

Summary
P66, a B orrelia burgdorferi surface protein with porin and integrin‐binding activities, is essential for murine infection. The role of P66 integrin‐binding activity in B . burgdorferi infection was investigated and found to affect transendothelial migration. The role of integrin binding, specifically, was tested by mutation of two amino acids (D205A,D207A) or deletion of seven amino acids (Del202–208). Neither change affected surface localization or channel‐forming activity of P66, but both significantly reduced binding to αvβ3. Integrin‐binding deficient B . burgdorferi strains caused disseminated infection in mice at 4 weeks post‐subcutaneous inoculation, but bacterial burdens were significantly reduced in some tissues. Following intravenous inoculation, the Del202–208 bacteria were below the limit of detection in all tissues assessed at 2 weeks post‐inoculation, but bacterial burdens recovered to wild‐type levels at 4 weeks post‐inoculation. The delay in tissue colonization correlated with reduced migration of the Del202–208 strains across microvascular endothelial cells, similar to Δp66 bacteria. These results indicate that integrin binding by P66 is important to efficient dissemination of B . burgdorferi, which is critical to its ability to cause disease manifestations in incidental hosts and to its maintenance in the enzootic cycle.
Introduction
Borrelia burgdorferi, the causative agent of Lyme disease, routinely causes disseminated infection in immunocompetent hosts. Dissemination of B. burgdorferi throughout the host has long been thought to involve entry into the vasculature near the site of the tick bite and exit from the vasculature in distant tissues, and for brief periods bacteria can be found in the bloodstream. Three types of interactions within the microvasculature, designated tethering, dragging and stationary interactions, have been identified and modelled (Norman et al., 2008; Moriarty et al., 2012). Tethering interactions are brief contacts between the spirochete and the lumen of the vessel, taking less than 1 s to traverse 100 μm along the vessel wall, whereas dragging interactions are defined as a longer contact, 3–20 s to traverse 100 μm along the vessel wall, with the length of the spirochete generally contacting the vessel wall (Norman et al., 2008; Moriarty et al., 2012). Stationary adhesions typically occur at endothelial cell junctions and are thought to be required for eventual transmigration of the spirochete into the surrounding tissue (Norman et al., 2008; Moriarty et al., 2012). An outer surface protein of B. burgdorferi, BBK32, possesses two distinct adhesin functions that have been demonstrated to be involved in two distinct types of vascular interactions. BBK32‐fibronectin binding is required for tethering interactions and BBK32‐glycosaminoglycan (GAG) binding is involved in dragging interactions (Moriarty et al., 2012). Proteins required for stationary interactions or transmigration of the bacteria to escape from the vasculature and invade surrounding tissues have yet to be mechanistically identified.
P66 is an outer surface protein of B. burgdorferi that was identified in vitro as a ligand for β3 and at least some β1 integrins (Coburn et al., 1993; 1998; Behera et al., 2008) and as a porin (Skare et al., 1997; Pinne et al., 2007; Bárcena‐Uribarri et al., 2010). P66 is required for infectivity in mice (Ristow et al., 2012), but not ticks, consistent with its expression pattern (Cugini et al., 2003). Structural predictions as well as detergent phase partitioning and efflux of fluorescent dye from liposomes treated with P66 suggest formation of a transmembrane β‐barrel (Kenedy et al., 2014), although the crystal structure has not yet been solved. The Δp66 bacteria inoculated by subcutaneous or intradermal injection are cleared from the site of inoculation within 48 h post‐inoculation and cannot be recovered from any disseminated sites of infection even within short time periods after infection (Ristow et al., 2012). This rapid clearance suggests involvement of the innate immune system, but the infectivity of Δp66 bacteria was not rescued in mice deficient in either TLR2 or MyD88, signalling molecules known to participate in control of B. burgdorferi infection (Ristow et al., 2012). The numbers of monocytes, neutrophils and other cells of the host defence system at the inoculation site did not differ in mice inoculated with Δp66 bacteria as opposed to the wild‐type (WT) parental strain (Ristow et al., 2012), suggesting that the clearance of the Δp66 bacteria is mediated through host defences already in place, as opposed to defence cells that migrate to the site of infection. Microarray studies comparing endothelial cell responses to B. burgdorferi revealed an up‐regulation of the vascular endothelial growth factor/vascular permeability factor pathway in cells incubated with WT bacteria compared with those incubated with Δp66 bacteria (LaFrance et al., 2011). This result suggested that one function of P66 might be to facilitate bacterial transmigration across endothelial barriers.
Previous work identified a portion of P66 [amino acids (a.a.) 142–384] that contains all the sequences required for integrin‐binding activity, and a synthetic peptide corresponding to a.a. 203–209 within that region significantly competed with intact B. burgdorferi for binding to αIIbβ3 (Defoe and Coburn, 2001). No other peptides covering the region of a.a. 142–384 significantly or reproducibly inhibited integrin binding. A synthetic peptide representing a.a. 203–209 in content, but in scrambled order, also did not affect integrin binding of B. burgdorferi. Although P66 does not contain the traditional consensus arginine‐glycine‐aspartic acid (RGD) tripeptide sequence of mammalian integrin ligands, in which the aspartic acid residue is critical, a.a. 205 and 207 are both aspartic acids. Synthetic peptides generated with mutations of either a.a. 205 or 207 to alanine were unable to compete with intact B. burgdorferi for binding to αIIbβ3 (Defoe and Coburn, 2001). Maltose‐binding protein (MBP)‐P66 fusion proteins were constructed with mutations at a.a. 205 or 207 (LaFrance et al., 2011). A severe defect of MBP‐P66 fusion binding to purified αIIbβ3 was observed when a.a. 207 was mutated to alanine, and to a lesser extent, binding was reduced when a.a. 205 was mutated to alanine (LaFrance et al., 2011). As the tools to perform genetic mutations in B. burgdorferi advanced, disruption of p66 expression in a high‐passage, non‐infectious strain background of B. burgdorferi was performed by replacement of a portion of p66, including the putative integrin‐binding region, with a kanamycin resistance cassette (Coburn and Cugini, 2003). These mutants were defective in binding to a cell line stably transfected to express αvβ3 as compared with WT bacteria.
