

Abstract
Transdermal delivery of therapeutics is restricted by narrow limitations on size and hydrophobicity. Nanotopography has been shown to significantly enhance high molecular weight paracellular transport in vitro. Herein, we demonstrate for the first time that nanotopography applied to microneedles significantly enhances transdermal delivery of etanercept, a 150 kD therapeutic, in both rats and rabbits. We further show that this effect is mediated by remodeling of the tight junction proteins initiated via integrin binding to the nanotopography, followed by phosphorylation of myosin light chain (MLC) and activation of the actomyosin complex, which in turn increase paracellular permeability.
Keywords: Nanotopography, transdermal, drug delivery, etanercept, integrin
Skin serves as the critical boundary between the external world and our viscera. The stringency of the epidermal barrier is reflected by the dearth of transdermally delivered drugs; currently only 17 drugs have been approved for this delivery route by the Food and Drug Administration (FDA), and all of these agents fall within a very narrow parameter space (<500 Da in size and highly lipophilic). The limitations of transdermal technology appear even starker with the increasingly frequent development of high molecular weight, antibody-based therapeutics, many of which have promising biologic effects but are hampered by limited delivery options.
Until recently, skin barrier function was thought to be mediated entirely by the outermost stratum corneum, a defined meshwork of anucleate keratinocytes, lipids, and proteins. However, recent studies have discovered the presence of the epidermal tight junction (TJ) and multiple TJ proteins, including occludin, zonula occludins-1 (ZO-1), and in particular claudins, which are critical for epidermal barrier function.1–8 TJ complexes function as the paracellular barrier in simple epithelial cell types such as gastrointestinal mucosal cells and offer an additional well-conserved mechanism through which the skin can regulate its permeability.9–11 While multiple technologies have attempted to increase the permeability of the stratum corneum, less emphasis has been placed on modulation of the skin TJ barrier.12–15 In a recent study, Kam et al. demonstrated that nanotopography loosens tight junctions in simple epithelia and dramatically increases transepithelial transport of etanercept, a high molecular weight therapeutic that is used clinically for rheumatoid arthritis (RA) and severe psoriasis and psoriatic arthritis.16 However, the extent to which nanotopography loosens tight junctions in more stratified squamous epithelium such as skin or in vivo is not known.
In this study, we explore nanotopography as a novel strategy to enhance transdermal drug delivery. In both rat and rabbit animal models, we demonstrate for the first time that the addition of nanotopography dramatically enhances transdermal delivery of etanercept through microneedle arrays (MNAs). To explore the molecular mechanisms underlying this phenomenon, we examined TJ protein expression in cultured human primary multilayered keratinocytes and found that levels of claudin-1 and claudin-4 are significantly and reversibly reduced with nanotopography. This phenomenon is conserved in intestinal epithelial Caco-2 cells and furthermore is dependent on upstream integrin binding and MLC phosphorylation. These findings demonstrate that nanotopographic surfaces provide a new approach to significantly expand the scope of drugs that can be administered transdermally, including agents with a size range that includes the emerging and expanding class of antibody-based therapeutics.
The effect of nanotopography on microneedle-based transdermal delivery of etanercept was assessed in vivo in both rabbits and rats. Transdermal devices consisting of two different permeability enhancing components were fabricated. The first component was a 25 mm by 25 mm array of microneedles (Figure 1a). Each microneedle on this array was 290 μm in length and 100 μm wide, with a pitch of 400 μm; drug flowed out of each microneedle via longitudinal grooves along its sides. The second component was a polymeric film with nanostructures imprinted onto its surface (Figure 1a, inset). This film was draped over the microneedle array (MNA), effectively giving each needle a nanostructured coating. Depth of penetration of the MNAs was assessed in rats by soaking the skin samples in methylene blue following application of the MNA. The skin was then cryosectioned, and the depth of dye penetration was measured for each point in the array. The average depth of penetration in rat skin was 58 μm, placing the needles at the epidermal–dermal junction17 (Figure S1).
Figure 1.

