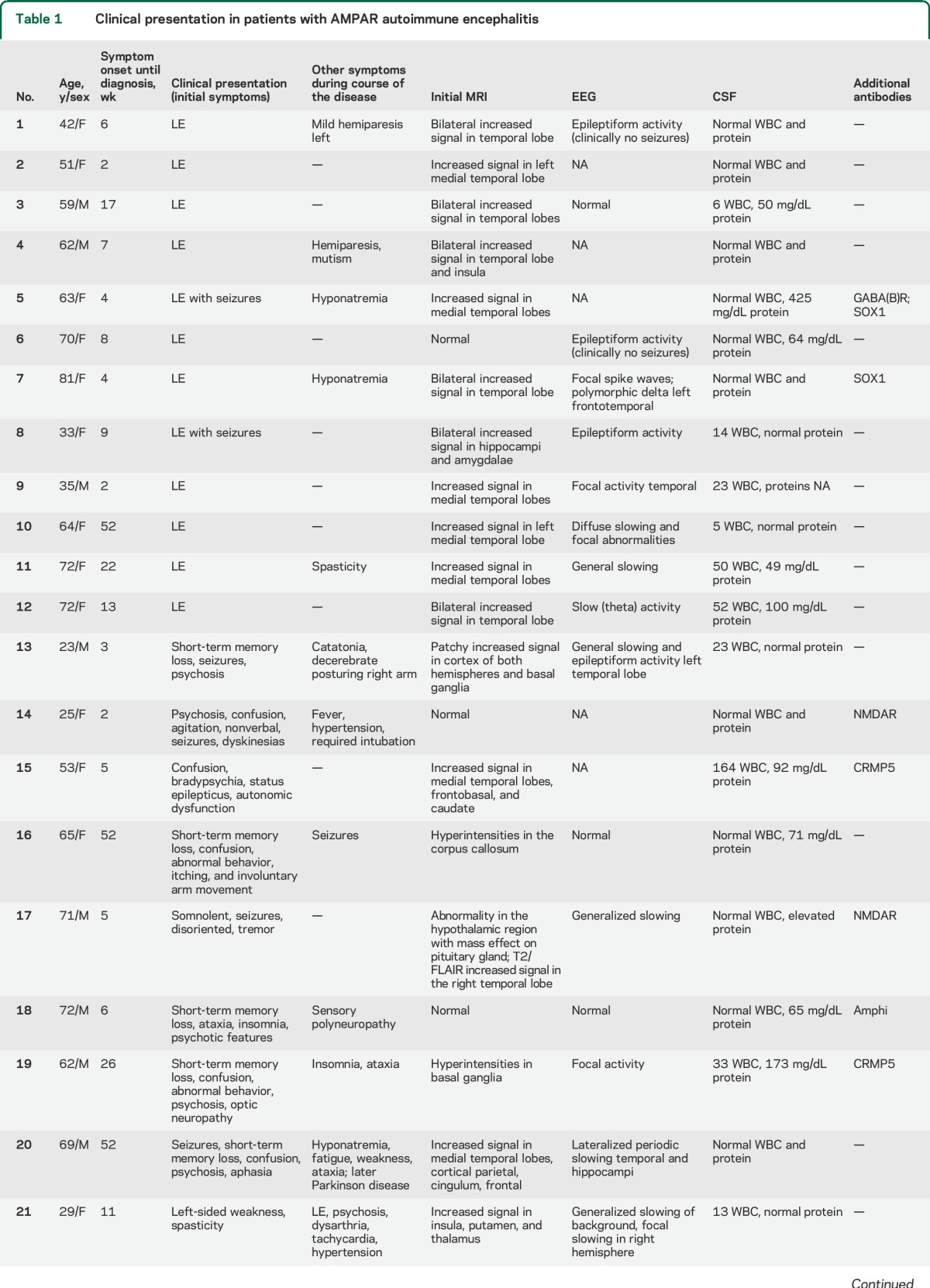

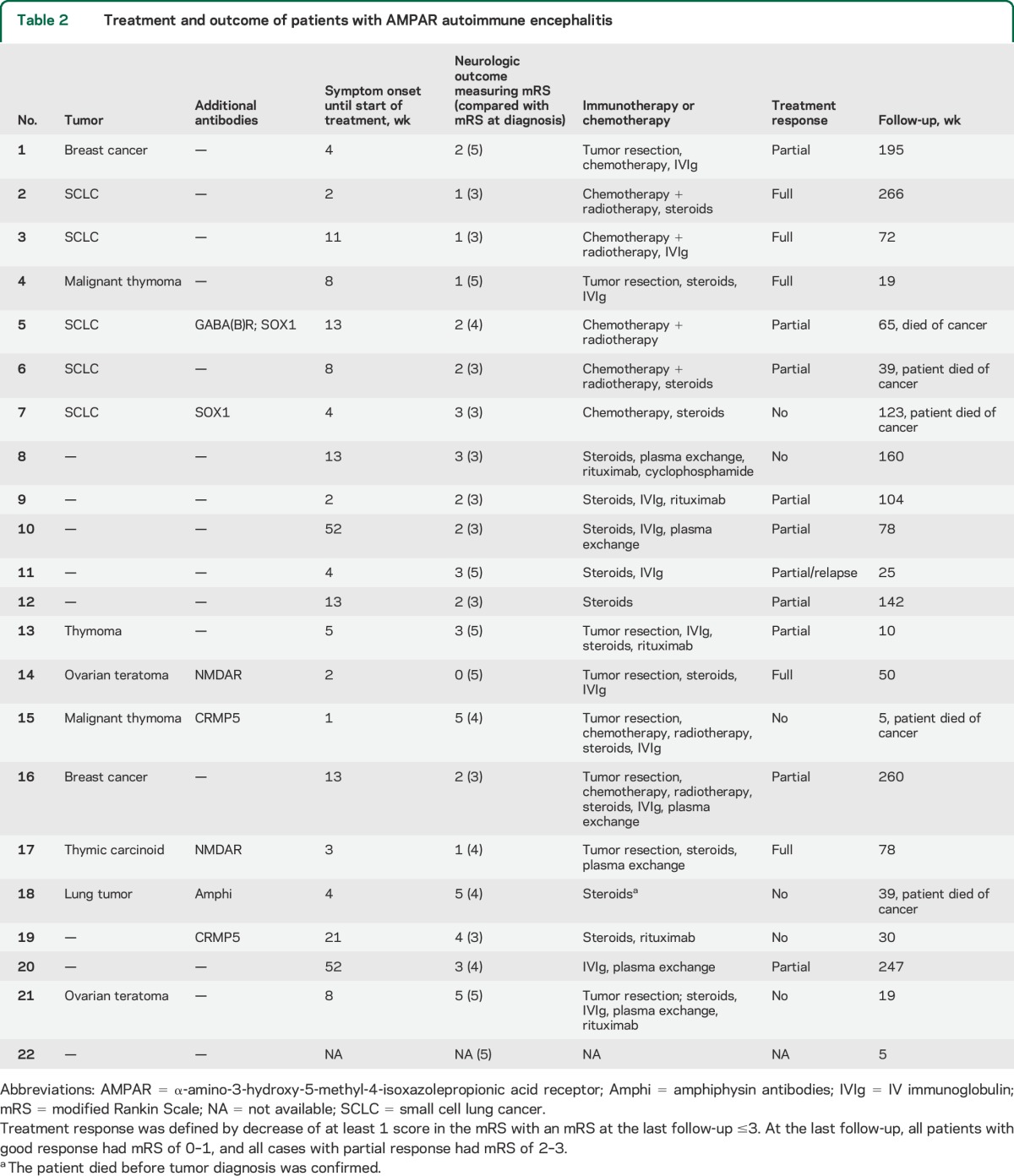
Table 2.
Treatment and outcome of patients with AMPAR autoimmune encephalitis
The median time from symptom onset until diagnosis of autoimmune encephalitis was 6.5 weeks (IQR 4–18.3 weeks), and was not different between patients with or without tumor (5.5 weeks [IQR 3.8–8.8] vs 13 weeks [5–52], respectively; p = 0.11). In 12 patients, the neurologic symptoms occurred before tumor diagnosis or led to tumor screening (median 5 weeks [IQR 2.3–37.5], range 1.5–56 weeks) and in 2 patients the tumor was identified before developing encephalitis (1 year and 6 months, respectively). The CSF analysis showed lymphocytic pleocytosis in 11/22 patients (median leukocyte count 23 cells/μL, range 5–820), and elevated protein concentration in 10/20 (median 71 mg/dL, range 49–425); only 5/22 patients had an elevated protein concentration as an isolated finding (table 1).