The goal of this work was to determine if the integrin‐binding function of P66 contributes to B. burgdorferi infection in vivo. We examined the role of P66 in transendothelial migration in vitro and in dissemination in vivo, and found that integrin binding by P66 is important to both activities of B. burgdorferi. The role of P66 in efficient dissemination is biologically significant in light of the natural transmission cycle required to maintain the bacteria in wildlife reservoirs, and is relevant to the development of disseminated disease in accidental hosts such as humans.
Results
Infectivity of WT or Δp66 bacteria is not altered in β3 chain integrin‐deficient mice
P66 has been shown to bind both β1 and β3 chain integrins in vitro (Coburn et al., 1993; 1998; Coburn and Cugini, 2003; Behera et al., 2008). To assess the importance of the interaction of P66 with β3 chain integrins in vivo, mice deficient in β3 chain integrin expression (β3 −/−) were utilized. Unfortunately, β1 integrin chain deficiency results in early embryonic lethality (Stephens et al., 1995), precluding testing of mice deficient in this subfamily of integrins. Although β3 −/− mice are viable, they have increased vascular permeability and prolonged bleeding times compared with WT mice due to defects in platelet aggregation (Hodivala‐Dilke et al., 1999). We hypothesized that if the interaction of P66 with β3 chain integrins was critical for B. burgdorferi infection and dissemination, WT B. burgdorferi would be non‐infectious in β3 −/− mice. Alternatively, if the interaction of P66 with β3 chain integrins serves to down‐regulate a negative host response to the bacteria, in the absence of β3 chain integrins, the presence of P66 should not be required. To test these hypotheses, the ID50 of B31‐A3 (WT) or Δp66 bacteria was determined in β3 +/+ or β3 −/− mice after subcutaneous inoculation at a range of doses. At 4 weeks post‐inoculation, the site of inoculation, heart, tibiotarsal joint and ear were collected for analysis by culture and qPCR. ID50 determinations were based on B. burgdorferi outgrowth in the cultures and were calculated as previously described (Supporting Information Table S1) (Reed and Muench, 1938). Wild‐type bacterial loads were similar in both β3 +/+ and β3 −/− mice, whereas Δp66 bacteria were undetectable in either mouse strain (Supporting Information Fig. S1). These results demonstrate that B. burgdorferi interaction with β3 chain integrins, specifically, is not critical to infection in mice.
Amino acids 202–208 of P66 are important for binding to β3 chain integrins but are not critical to porin function
Although the interaction of P66 with β3 chain integrins is not required for infection of mice, the function of P66 as an integrin ligand may still be critical to its role in the ability of B. burgdorferi to establish infection, given that the recombinant protein and the bacteria do bind to additional integrins. To further investigate the roles of the P66 sequences previously implicated in adhesion in vitro, recombinant MBP‐P66 fusion proteins were constructed with mutations in the putative integrin‐binding region at aspartic acid residues 205 and 207 to alanine (D205A,D207A), or a deletion of seven amino acids (Del202–208), spanning the critical portion of the putative integrin‐binding region (Fig. 1A).
Figure 1.
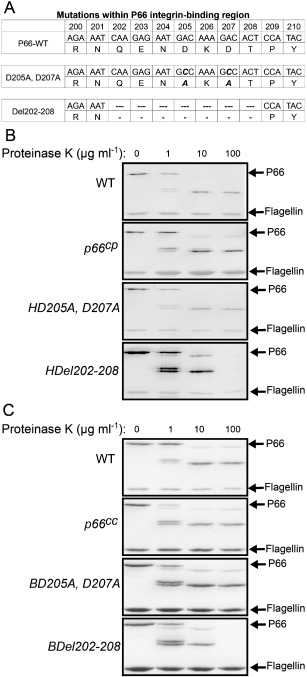
Genotype and phenotype of P66 integrin‐binding region mutants.
A. Nucleotide and amino acid sequence of WT, D205A,D207A and Del202–208 strains. Mutations in the nucleotide sequence that resulted in altered amino acids are in bold and italics for mutations at a.a. 205 and 207. Nucleotides removed from the coding region are represented as dashes for Del202–208.
B and C. Surface expression of P66 in various B orrelia burgdorferi strains (B: HB19 strains, C: B31‐A3 strains). The bacterial strains were incubated with varying concentrations of proteinase K (1, 10, 100 μg ml−1) for 1 h. Untreated bacteria were used as a negative control. Bacterial lysates were run on a 10% SDS‐PAGE gel and transferred to an Immobilon membrane that was probed with antibodies to P66 and flagellin. Colorimetric methods were used for visualization. cp = p66 gene restored on a shuttle plasmid, cc = p66 gene restored to endogenous locus on chromosome.
In parallel, the same mutations were introduced into non‐infectious (high‐passage HB19 clone 1) and infectious (B31‐A3) B. burgdorferi strain backgrounds. Borrelia burgdorferi HB19 clone 1 (HB19‐1) is a high‐passage, non‐infectious derivative of a clinical isolate of B. burgdorferi with significant loss of genomic elements compared with infectious strain backgrounds. This results in the expression of many fewer adhesins and lower background ELISA signals in adhesion assays compared with infectious strain backgrounds and is a useful tool for in vitro studies. Previous work had demonstrated that HB19‐1 Δp66 binds less efficiently than the parental strain to HEK293 cells stably expressing human integrin αvβ3 (293 + αvβ3) (Coburn and Cugini, 2003). Surface expression of the mutant forms of P66 in both the non‐infectious (HB19‐1, Fig. 1B) and infectious (B31‐A3, Fig. 1C) strain backgrounds was confirmed by Western blot after proteinase K treatment of the bacteria. Notably, both mutant forms were very similar to the WT form of P66 in terms of the fragments generated as determined by the banding pattern of the digested protein, with a doublet formed at approximately 54 and 55 kDa for the mutant as well as WT forms of the protein. The Del202–208 protein did however appear somewhat more sensitive to proteolysis, as the P66 bands were undetectable at the highest proteinase K concentration. Nevertheless, the proteinase K digestion of all P66 variants demonstrated that the protein was exposed on the surface of the bacteria. Flagellin was used as a control for intact bacteria, as the protein is periplasmic in B. burgdorferi, and therefore is not accessible to the protease in intact bacteria.