Nanotopography significantly enhances transdermal delivery of etanercept in vivo. (a) Schematic representation of the transdermal delivery devices used in this study. Progressing from top to bottom: each device is made of an impermeable backing (tan), drug reservoir (green), rate-controlling membrane (yellow), and silicon microneedle array (MNA) (gray). Each 290 μm long, 100 μm wide microneedle has longitudinal perforations along the side, through which drug flows out. Drug flow from the reservoir down the grooves of the microneedles is indicated by a green dashed arrow. Perforations are denoted with a white arrowhead. “Smooth” microneedles were not coated with a film, while “nanostructured” microneedles were coated with a nanostructured film (scale bar represents 300 μm). Inset on furthest right depicts an SEM image of the nanostructures coated onto each microneedle (scale bar = 3 μm). (b) Nanostructured MNAs deliver significantly more etanercept transdermally than either drug reservoir patch alone or unstructured, smooth MNA controls in rats. After 72 h, the nanostructured MNA cumulatively delivered 10.6 times more etanercept (*p < 0.01) and achieved a maximal serum concentration (Cmax) 13.9 times higher (*p < 0.01) than the smooth microneedles (n = 4 animals for each category). (c) Nanostructured MNAs deliver significantly more etanercept transdermally than unstructured, smooth MNA controls in rabbits. After 72 h, the nanostructured MNA devices cumulatively delivered 35 times more etanercept (*p < 0.01) and achieved a Cmax 10.2 times higher than smooth MNA controls (*p < 0.01) (n = 4 animals for each category).
For drug delivery characterization, 12.5 mg of etanercept (0.25 mL at 50 mg/mL) was administered transdermally to rats, or 1.85 mg was administered to rabbits through one of three devices: the drug reservoir alone; an MNA array with no nanostructured coating (“smooth”); or an MNA array coated with nanostructured film (Figure 1b,c). The serum concentration of etanercept via each delivery route was then measured over 72 h. After 72 h in rats, the nanostructured MNA cumulatively delivered 10.6 times more etanercept (p < 0.01) and achieved a maximal serum concentration (Cmax) 13.9 times higher (p < 0.01) than the unstructured, smooth microneedles. In rabbits, the nanostructured MNA devices cumulatively delivered 35 times more etanercept (p < 0.01) and achieved a Cmax 10.2 times higher than unstructured, smooth MNA controls (p < 0.01). Regarding the kinetics of drug delivery, the time to maximal serum concentration (Tmax) in rabbits was equivalent between nanostructured and smooth MNAs, while in rats the Tmax was shorter for the nanostructured MNAs (30 h) than with the smooth MNAs (42 h). Examination of the skin following application of the devices showed minimal erythema and inflammation after 72 h (Figure S2).
To explore the molecular mechanisms through which nanotopography increases epidermal permeability, we directly exposed primary human cultured keratinocytes to the nanostructured coating. This culture system forms stratified layers and tight junctions that recapitulate skin development in vivo.1 Keratinocytes were allowed to develop to day 8, at which point they develop only a minimal stratum corneum layer. We elected this time point in order to provide a differentiated cell system that still enables maximal contact of the nanostructured film to the epidermal TJs, which lie beneath the stratum corneum layer of skin. After 24 h of exposure to the nanotopography, keratinocytes showed marked downregulation of the TJ proteins claudins-1 and -4, compared to control keratinocytes that were either unexposed or placed in contact with unstructured, flat control film of the same material (p < 0.01) (Figure 2a,b).
Figure 2.

Nanotopography leads to reversible downregulation of claudin-1 and -4 expression in cultured human keratinocytes. (a,b) Day 8 primary human keratinocytes showed marked diminishment in claudin-1 and -4 expression after 24 h incubation with nanotopography, compared to controls that were exposed to no device or to unstructured, flat films. Claudin-1 was decreased by 58% with polyether ether ketone (PEEK)-based nanostructured films (NS PEEK) and by 36% with polypropylene-based nanostructured films (NS PP), relative to no device controls (**p < 0.01). Claudin-4 was decreased by 49% with PEEK nanostructured films and by 39% with polypropylene nanostructured films, relative to no device controls (**p < 0.01). Exposure to unstructured, flat films (Flat PP) had no effect on claudin-1 or -4 expression relative to no device controls (scale bar = 10 μm). Fluorescence intensity was normalized by area. (c,d) Twenty four hours following removal of nanotopography, the decrease in claudin-1 and -4 expression was reversed, and there was no statistically significant difference between nanotopography-treated keratinocytes (NS PP and NS PEEK) versus no device or unstructured, flat controls (scale bar = 10 μm).