Clinical presentation and MRI features.
Four clinical presentations were identified: 12 patients (55%) developed symptoms of LE, defined by the presence of short-term memory loss, confusion, and abnormal behavior (table 1, patients 1–12). Two of these patients developed seizures (nos. 5 and 8) and one of them (no. 5) had additional GABA(B)R and SOX1 antibodies. Hyponatremia occurred in 2 patients, both with SCLC. In 11 of the 12 patients, the clinical diagnosis of LE was confirmed by the MRI findings of unilateral (2 patients) or bilateral (9 patients) mesiotemporal increased fluid-attenuated inversion recovery (FLAIR)/T2 signal abnormalities (figure 1, A and B). In patient 6 (table 1), the brain MRI was normal.
Figure 1. Brain MRI findings of anti-AMPAR encephalitis.

Brain MRI obtained 6 weeks after symptom onset (patient 4, table 1) shows increased T2/fluid-attenuated inversion recovery (FLAIR) signal abnormalities involving medial temporal lobes (A) and insular cortex (B). In addition, the frontal and parieto-occipital cortex and claustrum show mild focal hyperintensity (B). Diffusion-weighted images of patient 13 (table 1) show widespread involvement of the temporal cortex (C). Similar abnormalities are shown on FLAIR sequences involving the left frontal and right temporal cortex as well as the right putamen (D). AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor.
Eight patients (table 1, cases 13–20) had diffuse encephalitis with clinical and/or MRI evidence of involvement of multiple areas of the CNS (figure 1, C and D). All developed limbic dysfunction along with one or more of the following symptoms: 6 had seizures, 5 prominent psychiatric manifestations, 3 ataxia, 2 abnormal movements, and 1 each optic neuropathy and aphasia. Hyponatremia occurred in one patient without cancer. Two patients in this group had NMDAR antibodies (further described in Immunologic studies).
Another patient (table 1, patient 21) was a 29-year-old woman who first presented with left-sided weakness and spasticity involving face, arm, and leg, and 2 months later developed memory loss, confusion, abnormal behavior, psychiatric symptoms, visual hallucinations, autonomic dysfunction, and dysarthria. The MRI showed increased FLAIR/T2 signal in the right thalamus, bilateral putamen, and cerebellum. Tumor screening revealed an ovarian teratoma but NMDAR antibodies were negative.
The remaining case (patient 22, table 1) was a 38-year-old woman who presented with new-onset psychosis with bipolar features. She was started on antipsychotic medication and 1 week later developed nystagmus and neuroleptic malignant syndrome (rigidity, tonic fencing-like posture, and autonomic instability), requiring intubation for airway protection. The brain MRI was normal, EEG demonstrated moderate generalized slowing, CSF showed 90 leukocytes/mm3, and the tumor screening was negative. Her mental status did not improve and she was lost to follow-up after 5 weeks.
Immunologic studies.
All patients' serum or CSF samples showed intense neuropil staining on brain tissue immunohistochemistry and reacted with HEK cells coexpressing GluA1/2 subunits of the AMPAR.2 From 5 patients only CSF was available and from another 3 only serum was available; paired samples were available from 14 patients, all CSF were antibody-positive but only 10/14 sera were positive (p = 0.13). Four patients had antibodies only against the GluA1 subunit, 7 only against the GluA2 subunit, and 9 against both subunits (figure 2); in 2 cases, the limited amount of sample did not allow for independent subunit assessment. There were no significant differences among clinical presentation, association with a tumor, or prognosis in patients with antibodies recognizing different subunits.
Figure 2. Reactivity of patients' antibodies with GluA1 or GluA2 subunits of AMPAR.

Patients' antibodies were identified on HEK293 cells transfected with GluA1 or GluA2 subunits of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). Examples of patients with antibodies targeting only GluA1 (patient 9), or GluA2 (patient 15), or both subunits (patient 3) are shown. Patients' antibodies are shown with green fluorescence; commercial monoclonal antibodies against GluA1 or GluA2 are shown with red fluorescence; the blue nuclear staining is shown with 4',6-diamidino-2-phenylindole (DAPI). All panels: ×400.