Although the mutations resulting in a substitution of aspartic acid to alanine at a.a. 205 and 207 are predicted to be relatively minor in terms of effects on overall protein structure, the deletion of seven amino acids to create the Del202–208 mutants has the potential to cause a significant change in the protein structure. To determine whether these mutations affected the other known function of P66, i.e. channel formation, a planar lipid bilayer assay was used to assess the ability of P66 purified from strains expressing targeted P66 mutants to assemble and form a functional porin. Previous work described the loss of the P66 channel‐forming activity (∼11 ns in 1 M KCl) in B. burgdorferi when outer membrane porins were purified from Δp66 bacteria compared with WT bacteria (Pinne et al., 2007). For this work, the native proteins were purified from the overexpressing, non‐infectious HB19 strain background for assessment. No differences were observed in the channel size between P66 with targeted mutations and WT P66 (Fig. 2), although fewer pores were formed by the D205A,D207A mutant. In addition, the activity of a different channel at ∼5 ns may have been affected in the D205A,D207A mutant. The reason for this is unclear, given that the deletion of a.a. 202–208 did not affect either the number of channels formed or the activity of the other former channel. However, both site‐directed mutant versions of P66 formed the 11 ns channels in the planar lipid bilayer system, demonstrating that the structures of the proteins were not grossly disrupted in the context of the B. burgdorferi outer membrane. These data are supported by the appearance of a band of ∼480 kDa that co‐migrated with the WT protein when the site‐directed mutant proteins were analysed by blue native gel electrophoresis (data not shown). Structural predictions using PRED‐TMBB (Bagos et al., 2004) did not suggest gross alterations of the protein due to the site‐directed changes, although definitive analyses of possible effects on the overall structure of the mutated versions of P66 await crystal structure determination.
Figure 2.
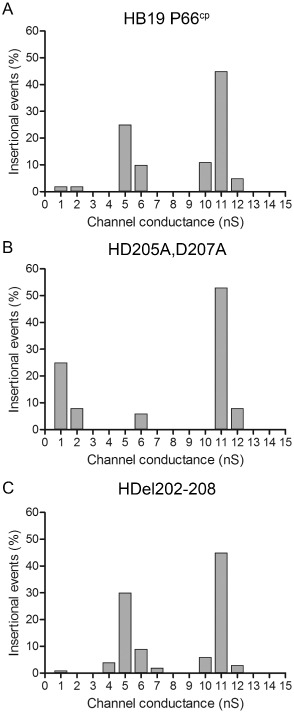
Targeted mutation of the integrin‐binding region of P66 does not affect pore formation. Histograms show pore‐forming activities of P66 isolated from B orrelia burgdorferi HB19 p66cp (A), HD 205A, D 207 A (B), or HDel202–208 (C) in a diphytanoyl phosphatidylcholine/n‐decane (PC) membrane. The bathing solution was 1 M KCl. The total number of insertional events was 100 for HB19 p66cp, 100 for HD el202–208 and 36 for HD 205A, D 207 A. The experiment was performed in triplicate for all strains, with one representative shown. The typical single channel conductance activity for P66 (11 nS) was observed in outer membrane protein preparations from all three B . burgdorferi strains.
The abilities of WT and mutant recombinant forms of the integrin‐binding domain of P66 (P66M) to bind αvβ3 were assessed in vitro using both purified αvβ3 and cells overproducing this integrin. The binding affinity (K D) of human integrin to WT MBP‐P66 was ∼1370‐fold greater than that of the Del202–208 protein and ∼225‐fold greater than that of the D205A,D207A protein as assessed by surface plasmon resonance (SPR) (Table 1). Similarly, the binding of mouse integrin to WT P66 indicated by K D values was ∼1320‐fold more efficient than that of the Del202–208 mutant and ∼100‐fold more efficient than that of the D205A,D207A protein (Table 1). However, there were no significant differences between mouse and human integrins in binding to the same recombinant P66 proteins (P = 0.92) (Table 1). Similar trends for the changes in K Ds were also seen using an ELISA‐based approach (data not shown). These results suggest that the D205A,D207A and the Del202–208 mutant proteins each had reduced binding to purified integrin αvβ3, and that the reduction in integrin binding to the D205A,D207A mutant protein was less severe than to the Del202–208 mutant protein.
Table 1.
MBP‐P66 site‐directed mutant proteins differ in binding to integrin αvβ3.
| MBP‐P66 variant | Ligand | K D (μM) | kon (103 s−1 M−1) | koff (10−1 s−1) |
|---|---|---|---|---|
| P66M | Human αvβ3 | 0.13 ± 0.06a | 321.33 ± 115.18 | 0.31 ± 0.05 |
| Mouse αvβ3 | 0.20 ± 0.04a | 111.56 ± 32.49 | 0.20 ± 0.01 | |
| D205A,D207A | Human αvβ3 | 29.28 ± 6.76a, b | 0.71 ± 0.18a | 0.19 ± 0.10a |
| Mouse αvβ3 | 20.34 ± 5.90a, b | 1.45 ± 0.39a | 0.25 ± 0.04a | |
| Del202–208 | Human αvβ3 | 178.33 ± 27.35a, b | 0.09 ± 0.01a | 0.17 ± 0.01a |
| Mouse αvβ3 | 264.29 ± 147.34a, b | 0.13 ± 0.06a | 0.18 ± 0.01a | |
| MBP‐βgal | Human αvβ3 | n.b.c | n.b.c | n.b.c |
| Mouse αvβ3 | n.b.c | n.b.c | n.b.c |
The K D values were obtained from the average of every koff divided by every kon from each run.