To assess whether this effect on claudins was reversible, we removed nanotopography for 24 h after exposure and again assessed for claudin-1 and -4 protein expression. After removal of the device, claudin-1 and -4 levels were equivalent in keratinocytes alone, keratinocytes exposed to unstructured control films, and keratinocytes exposed to nanotopography, suggesting that alterations in TJ morphology by nanotopography are reversible (Figure 2c,d).
To explore whether down regulation of claudins in keratinocytes is a robust and well-conserved mechanism, we performed analogous experiments in Caco-2 epithelial cells cultured on transwell permeable supports. Cells were either untreated or placed in contact with either an unstructured control film or a nanostructured film. Similar to keratinocytes, staining for claudins-1 and -4 in Caco-2 cells showed reduced localization at cell–cell junctions when cells were in contact with the nanostructures, in comparison to either the cells alone or cells in contact with an unstructured film, with claudin-1 being reduced by the greatest extent (Figure 3a). In contrast to claudins-1 and -4, immunostaining of the TJ protein occludin was preserved. However, instead of the stereotypical cobblestone pattern demonstrated by the control cells, occludin staining demonstrated a ruffled pattern when cells were exposed to nanostructured films, a pattern that has previously been reported with disruption of other TJ proteins such as ZO-116 (Figure 3a).
Figure 3.

Nanotopography-induced disruption of TJ structure is conserved among different epithelia. (a,b) In Caco-2 cells, nanostructured films (NS) induce decreased expression of claudin-1 and -4 at cell borders, relative to controls exposed to no film or to unstructured, flat film (Flat). Claudin-1 was decreased 50% compared to no film control and 73% compared to the flat film control (**p < 0.01). Claudin-4 was decreased 76% and 66% compared with the no film or flat film controls, respectively (**p < 0.01). Occludin immunostaining reveals a ruffled pattern when Caco-2 cells are in contact with the nanostructured film, in contrast to the stereotypical cobblestone pattern in control cells exposed to no film or unstructured, flat film (scale bar = 20 μm). Occludin ruffling, as measured by cell perimeter, was increased with nanotopography by 87% over cells alone and 118% over flat (**p < 0.01). Fluorescence intensity was normalized by number of cells. (c) TEM imaging of the tight junction (TJ) (solid black arrow), adherens junction (AJ) (dashed black arrow), and desmosome (solid white arrow). Cells exposed to unstructured, flat films (Flat) show a partial loss of electron density in the TJ and AJ, relative to no film controls. Cells exposed to nanostructured films (NS) show near complete loss of electron density in the TJ and AJ, as well as partial loss in the underlying desmosome, relative to no film controls (scale bar = 200 nm).
To further explore the structural effect of nanotopography on TJs, we characterized TJ structure in Caco-2 cells by transmission electron microscopy (TEM) (Figure 3c). Cells in contact with no film showed canonical cellular junction morphology, consisting of an apically located electron-dense TJ and a subjacent adherens junction (AJ) and desmosome. Cells in contact with the unstructured, flat film showed partially decreased electron density in both the TJ and AJ, as well as blurring of the boundaries between these two types of junctions. In cells treated with nanostructured film, the electron density of both the TJ and AJ were completely abrogated, suggesting significant loss of proteins and cytoskeleton within these complexes in response to nanotopography. In addition, there is loss of intermediate filaments near the desmosome with nanotopography. These data collectively demonstrate that nanotopography induces dramatic remodeling and diminishment of epithelial TJs, as well as other cell–cell adhesions.