Additional neuronal antibodies were found in 7 patients (3 onconeuronal-intracellular, 1 SOX1 [tumor-associated], 2 cell surface/synaptic, and 1 SOX1 and cell surface/synaptic), 6 of them with an associated tumor or cancer (table 2). The 3 patients with onconeuronal antibodies (2 CRMP5, 1 amphiphysin) developed a clinical phenotype, outcome, or tumor association that were more characteristic of the additional immune response than that of the AMPAR antibody syndrome (tables 1 and 2). Two of the 3 patients with additional antibodies to cell surface/synaptic proteins [1 GABA(B)R, 2 NMDAR] developed syndromes influenced by the additional antibody: the patient with GABA(B)R antibodies had seizures and SCLC, both typical of this autoimmune syndrome, and one patient with NMDAR antibodies developed psychosis, confusion, agitation, decrease of verbal output, seizures, and dyskinesias, all typical of anti-NMDAR encephalitis. In contrast, the second patient with NMDAR antibodies was atypical in many respects, including demographics (71-year-old man), symptoms (somnolence, seizures), and tumor (thymic carcinoid). The presentation with somnolence is atypical of NMDAR autoimmunity, and it was likely related to an inflammatory lesion in the hypothalamus (table 1); this case probably represents an overlap syndrome of 2 or more autoimmune disorders.
Treatment and follow-up.
Clinical follow-up was available from 21 patients (median 72 weeks, range 5–266). The remaining patient (patient 22) was lost to follow-up shortly after diagnosis. Twenty patients received first-line immunotherapy (steroids, IV immunoglobulins, or plasma exchange) and 5 of them also received second-line immunotherapy (rituximab, cyclophosphamide) (table 2). Thirteen patients had one or more of the following oncologic treatments: 7 tumor resection, 8 chemotherapy, and 6 radiation therapy (table 2). Five patients showed good neurologic response to immunotherapy or oncologic therapy with a modified Rankin Scale (mRS) score between 0 and 1 at the last follow-up; 10 patients had partial response with an mRS score between 2 and 3, and 6 patients had poor or no response to treatment (table 2). One of the patients with partial response (patient 11, table 2) relapsed 2 months later; this patient was lost to follow-up after relapse. Of the 6 patients with poor or no response to treatment, 4 had a malignant tumor or additional antibodies (1 SOX1, 1 amphiphysin, and 2 CRMP5) with superimposed paraneoplastic symptoms (see details in appendix e-1 on the Neurology® Web site at Neurology.org).
There was no significant change in survival for patients with or without a tumor (median 123 weeks vs not met, all 6 without tumor still alive; p = 0.086; figure 3A; the patient with onconeuronal CRMP5 antibodies but without tumor was excluded from this as well as from the following analysis). On the other hand, the median survival of the 4 patients with tumor and onconeuronal or SOX1 antibodies (cases 5, 7, 15, and 18) was 52 weeks (range 5–123 weeks; all died), while that of the 10 patients with tumor but without additional onconeuronal antibodies was not met (median follow-up 61 weeks, range 10–266 weeks; 9 of 10 alive at last follow-up; p = 0.009; figure 3B).
Figure 3. Survival of patients with paraneoplastic or idiopathic anti-AMPAR encephalitis.