Because the binding of D205A,D207A or Del202–208 to mouse or human integrin αvβ3 is weak, the determined values should be considered estimates.
No binding activity was detected.
All values represent the mean ± SEM of three experiments.
To assess binding of the mutant forms of P66 to integrin αvβ3 in the context of intact host cells, confluent monolayers of 293 + αVβ3 cells were probed with the recombinant forms of P66. After unbound proteins were removed by washing, bound proteins were fixed and quantified by ELISA. ELISA signal was normalized to cell retention assessed by crystal violet staining, and signals were normalized to P66M (WT). MBP‐fusion proteins D205A,D207A and Del202–208 bound significantly less to 293 + αVβ3 cells when compared with WT P66M binding to 293 + αVβ3 cells (Fig. 3A).
Figure 3.
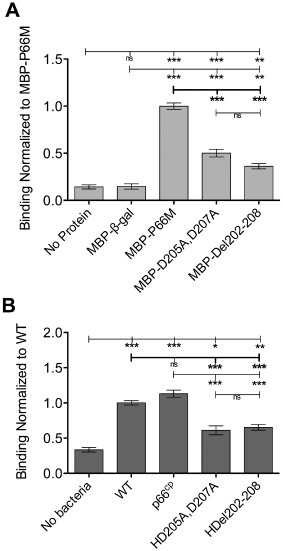
Targeted mutation of the integrin‐binding region of P66 significantly reduces binding to cells expressing αvβ3 integrin.
A. Up to 0.1 μM recombinant protein was incubated with 293 + αvβ3 cells grown to confluence in 96‐well plates for 1 h before washing and fixation of bound protein. Bound protein was quantified by ELISA using anti‐MBP as primary antibody, and anti‐rabbit‐HRP conjugate as secondary. Developed plates were read at 655 nm. ELISA signal was normalized to crystal violet staining of the wells and results were normalized to MBP‐P66M.
B. 1.25 × 106 HB19‐1 (wild‐type) and derivative bacteria per well were incubated with cells for 1 h before completing the assay as above, substituting anti‐Lyme spirochete as primary antibody. Results were normalized to wild type. Mean and standard error of measurement are indicated. Statistical analyses were performed using one‐way ANOVA Kruskal–Wallis test with Dunn's multiple comparison post‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
The deficiency in integrin binding observed with targeted mutants of P66 in recombinant form was also investigated in the context of intact bacteria. HB19‐1Δp66 bacteria were previously shown to be deficient in binding to integrin αVβ3 (Coburn and Cugini, 2003). In the current study, both the HB19‐1 (WT) and complemented derivatives expressing WT P66 (WT and p66cp) bound to 293 + αVβ3 cells at similar levels. In contrast, HD205A,D207A and HDel202–208 bacteria both bound significantly less to 293 + αVβ3 cells than did WT bacteria or p66cp bacteria (Fig. 3B), and bound to the cells at efficiencies similar to that of the HB19‐1Δp66 bacteria relative to the parental strain (Coburn and Cugini, 2003). Together with the channel‐forming results presented above, these results demonstrated that the two in vitro identified functions of P66 (integrin binding and 11 ns pore formation) can be separated to allow specific assessment of the integrin‐binding function without grossly affecting porin activity.
The integrin‐binding region of P66 is not critical for establishment of B . burgdorferi infection after subcutaneous inoculation
The infectivity phenotype of the site‐directed mutant forms of P66 was examined by ID50 determinations. C3H/HeN mice were inoculated with B31‐A3 (WT), Δp66, BD205A,D207A or BDel202–208 bacteria at a range of doses. At 4 weeks post‐inoculation, tissues, including the heart, tibiotarsus, inoculation site skin and ear, were harvested for culture and qPCR determination of bacterial load. ID50 determinations were based on infection status of cultures and were calculated as previously described (Reed and Muench, 1938). All strains bearing mutations in p66 had ID50 values comparable to WT bacteria with the exception of one clone of BDel202–208 (Table 2). That clone may have had additional mutations in the genome beyond the mutant P66, but we did verify that all genomic plasmids present in the parental strain were present in the mutant as detected by end point PCR on the day of inoculation into mice. Despite the aberrant ID50 of one mutant clone, these results clearly demonstrate that WT levels of β3 chain integrin binding are not required for B. burgdorferi to establish disseminated infection at 4 weeks post‐inoculation. More importantly, these results demonstrate that the complete loss of infectivity of the Δp66 mutant clones (Ristow et al., 2012) is due to at least one additional function of P66 (e.g. pore formation, specifically when the bacteria are not in a protected environment) that is important for B. burgdorferi infection.
Table 2.
Subcutaneous ID 50 determinations of p66 site‐directed mutants 4 weeks post‐inoculation
| Bacterial strain | Doses (number of motile bacteria per mousea) | ID50 value |
|---|---|---|
| B31‐A3 (wild type) | 101–109 | 4.64 × 102–2.37 × 104 |
| Δp66 C3–14 | 101–109 | >109 |
| BD205A,D207A clone 2–9 | 101–109 | 1.78 × 104 |
| BD205A,D207A clone 2–30 | 101–109 | 5.62 × 103 |
| BD205A,D207A clone 3–32 | 101–109 | 5.62 × 103 |
| BDel202–208 clone 3 | 101–109 | 1 × 104 |
| BDel202–208 clone 29 | 101–109 | 1 × 104 |
| BDel202–208 clone 78 | 101–109 | 4.64 × 106 |
Five mice were inoculated per dose per bacterial strain.