To explain the structural changes in TJs discussed above, we hypothesized that epithelial cell interaction with nanotopography mediates changes in the actin cytoskeleton underlying TJs. It is well established that cells can bind to nanotopography,18–20 typically by recognizing conserved binding sequences in proteins absorbed onto the biomaterial.21 This protein recognition is most often mediated by integrins, transmembrane cell surface receptors that serve as a link between the ECM and the actin cytoskeleton.22 Integrin binding triggers a phosphorylation cascade that recruits and assembles a focal adhesion complex and culminates in phosphorylation and activation of myosin.23 Activation of myosin, in turn, alters the actin cytoskeleton, which can lead to increased paracellular permeability.24,25
Immunofluorescence was used to investigate the effects of nanotopography on focal adhesion complexes and myosin activation within cells. Cells in contact with the nanostructures showed increased punctate staining of phosphorylated focal adhesion kinase (pFAK), which is phosphorylated upon integrin-ligand binding and subsequently recruits other components of the adhesion complex26 (Figure 4a). Consistent with this increase in pFAK, clustering of vinculin, another downstream component of the focal adhesion complex, was also increased in response to nanotopography (Figure 4b). To assess whether these increases in focal adhesion proteins were associated with myosin activation, immunostaining for phosphorylated myosin light chain was performed. As with pFAK and vinculin, pMLC clustering was increased exclusively in cells exposed to the nanostructured films (Figure 4c). These data collectively demonstrate that integrin-binding to nanotopography induces formation of focal adhesion complexes and activation of actomyosin contractility.
Figure 4.

Nanotopography leads to clustering of focal adhesion proteins and activation of myosin light chain. (a–c) In Caco-2 cells, exposure to the nanostructured film (NS) leads to increased staining of pFAK, vinculin, and pMLC, relative to controls cells exposed to either no film or to flat, unstructured film (Flat) (scale bar = 20 μm). Nuclear staining is highlighted by DAPI in blue, while protein immunostaining is highlighted in green. (d–f) pFAK was increased by 112% and 43% compared with the no film or flat film controls, respectively (**p < 0.01). Vinculin was increased by 117% and 91% compared with the no film or flat film controls, respectively (**p < 0.01). pFAK was increased by 263% and 161% compared with the no film or flat film controls, respectively (**p < 0.01).
To investigate whether integrin engagement with the film and subsequent focal adhesion complex formation is suficient for drug transport facilitated by nanotopography, we first treated cells with RGD peptide, which is the recognition sequence for some integrins and can artificially induce integrin clustering.27 RGD treatment of the cells was found to fully mimic the drug-enhancing effects of nanotopography and nullified the previously observed differences in drug delivery between unstructured and nanostructured films (Figure 5a).
Figure 5.

Nanotopography-mediated drug delivery and down-regulation of TJs requires integrin activation and phosphorylation of myosin light chain. (a) The integrin-binding RGDS peptide is suficient to recapitulate the effect of nanotopography on Caco-2 permeability. As expected, cells exposed to nanostructured films without RGDS (open diamond) showed decreased transepithelial electrical resistance (TEER) relative to controls exposed to flat, unstructured film without RGDS (solid square). The addition of RGDS led to diminishment of TEER in both nanostructured and flat film groups, abrogating the previous difference observed in the absence of RGDS (n = 4). (b) Integrin function is necessary for nanotopography-mediated drug transport. In the absence of integrin blockade, nanostructured films (NS) significantly increase transport of FITC-BSA (left panel) (*p < 0.05). In the presence of integrin blockade, this nanotopography-induced increase in BSA transport is abolished, and there are no differences between the nanotopography and unstructured, flat film groups (n = 4). (c) Myosin light chain kinase (MLCK) function is necessary for nanotopography-mediated drug transport. In the absence of MLCK blockade, nanostructured films (NS) significantly increase transport of FITC-BSA (left panel) (*p < 0.05). In the presence of MLCK blockade, this nanotopography-induced increase in BSA transport is abolished, and there are no differences between the nanotopography and unstructured, flat film groups (n = 4). (d) Myosin light chain kinase (MLCK) function is necessary for nanotopography-mediated changes in TJ structure. ZO-1 immunostaining adopts a ruffled pattern in Caco2 cells exposed to nanostructured films (NS), indicating a disruption of the TJ complex. After addition of MLCK inhibitor, this ruffling is abolished, and cells exposed to nanotopography resume the canonical ZO-1 pattern shown in control cells exposed to either no film or flat, unstructured films.