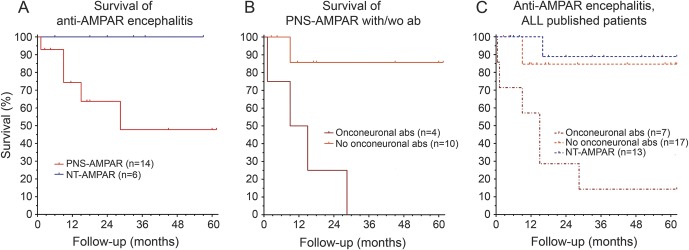
(A) Kaplan-Meier survival curves for patients with paraneoplastic anti–α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) encephalitis (with or without onconeuronal antibodies, red) and patients with idiopathic anti-AMPAR encephalitis (blue). (B) Kaplan-Meier survival curves for 4 patients with paraneoplastic anti-AMPAR encephalitis plus additional onconeural antibodies (dark red) and 10 patients with paraneoplastic anti-AMPAR encephalitis without onconeuronal antibodies (orange). (C) Kaplan-Meier survival curves for all assessable reported patients with paraneoplastic anti-AMPAR encephalitis and onconeuronal antibodies (dark red), paraneoplastic anti-AMPAR encephalitis without onconeuronal antibodies (orange), and idiopathic anti-AMPAR encephalitis (blue).2,5,6,8
In order to further assess the significance of the presence of an underlying tumor and onconeuronal antibodies in patient outcome, we reviewed the data of all oncologic cases of the current study together with those previously reported.2,5,6,8 Of 24 patients identified, 7 had onconeuronal, GAD65, or SOX1 antibodies and 17 had no additional antibodies. Six out of 7 patients with paraneoplastic syndromes and additional antibodies have died (median survival 65 weeks); in contrast, 15 of 17 patients with tumor but without additional onconeuronal antibodies were alive (median survival not met; median follow-up 60 weeks, range 10–516 weeks; p = 0.003). Eleven out of 13 patients without tumor are alive (median follow-up 104 weeks; range 25–390), which was not significant compared to patients with tumor (p = 0.079; figure 3C).
To determine whether clinical relapses were associated with less aggressive therapy, we reviewed the data of 21 patients of the current study and those of 16 previously reported cases for which information was available.2,5,6,8 Overall, relapses occurred in 6/37 patients (16%, 1 case in the current study). While 0/19 patients who received aggressive therapy (chemotherapy or rituximab) had relapses, 6/18 who did not receive aggressive therapy had relapses (p = 0.008).
DISCUSSION
We report 22 newly identified patients with anti-AMPAR encephalitis and provide several clinically relevant findings, including (1) the frequent presentation of the disorder as LE, sometimes with prominent psychiatric features, psychosis, or hyponatremia that can mislead the initial diagnosis; (2) the presence of an underlying tumor in 64% of the patients; (3) the coexistence of onconeuronal and cell surface autoantibodies in 32% of the patients, all with symptoms or tumors that reflect the concurrent autoimmunity; and (4) the frequent but limited response to treatment, with long-term outcome influenced by the presence of onconeuronal antibodies and related paraneoplastic symptoms or tumors.
The term LE refers to an inflammatory process of the brain restricted clinically or radiologically to the limbic system, which includes the medial temporal lobes and frontobasal and cingular regions. Despite a well-defined syndrome, it is frequently poorly recognized, misnaming as LE any type of autoimmune or paraneoplastic process above the foramen magnum. Among the many antigens of paraneoplastic and autoimmune encephalitis, there are 3 that typically associate with LE, including LGI1, GABA(B)R, and AMPAR.2,13,16–18 At least 60% of the patients with any of these disorders develop a typical clinical or radiologic picture of LE, with additional symptoms or associations that can suggest the antigen. For example, while LE with LGI1 antibodies frequently associates with hyponatremia but almost never SCLC,13 LE with GABA(B)R frequently occurs with early and prominent seizures and 50%–60% of the patients have SCLC.16,18 Recognition of each autoantigen is important because all 3 disorders are potentially treatable but for the appropriate treatment one should consider the associated comorbidities or tumors (further discussed later). Our findings show that LE with hyponatremia not only occurs in patients with LGI1, but also in some patients with antibodies to AMPAR or GABA(B)R, either as part of SCLC-related syndrome of inappropriate antidiuretic hormone secretion (which was not further investigated in 2 of our patients) or as a primary effect of the immune response (one patient did not have cancer).
In the initial report of 10 patients with anti-AMPAR encephalitis, the main findings were that all developed LE, 7 had a tumor (lung, breast, or thymoma), and the disorder frequently responded to treatment but had tendency to relapse. The current data confirm some of these findings and expand on others. Ten of the current patients did not initially present with classical LE, although most of them had limbic dysfunction concurrent or heralded by other symptoms. Importantly, 6 of these patients had prominent psychiatric symptoms, one of them manifesting as pure psychosis for 1 week before developing a neuroleptic malignant syndrome induced by treatment along with mild additional neurologic symptoms. The presentation of anti-AMPAR encephalitis as pure psychosis (without additional neurologic symptoms) has been previously reported in 2 cases,6 indicating that this type of encephalitis should be included in the differential diagnosis of autoimmune psychosis.