In agreement with the ID50 values, inoculation with BD205A,D207A clones resulted in similar bacterial burdens in most tissues assessed compared with WT inoculated mice (Fig. 4). Bacterial burdens in the hearts of mice inoculated with BD205A,D207A or BDel202–208 were significantly lower than those of WT inoculated mice. In addition, mice inoculated with BDel202–208 bacteria had significantly lower bacterial burdens in the tibiotarsal joint compared with WT or BD205A,D207A inoculated mice. At the site of inoculation, BD205A,D207A bacteria were observed at levels similar to WT bacteria, but were significantly higher than those of BDel202–208 inoculated mice. The differences in bacterial burdens between the strains producing the two different mutated versions of P66 indicate that there is a more substantial colonization defect in the BDel202–208 mutant. This result suggests that although pore formation is not affected, the difference in affinities of the mutant proteins for purified mouse αvβ3 observed in vitro correlates with the ability of B. burgdorferi to colonize tissues in mice.
Figure 4.
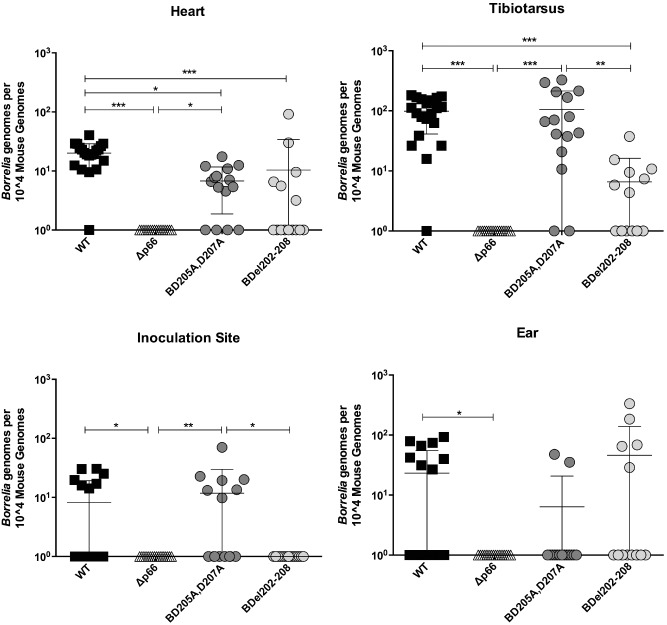
Targeted mutation of P66 alters dissemination of B orrelia burgdorferi following subcutaneous inoculation. C3H/HeN mice were inoculated with a range of doses (101–109 per mouse) of B31‐A3 (wild‐type), Δp66, BD 205A, D 207 A or BD el202–208 bacteria. Tissues were harvested 4 weeks post‐inoculation and total DNA was purified and analysed by qPCR with primers to detect B orrelia genomes or mouse genomes. Bacterial burdens from mice inoculated with 105 bacteria are shown. Mean bacterial load is indicated with error bars representing standard deviation. Statistical analyses were performed using a one‐way ANOVA Kruskal–Wallis test with Dunn's multiple comparison post‐test. *P < 0.05, **P < 0.01, ***P < 0.001. n ≥ 5 mice per strain.
The integrin‐binding region of P66 promotes endothelial transmigration of B . burgdorferi in vitro
The effect of the Del202–208 mutation on B. burgdorferi burdens in some tissues suggests a potential defect in at least some steps in the complex dissemination process. The spread of B. burgdorferi to tissues distant from the site of inoculation may occur by migration through tissues without entry into the vasculature, but is likely to be facilitated by entry into and dissemination through the vasculature. Colonization of distal sites would then involve crossing endothelial barriers while the bacteria are entering and exiting the vascular system. The potential role of the integrin‐binding function of P66 in the ability to cross endothelial cell monolayers was therefore investigated in vitro based on the Boyden chamber assay (Boyden, 1962). A microvascular endothelial cell line (HMEC‐1) was used to represent the cells present in small vessels or capillary beds in vivo, where B. burgdorferi can successfully exit the vasculature (Moriarty et al., 2008; 2012). Bacterial transmigration was assessed using confluent layers of HMEC‐1 cells in Transwell inserts. Transwells without cells were used as controls for bacteria crossing the membrane alone. The role of P66 in this activity was quantified by comparing the ability of B31‐A3 (WT), Δp66, p66cc, BD205A,D207A and BDel202–208 bacteria to cross the cell monolayer at a multiplicity of infection (MOI) of 20. Migration of all B. burgdorferi strains in wells without cells was significantly higher than that of any strain crossing wells with a HMEC‐1 cell monolayer at time points ≥24 h (data not shown). By 48 h, there was a significant defect in the ability of Δp66 bacteria to cross the cell monolayer compared with WT and p66cc bacteria (Fig. 5). A similar defect was observed in BDel202–208 bacterial transmigration of the cell monolayer compared with WT and p66cc bacteria. The ability of the BD205A,D207A bacteria to cross the cell layers was not significantly different from that of WT bacteria, consistent with the decreased severity of phenotypic effects in vivo in this mutant as compared with the BDel202–208 bacteria and with the in vitro integrin‐binding results. Transmigration was analysed up to 72 h post‐inoculation, but the health of all B. burgdorferi strains diminished after 48 h in the cell growth medium, which is relatively nutrient poor compared with the bacterial culture medium, and results were not reproducible after 48 h.
Figure 5.