To explore whether integrins were necessary for nanotopography-induced TJ remodeling, we used antibody blockade to prevent integrin binding and downstream clustering of focal adhesions. Specifically, integrins alpha V and beta 1 were Specifically inhibited because these integrins are common to the majority of RGD sequence binding integrin pairs.28 As expected, nanotopography significantly enhanced drug delivery in the absence of integrin blockade. With integrin blockade, however, this finding was completely abrogated, with no difference in drug transport between the nanostructured film and the unstructured films (Figure 5b). Therefore, integrin binding appears to be both suficient and necessary for nanotopography-mediated enhancement of drug delivery.
To assess whether downstream myosin phosphorylation and activation is also necessary for nanotopography-induced changes, cells were treated with permeant inhibitor of myosin light chain kinase (PIK). As with inhibition of integrin clustering, inhibition of myosin phosphorylation abolished the enhancement of drug delivery by nanotopography (Figure 5c). To investigate whether myosin phosphorylation was also required for nanotopography-induced changes in TJ structure, cells were treated with PIK, and TJ morphology was assessed by immunostaining for ZO-1. Treatment with PIK abolished the previously observed ruffling pattern of ZO-1 when cells were placed in contact with the nanostructured film (Figure 5d). Together, the above functional studies demonstrate that both integrin and myosin activation are requisite for nanotopography-mediated remodeling of the epithelial TJ and increase in epithelial permeability.
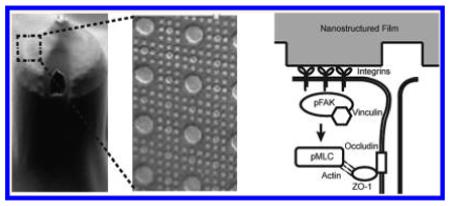
Materials with controlled nanotopography have recently been explored in the drug delivery field. Other groups have shown that nanotopography can increase drug delivery via endocytosis.29,30 We have shown previously that a purely topographical cue can significantly increase the transport of high molecular weight molecules across a simple epithelial monolayer by paracellular transport.16 In this study, we show for the first time that nanotopography significantly enhances transdermal delivery of a high molecular weight therapeutic in vivo. To better understand these phenomena, we have identified a molecular mechanism in which nanotopography binds to cell surface integrins, induces clustering of focal adhesion proteins and actomyosin activation, and ultimately remodels components of the tight junction barrier to increase paracellular permeability (Figure 6).
Figure 6.

Nanotopography downregulates epidermal tight junctions via integrin activation and myosin light chain phosphorylation. Proposed mechanism for nanotopography-mediated enhancement of transdermal drug delivery: Integrins bind to adsorbed proteins on the nanotopography and induce downstream clustering of focal adhesion proteins such as pFAK and vinculin. Formation of focal adhesions, in turn, leads to phosphorylation of myosin light chain and increased myosin contraction of the actin cytoskeleton. Actin dynamics ultimately remodel and downregulate proteins at the epidermal tight junction, thereby allowing increased paracellular transport within the epidermis. By contrast, we propose that the lack of topography on the flat surfaces fails to induce integrin clustering and the downstream initiation of focal adhesions and actomyosin activation (right lower image).
What specific characteristics of the nanotopography are driving these effects? Although all of the nanostructured films we tested in this study effectively remodeled tight junctions, we found material-dependent differences between PEEK and polypropylene in human multilayered keratinocytes, with PEEK inducing more downregulation of claudins-1 and -4. As flat controls for both materials did not induce any changes in tight junctions, this material difference is unlikely to be due to chemical effects on the keratinocytes. Rather, the discrepancy is more likely attributable to differences in pliability and stiffness of the material, leading to differences in tension experienced within the cells. Other variables that could potentially have effects on drug delivery and be variably tuned include the aspect ratio of the fibers, the pitch between different fibers, and the surface area of the nanostructures.31–36 It will be interesting in future studies to systematically define these parameter windows to maximize the biological effect of nanotopography. In addition to varying the parameters of the devices themselves, it would be valuable to explore lower molecular weight therapeutics as well, in order to delineate the relationship between molecular weight, topography, and delivery rate.