The histologic types of tumors were similar to those previously reported in anti-AMPAR encephalitis,2 but 2 patients had ovarian teratoma (one without evidence of NMDAR antibodies), an association not previously reported. We do not know why patients in the current study had fewer neurologic relapses than those in previous studies (1/21 vs 5/16)2,5,6,8; however, the approach to therapy may be a reason. Indeed, when considering all current and previous cases for which treatment information is available (n = 37), all clinical relapses were in the group who did not receive aggressive therapy (6 of 18 patients, p = 0.008). Aggressive treatment has been associated with lower relapse risk in anti-NMDAR encephalitis.19 Another possible explanation could be a diagnosis bias: at the time of the initial study, patients with monophasic disease were often not sent for antibody testing and were missed, resulting in an overrepresentation of relapsing patients.
Overall, 71% of the patients responded to immunotherapy or treatment of the tumor, most of them showing a partial neurologic response (48%). These data suggest that patients with AMPAR antibodies have less substantial recoveries than those with other types of autoimmune encephalitis [NMDAR, LGI1, or GABA(B)R]. Despite this, substantial and sometimes unexpected recoveries do occur, but require aggressive therapy. An example is patient 13, who presented with severe encephalitis, refractory seizures, and a large thymoma, but his Karnofsky Performance Status was considered too low (30/100) for treatment. The patient was transferred to our institution (Hospital Clinic, Barcelona) under pharmacologically induced coma and mechanical ventilation. The brain MRI showed changes suggestive of widespread cortical damage (figure 2). He underwent intensive immunotherapy and tumor removal, and currently is at home, free of seizures, and rapidly improving. This case suggests caution in clinically assessing autoimmune encephalitis using Karnofsky or similar neurologic scales for treatment decisions.
The presence of concurrent antibodies, mainly onconeuronal (CRMP5, amphiphysin) or tumor biomarkers linked to paraneoplastic autoimmunity (e.g., SOX1), is associated with additional paraneoplastic symptoms and a poorer prognosis. The 5 patients who died had one or more of these features along with an underlying tumor. These findings along with the review of previously reported cases with tumor (either with or without an additional paraneoplastic neurologic autoimmunity)2,5,6,8 suggest that the presence of an additional paraneoplastic autoimmunity is the main prognostic factor for a poor outcome. Indeed, the survival of patients with tumor but without additional paraneoplastic autoimmunity was similar to that of patients without tumor, but the survival of patients with tumor and additional paraneoplastic autoimmunity was significantly worse than that of the other subgroups. Similar comorbidities linked to a poor prognosis have been reported in patients with anti-GABA(B)R encephalitis, a disorder that frequently occurs with SCLC and may associate with concurrent paraneoplastic immune responses (e.g., amphiphysin, Ri, SOX1).18
This and previous studies suggest an interesting molecular link between the symptoms of LE and the effects of LGI1 and AMPAR antibodies. Indeed, LGI1 is a secreted neuronal protein that forms a trans-synaptic complex interacting at the postsynapse with ADAM22 and AMPAR and at the presynapse with ADAM23 and the shaker potassium channel Kv1.20 There is evidence that LGI1 antibodies prevent the binding of LGI1 to ADAM22 and by unclear mechanisms result in a decrease of AMPAR (the effects on the presynapse have not been determined).21 On the other hand, the antibodies of patients with anti-AMPAR encephalitis bind directly to the GluA1/2 subunits, causing a decrease of the levels of receptors by antibody-mediated internalization.2 Therefore, 2 seemingly different immune responses against different synaptic proteins result in a common downstream effect (decrease of postsynaptic AMPAR clusters), providing a potential explanation for the frequent association of both disorders with classical LE.
The current findings taken together with those of previous studies have several practical implications. First, in patients with classic LE with or without tumor association, determination of AMPAR antibodies should be considered. These antibodies may also occur in patients with multifocal encephalitis usually involving the limbic system, and less frequently can present as pure psychosis. Second, detection of GluA1/2 AMPAR antibodies should lead to an extensive screening, including underlying tumors, classical paraneoplastic antibodies, and other cell surface antibodies. Third, prompt treatment of the tumor and immunotherapy are important, because the disorder is potentially reversible, but the outcome seems related to the presence of concurrent paraneoplastic autoimmunity linked to an underlying tumor.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Mercè Alba, Eva Caballero, Esther Aguilar, and Esther Hulsenboom for technical support.