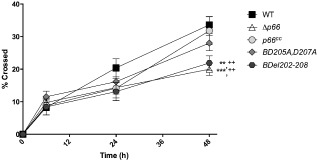
Mutation of P66 affects the ability of B orrelia burgdorferi to cross an endothelial monolayer of cells. HMEC‐1 human microvascular endothelial cells were plated in 3 μm pore size Transwell inserts and grown to confluence in antibiotic free medium. B31‐A3 (wild‐type), Δp66, p66cc, BD 205 A , D 207 A or BD el202–208 bacteria were added to the insert at an MOI of 20 and the ability of bacteria to cross the monolayer of cells was assessed over 48 h by taking an aliquot from the insert and from the well below. Bacteria were counted manually using a Petroff‐Hausser counting chamber under dark‐field microscopy, and the percentage of bacteria that crossed the cell monolayer was calculated. Inserts without a cell monolayer were assessed as controls for bacterial crossing of the membrane alone. A two‐way repeated measures ANOVA with Bonferroni's post‐test was performed. Asterisks indicate comparison to wild‐type bacteria, plus signs indicate comparison to p66cc bacteria. **/++ P < 0.01, ***P < 0.001, n = 5.
Decreased integrin‐binding activity of P66 delays B . burgdorferi dissemination from the vasculature
In the context of infectivity and dissemination, the ability of integrin‐binding deficient mutants to cross endothelial layers in vivo and exit the vasculature was quantified in multiple tissues by performing an ID50 determination after intravenous inoculation. C3H/HeN mice were retro‐orbitally inoculated with a range of doses of B31‐A3 (WT), Δp66, p66cc or BDel202–208 bacteria. After 2 weeks, tissues were harvested for culture and qPCR, including the heart, tibiotarsal joint, skin between the scapulae and ear. ID50 determinations were based on bacterial outgrowth in cultures and were calculated as previously described (Table 3). The ID50 of WT bacteria was slightly higher when the bacteria were inoculated via the intravenous route as compared with subcutaneous inoculation (compare Tables 2 and 3). Within the intravenous inoculation model, p66cc bacteria had a similar ID50 to WT bacteria, but the Δp66 and BDel202–208 bacteria were not recovered at 2 weeks post‐inoculation (Table 3). Bacterial loads assessed by qPCR confirmed culture results in that mice inoculated with WT or p66cc bacteria had similar bacterial loads in all tissues observed, while Δp66 and BDel202–208 bacteria were not detected by either culture or qPCR at 2 weeks post‐inoculation (Table 3 and Fig. 6A). Interestingly, when infections were allowed to progress for 4 weeks post‐intravenous inoculation, the BDel202–208 bacteria and BD205A,D207A bacteria were as infectious as WT bacteria by ID50 determination (Table 3). Although infectivity was similar between all strains at 4 weeks post‐inoculation, bacterial burdens were variable in some tissues, as determined by qPCR (Fig. 6B). Unlike the defect in tissue colonization observed in the tibiotarsus and ear after subcutaneous inoculation, BDel202–208 bacteria were observed at WT levels in all tissues except the heart, where the bacterial load was significantly lower than that of WT bacteria following intravenous inoculation. In the tibiotarsal joint, the BD205A,D207A bacterial burden was significantly higher than Δp66 and BDel202–208 bacterial burdens, but not significantly different from WT bacterial burdens. In the skin between the scapulae, the p66cc bacterial burden was significantly higher than Δp66 and BD205A,D207A bacterial burdens, but not significantly different from WT bacterial burdens. The observed delay in colonization of tissues by the BDel202–208 bacteria following intravenous inoculation may explain the variation in bacterial burdens, as BDel202–208 and potentially BD205A,D207A bacteria may have yet to establish a stable bacterial burden for maintenance of the bacteria. The more severe colonization defect of the BDel202–208 mutant as compared with the BD205A,D207A strain also correlates with the more severe loss of affinity of the recombinant proteins in integrin binding as determined by SPR.
Table 3.
Intravenous ID 50 determinations of p66 site‐directed mutants
| Bacterial strain | Doses (number of motile bacteria per mousea) | Weeks post‐inoculation | ID50 value |
|---|---|---|---|
| B31‐A3 (wild type) | 103–107 | 2 | 4.64 × 104 |
| 4 | 2.31 × 104 | ||
| Δp66 C3–14 | 103–107 | 2 | >107 |
| 4 | >107 | ||
| C3–14cc clone 23 | 103–107 | 2 | 2.15 × 105 |
| 4 | 5.62 × 105 | ||
| BD205A,D207A clone 2–30 | 103–107 | 4 | 2.15 × 105 |
| BD205A,D207A clone 3–32 | 103–107 | 4 | 4.64 × 104 |
| BDel202–208 clone 3 | 103–107 | 4 | 5.62 × 105 |
| BDel202–208 clone 29 | 103–107 | 2 | >107 |
| 4 | 1 × 106 |
Five mice were inoculated per dose per bacterial strain.
Figure 6.
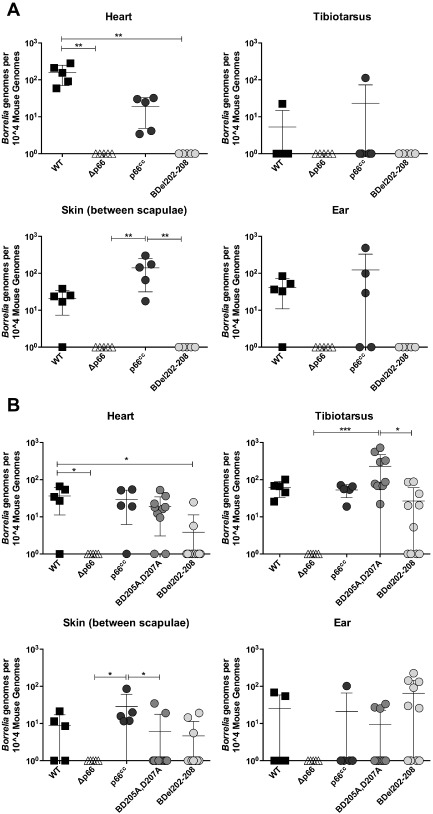
Targeted mutation of residues involved in integrin binding by P66 delays dissemination of B orrelia burgdorferi following intravenous inoculation. C3H/HeN mice were inoculated retro‐orbitally with a range of doses (103–107 per mouse) of B31‐A3 (wild‐type), Δp66, p66cc, BD 205 A , D 207 A or BD el202–208 bacteria. Tissues were harvested 2 weeks (A) or 4 weeks (B) post‐inoculation and total DNA was purified and analysed by qPCR with primers to detect B orrelia genomes or mouse genomes. Bacterial burdens from mice inoculated with 107 bacteria are shown. Mean bacterial load is indicated with error bars representing standard deviation. Statistical analyses were performed using a one‐way ANOVA Kruskal–Wallis test with Dunn's multiple comparison post‐test. *P < 0.05, **P < 0.01, ***P < 0.001. n = 5 mice per group.