Although we demonstrated that focal adhesion clustering and myosin activation are necessary for nanotopography-mediated drug delivery, it would be valuable to explore other mechanisms that may be acting in parallel with this. In particular, cells that are not in direct contact with the nanotopography also demonstrate dramatic reductions in tight junction proteins, suggesting that there is paracellular signaling from upper cells of the stratum granulosum downward to the lower skin layers. This indirect effect on cells that are not in contact with the microneedles is further suggested by our in vivo rabbit data in which microneedles likely do not penetrate to the lower epidermis yet still deliver etanercept as effectively as in rats. Interestingly, preliminary mRNA expression data from our laboratory identify a cellular adhesion/polarity pathway that is downregulated in response to nanotopography. Key signaling molecules in the establishment of cell polarity, such as Tiam1, Rac1, and aPKC, are all suppressed in Caco-2 cells after exposure to nanotopography. Such loss of polarity could provide another mechanism through which nanotopography alters epithelial permeability. For example, nanotopography may be promoting dedifferentiation or trans-differentiation of epithelial cells, similar to what occurs in epithelial to mesenchymal transition (EMT).37,38
Regarding clinical usage, the impact of transdermally delivered biologic therapeutics such as etanercept on rheumatoid arthritis and psoriasis would be significant. Currently, medications within this family are administrated subcutaneously, intramuscularly, or intravenously, all of which are inconvenient and painful for the patient. Exchanging this current standard of care with a longer lasting transdermal device would likely improve patient compliance, as well as offer more biologically effective dosing with fewer peaks and troughs and less hepatic metabolism. Transdermal delivery also opens up the possibility of using biologics for localized, as opposed to systemic, therapy. For example, local topical agents for psoriasis are currently limited in scope, comprising primarily corticosteroids and vitamin D analogues. Being able to deliver small doses of the appropriate biologic locally to the skin would expand upon this repertoire significantly.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Jessica Allen, Dr. Jennifer Wade, and the UCSF Nikon Imaging Center. Funding for this work was kindly provided by the National Institutes of Health (R01-EB018842) and Kimberly-Clark Corporation. The authors would like to acknowledge the National Psoriasis Foundation (NPF) for their support of this project through an NPF Discovery Grant.
Footnotes
Supporting Information
Experimental procedures; histogram summarizing depth of microneedle penetration; and pre- and postdevice application images of rat skin. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Celli A, Zhai Y, Jiang YJ, Crumrine D, Elias PM, Feingold KR, Mauro TM. Exp. Dermatol. 2012;21:798–801. doi: 10.1111/j.1600-0625.2012.01573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kirschner N, Houdek P, Fromm M, Moll I, Brandner JM. Eur. J. Cell Biol. 2010;89:839–842. doi: 10.1016/j.ejcb.2010.07.010. [DOI] [PubMed] [Google Scholar]
- (3).Roussel AJJ, Knol AC, Bourdeau PJ, Bruet VJ. Comp. Pathol. 2014;150:35–46. doi: 10.1016/j.jcpa.2013.06.008. [DOI] [PubMed] [Google Scholar]
- (4).Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita SJ. Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. J. Biol. Chem. 1998;273:29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- (6).Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita SJ. Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sugawara T, Iwamoto N, Akashi M, Kojima T, Hisatsune J, Sugai M, Furuse MJ. Dermatol. Sci. 2013;70:12–18. doi: 10.1016/j.jdermsci.2013.01.002. [DOI] [PubMed] [Google Scholar]
- (8).Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita SJ. Cell Biol. 2002;156:1099–1111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Deli MA. Biochim. Biophys. Acta, Biomembr. 2009;1788:892–910. doi: 10.1016/j.bbamem.2008.09.016. [DOI] [PubMed] [Google Scholar]