GLOSSARY
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- FLAIR
fluid-attenuated inversion recovery
- IQR
interquartile range
- LE
limbic encephalitis
- mRS
modified Rankin Scale score
- SCLC
small cell lung cancer
Footnotes
Supplemental data at Neurology.org
Editorial, page 2390
AUTHOR CONTRIBUTIONS
Design/conceptualization of the study: R.H., M.T., F.G., J.D.; analysis/interpretation of the data: R.H., A.v.S., F.L., M.G., J.G., M.P., D.H., L.S., A.S., M.T., F.G., J.D.; statistical analysis: M.T.; drafting/revising the manuscript: R.H., F.L., M.T., F.G., J.D. All authors give final approval of the version to be published.
STUDY FUNDING
Supported in part by grant PI12/00611 from the Fondo de Investigaciones Sanitarias, Madrid, Spain (F.G.), NIH RO1NS077851 (J.D.), RO1MH094741 (J.D.), Instituto Carlos III (FIS, Spain, 14/00203, J.D.), and Fundació la Marató de TV3 (J.D.). J.D. receives royalties from Athena Diagnostics for a patent for the use of Ma2 as autoantibody test, and licensing fees from Euroimmun for a patent for the use of NMDAR as autoantibody test. M.T. was supported by an ErasmusMC fellowship, by the Netherlands Organisation for Scientific Research (NWO, Veni incentive), and by the Dutch Epilepsy Foundation, project number 14-19.
DISCLOSURE
R. Höftberger, A. van Sonderen, F. Leypoldt, D. Houghton, M. Geschwind, J. Gelfand, M. Paredes, L. Sabater, A. Saiz, and F. Graus report no disclosures relevant to the manuscript. M. Titulaer received a travel grant for lecturing in India from Sun Pharma. J. Dalmau has a research grant from Euroimmun, and receives royalties from patents for the use of Ma2, GABA(B)R, and NMDAR as autoantibody tests. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sprengel R. Role of AMPA receptors in synaptic plasticity. Cell Tissue Res 2006;326:447–455. [DOI] [PubMed] [Google Scholar]
- 2.Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-AMPA receptor encephalitis antibodies. Ann Neurol 2015;77:381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gleichman AJ, Panzer JA, Baumann BH, Dalmau J, Lynch DR. Antigenic and mechanistic characterization of anti-AMPA receptor encephalitis. Ann Clin Transl Neurol 2014;1:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bataller L, Galiano R, Garcia-Escrig M, et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology 2010;74:265–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graus F, Boronat A, Xifro X, et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology 2010;74:857–859. [DOI] [PubMed] [Google Scholar]
- 7.Wei YC, Liu CH, Lin JJ, et al. Rapid progression and brain atrophy in anti-AMPA receptor encephalitis. J Neuroimmunol 2013;261:129–133. [DOI] [PubMed] [Google Scholar]
- 8.Dogan Onugoren M, Deuretzbacher D, Haensch CA, et al. Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J Neurol Neurosurg Psychiatry Epub 2014 Oct 9. [DOI] [PubMed]
- 9.Spatola M, Stojanova V, Prior JO, Dalmau J, Rossetti AO. Serial brain 18FDG-PET in anti-AMPA receptor limbic encephalitis. J Neuroimmunol 2014;271:53–55. [DOI] [PubMed] [Google Scholar]
- 10.Dalmau J, Gultekin SH, Voltz R, et al. Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain 1999;122:27–39. [DOI] [PubMed] [Google Scholar]
- 11.Hoftberger R, Dalmau J, Graus F. Clinical neuropathology practice guide 5-2012: updated guideline for the diagnosis of antineuronal antibodies. Clin Neuropathol 2012;31:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol 2010;257:509–517. [DOI] [PubMed] [Google Scholar]
- 13.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukata Y, Lovero KL, Iwanaga T, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci USA 2010;107:3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 2013;33:18161–18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.