Discussion
In this work, we tested the hypothesis that the infectivity defect of the Δp66 mutants is due to an inability to disseminate from the site of infection into other tissues, where the bacteria reside in extravascular connective tissue. The inability to escape the site of inoculation or from the bloodstream might effectively render the bacteria more susceptible to antimicrobial factors in the mouse skin or bloodstream. Although it is difficult to visualize bacterial entry into the circulation, we did demonstrate a defect in transendothelial migration by the Δp66 strains in vitro (three independent clones), and that B. burgdorferi strains producing a mutant P66 with drastically reduced affinity for integrin αvβ3 were phenotypically similar to the Δp66 strains. A new model, which allows in vivo assessment of transmigration of B. burgdorferi from the vasculature into the knee joint of mice, is described elsewhere (Kumar et al., submitted). Interestingly, when transmigration of Δp66 bacteria is compared with WT bacteria following intravenous inoculation, Δp66 bacteria were deficient in extravasation into the knee joint when assessed at 24 h post‐inoculation (Kumar et al., submitted). Consistent with our results, a defect in transendothelial migration into the knee joint of mice was also observed with integrin‐binding deficient strains BDel202–208 and BD205A,D207A strains using this in vivo method (Kumar et al., submitted). Furthermore, our work revealed that the observed defect in transendothelial migration of integrin‐binding deficient B. burgdorferi strains translates to a dissemination defect, as demonstrated in this work using both subcutaneous and intravenous inoculation to infect mice. Although the intravenous inoculation model does not recapitulate the natural route of transmission via tick bite, it does allow the delivery of known numbers of bacteria, and allows quantification of efficiency of crossing microvascular walls to colonize tissues distant from the inoculation site. Given the differences in the phenotypes of the D205A,D207A versus the Del202–208 strains, at least some of this function may be affected by the a.a. 202–208, or by the different affinities of the mutant versions of P66 for αvβ3 or other integrins. Because the Δp66 bacteria were previously shown to behave much like the WT parental strain when placed inside a dialysis membrane chamber in the peritoneum of rats (Ristow et al., 2012), we thought it unlikely that porin activity is essential to the function of P66 in this protected environment. In addition, no differences in in vitro growth have been detected for any of the p66 mutants in comparison to the WT parental strain. We did test the potential role of P66 in resistance of B. burgdorferi to serum (complement), and to phagocytosis by resident macrophages and dendritic cells in the skin (see Supporting Information Appendix S2), but the absence of P66 had no effect in any of these experiments. Together, our data suggest that either the porin activity or an as yet unknown function of P66 contributes to survival of B. burgdorferi in mouse infection when the bacteria are not sheltered.
In vivo function of P66 as an integrin ligand
To date, P66 is unique among bacterially encoded integrin ligands in that it interacts with the β3 chain and at least some β1 chain integrins. Of note, B. burgdorferi is an extracellular pathogen that has not been documented to occupy intracellular niches in vivo. P66 has been shown to interact with four integrins: αIIbβ3, αVβ3, α3β1 and α5β1 (Coburn et al., 1993; 1998; Behera et al., 2008). The mammalian ligands for these integrins contain an RGD (Arg‐Gly‐Asp) sequence (reviewed in Barczyk et al., 2010). Invasin, a Yersinia species β1 chain integrin ligand, does not contain an RGD sequence, but binds β1 chain integrins with 100‐fold higher affinity than the native ligand, fibronectin (Tran van Nhieu and Isberg, 1991). It has been demonstrated that invasin contains a critical aspartic acid residue required for entry into host cells (Leong et al., 1995). P66 does not encode an RGD sequence, but within the putative integrin‐binding region, the aspartic acid residues at a.a. 205 and 207 may contribute to the specific interaction with integrins. P66 binds to αvβ3 with an affinity on the order of that of a vitronectin‐derived 15‐amino‐acid peptide containing the RGD sequence (the K D for vitronectin was not measurable due to multiple interactions) (Orlando and Cheresh, 1991), and as shown in Table 1, the site‐directed mutant P66 proteins bind with considerably lower affinity.
As studies continue to carefully dissect the interactions of B. burgdorferi with the mammalian host, the parallels between B. burgdorferi exit from the vasculature and those observed in leukocyte interactions and exit from the vasculature are striking. During recruitment to sites of inflammation, neutrophils, monocytes and T‐cells engage integrins at various steps in vascular interactions that ultimately lead to transmigration out of the vasculature (reviewed in Herter and Zarbock, 2013). The initial steps in the extravasation of lymphocytes involve lectin–carbohydrate interactions, followed by interactions mediated by integrins of the β2 chain family (reviewed in Springer, 1994). Initial interactions of B. burgdorferi with the vasculature are mediated by BBK32 binding to fibronectin, then GAGs (Moriarty et al., 2012). Borrelia burgdorferi interactions with integrins would then be a subsequent step in transmigration through the vascular endothelium to cause infection in extravascular sites in diverse tissues. In both endothelial transmigration events, carbohydrate‐mediated interactions precede integrin‐mediated interactions. Both leukocyte and B. burgdorferi transmigration across the endothelium likely occur at endothelial cell‐cell junctions, as B. burgdorferi has been observed in the intercellular junctions between endothelial cells (Szczepanski et al., 1990; Moriarty et al., 2008). The P66‐integrin interaction may function to initiate signalling cascades to facilitate transmigration across the endothelium, including VEGF/VPF, as previously reported (LaFrance et al., 2011), which would represent a novel function for bacterial integrin ligands. Alternatively, B. burgdorferi may use the integrin interactions with P66 as traction in cooperation with the motility of the bacterium to drive through the endothelial barrier.