- (10).Findley MK, Koval M. IUBMB Life. 2009;61:431–437. doi: 10.1002/iub.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tsukita S, Furuse MJ. Cell Biol. 2000;149:13–16. doi: 10.1083/jcb.149.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Broderick KE, Chan A, Lin F, Shen X, Kichaev G, Khan AS, Aubin J, Zimmermann TS, Sardesai NY. Mol. Ther.–Nucleic Acids. 2012;1:e11. doi: 10.1038/mtna.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Elsabahy M, Foldvari M. Curr. Pharm. Des. 2013;19:7301–7315. doi: 10.2174/13816128113199990369. [DOI] [PubMed] [Google Scholar]
- (14).Brown MB, Traynor MJ, Martin GP, Akomeah FK. Methods Mol. Biol. 2008;437:119–139. doi: 10.1007/978-1-59745-210-6_5. [DOI] [PubMed] [Google Scholar]
- (15).Rao R, Nanda SJ. Pharm. Pharmacol. 2009;61:689–705. doi: 10.1211/jpp.61.06.0001. [DOI] [PubMed] [Google Scholar]
- (16).Kam KR, Walsh LA, Bock SM, Koval M, Fischer KE, Ross RF, Desai TA. Nano Lett. 2013;13:164–171. doi: 10.1021/nl3037799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Godin B, Touitou E. Adv. Drug Delivery Rev. 2007;59:1152–1161. doi: 10.1016/j.addr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- (18).Dalby MJ, Riehle MO, Sutherland DS, Agheli H, Curtis ASG. Eur. J. Cell Biol. 2004;83:159–169. doi: 10.1078/0171-9335-00369. [DOI] [PubMed] [Google Scholar]
- (19).Dalby MJ. Med. Eng. Phys. 2005;27:730–742. doi: 10.1016/j.medengphy.2005.04.005. [DOI] [PubMed] [Google Scholar]
- (20).Dalby MJ. Int. J. Nanomed. 2007;2:373–381. [PMC free article] [PubMed] [Google Scholar]
- (21).Anselme K, Davidson P, Popa AM, Giazzon M, Liley M, Ploux L. Acta Biomater. 2010;6:3824–3846. doi: 10.1016/j.actbio.2010.04.001. [DOI] [PubMed] [Google Scholar]
- (22).Biggs MJP, Richards RG, Dalby MJ. Nanomed.-Nanotechnol. Biol. Med. 2010;6:619–633. doi: 10.1016/j.nano.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jockusch BM, Bubeck P, Giehl K, Kroemker M, Moschner J, Rothkegel M, Rudiger M, Schluter K, Stanke G, Winkler J. Annu. Rev. Cell Dev. Biol. 1995;11:379–416. doi: 10.1146/annurev.cb.11.110195.002115. [DOI] [PubMed] [Google Scholar]
- (24).Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. J. Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- (25).Tinsley JH, de Lanerolle P, Wilson E, Ma WY, Yuan SY. Am. J. Physiol. Physiol. 2000;279:C1285–C1289. doi: 10.1152/ajpcell.2000.279.4.C1285. [DOI] [PubMed] [Google Scholar]
- (26).Schaller M, Hildebrand J, Shannon J, Fox J, Vines R, Parsons J. Mol. Cell. Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. J. Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. Pt 10. [DOI] [PubMed] [Google Scholar]
- (28).Ruoslahti E. Annu. Rev. Cell Dev. Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- (29).Adler AF, Leong KW. Nano Today. 2010;5:553–569. doi: 10.1016/j.nantod.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dalby MJ, Berry CC, Riehle MO, Sutherland DS, Agheli H, Curtis AS. Exp. Cell Res. 2004;295:387–394. doi: 10.1016/j.yexcr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- (31).Sjöström T, Dalby MJ, Hart A, Tare R, Oreffo ROC, Su B. Acta Biomater. 2009;5:1433–1441. doi: 10.1016/j.actbio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- (32).Takagi J, Petre BM, Walz T, Springer TA. Cell. 2002;110:599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- (33).Kim D-H, Kim P, Suh K, Kyu Choi S, Ho Lee S, Kim B. IEEE Eng. Med. Biol. Soc. Conf. 2005;4:4091–4094. doi: 10.1109/IEMBS.2005.1615362. [DOI] [PubMed] [Google Scholar]
- (34).Lee J, Kang BS, Hicks B, Chancellor TF, Jr., Chu BH, Wang H-T, Keselowsky BG, Ren F, Lele TP. Biomaterials. 2008;29:3743–3749. doi: 10.1016/j.biomaterials.2008.05.029. [DOI] [PubMed] [Google Scholar]
- (35).Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Nat. Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- (36).Besser A, Safran SA. Biophys. J. 2006;90:3469–3484. doi: 10.1529/biophysj.105.074377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ikenouchi J, Matsuda M, Furuse M, Tsukita SJ. Cell Sci. 2003;116:1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- (38).Yang ZB, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R. Cancer Res. 2005;65:3179–3184. doi: 10.1158/0008-5472.CAN-04-3480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.