Multiple functions of P66
Of the in vitro identified functions of P66 to date, this work strongly supports the hypothesis that the function of P66 as an integrin ligand is required for endothelial transmigration as a key step in the dissemination of B. burgdorferi. P66 is known to bind both of the β3 chain integrins and at least two β1 chain integrins. Based on ligand recognition by integrin family members, it is entirely possible that P66 also binds additional αv integrins. It is clear that, even in the absence of the integrin subunit β3, P66 would have multiple other targets for attachment. The discrepancy between WT ID50s of integrin‐binding deficient strains after subcutaneous inoculation, albeit with some reductions in tissue burdens, and avirulence of Δp66 bacteria implies an additional function(s) of P66 in B. burgdorferi survival in the mouse model. However, this work does highlight the importance of dissemination through the bloodstream in the ability of B. burgdorferi to colonize sites distal from the site of the tick bite. Future work will include determination of whether the Δp66 bacteria can enter the vasculature, and whether integrin binding is required for B. burgdorferi entry into the circulation, but the results presented here suggest the possibility of two mechanisms of dissemination that are functionally distinct. One, promoted by P66‐integrin interactions, would be more rapid and involve spread through the vasculature. The other, which does not involve integrin binding by P66, is slower and may rely more on the motility of the bacteria and interactions with the tissue matrix than on P66 integrins as seen in the intravascular stage. Future studies will be informed by the work presented here, in which we have demonstrated for the first time the requirement for integrin‐binding function in efficient bacterial dissemination, and in particular a role for P66 in dissemination of B. burgdorferi in vivo, a property that would facilitate maintenance of the bacteria in the enzootic cycle and that is significant to the pathogenesis of Lyme disease.
Experimental procedures
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this work are described in Supporting Information Table S2. Borrelia burgdorferi B31‐A3 strains and derivatives (Elias et al., 2002) were grown in BSK II medium (Barbour‐Stoenner‐Kelly II; Barbour et al., 1983). HB19 (human blood isolate 19) strains and derivatives were grown in MKP medium (modified Kelly‐Pettenkofer; Preac‐Mursic et al., 1986), which we had previously found to maximize binding to the platelet integrin αIIbβ3 (Coburn et al., 1994). Media were supplemented with 15 g l−1 agar (for Escherichia coli) or 6.8 g l−1 agarose (for B. burgdorferi; Samuels, 1995). The genomic plasmid content of all B. burgdorferi cultures to be injected into mice was assessed by PCR (Elias et al., 2002; Bunikis et al., 2011) to ensure retention of all plasmids in the parental strain immediately prior to the mouse infections. Growth of B. burgdorferi from mouse tissues was in BSK II medium supplemented with rifampicin to 50 μg ml−1, amphotericin B to 2.5 μg ml−1 and phosphomycin to 20 μg ml−1.
Mice
The Institutional Animal Care and Use Committee of the Medical College of Wisconsin approved all work with animals. The protocol numbers are AUA2263 and AUA0488. C3H/HeN mice were purchased at the age of 3 weeks from Charles River Laboratories. β3 −/− mice and β3 +/+ WT littermates were originally obtained from Dr Richard O. Hynes (Massachusetts Institute of Technology) and maintained at the Medical College of Wisconsin. Mice were fed and watered ad libitum throughout the experiments. Mice were euthanized by controlled CO2 administration followed by cardiac puncture.
Bacteria were prepared for syringe inoculation experiments as described previously (Ristow et al., 2012). Briefly, laboratory‐cultured B. burgdorferi were cultivated to a concentration of 1–5 × 107 bacteria per ml. For every experiment, plasmid content of each strain was assessed at the morning of infection by PCR as previously described (Bunikis et al., 2011), and strains retaining all parental plasmids were subjected to several rounds of centrifugation and washes to concentrate the bacteria and remove the protein‐rich medium in which the bacteria are grown. After suspension in phosphate‐buffered saline (PBS) + 0.2% normal mouse serum (NMS), the bacteria were counted and diluted to the desired concentrations for inoculation.
For ID50 determinations, mice were inoculated subcutaneously between the scapulae with varying doses of bacteria per mouse (as noted in Figure legends) in 100 μl. For i.v. ID50 determinations, mice were inoculated retro‐orbitally with varying doses of bacteria per mouse in 100 μl. In each experiment, control mice were sham inoculated with 100 μl PBS + 0.2% NMS. Two or 4 weeks post‐inoculation, mice were euthanized by CO2 narcosis and tissues including the heart, tibiotarsal joint, inoculation site skin (or skin between the scapulae in i.v. ID50 experiments) and ear were harvested for culture and for quantification of bacterial load by qPCR. Cultures were checked for the presence of B. burgdorferi by dark‐field microscopy for up to 8 weeks after which a final designation of positive or negative was assigned. ID50 values were calculated from the infection status of each mouse based on culture results. A mouse was designated as infected if any culture was positive.
Quantitative PCR
Bacterial burdens were quantified as described (Ristow et al., 2012). Briefly, DNA was harvested from tissue samples using the QIAGEN QIAamp DNA Mini Kit. The B. burgdorferi standard curve was generated in the presence of a constant amount of mouse DNA, using DNA isolated from B. burgdorferi grown in culture and mouse LA‐4 cells (ATCC #CCL 196) or uninfected mouse livers using the same kit. The mouse standard curve was generated from DNA isolated from mouse LA‐4 cells or uninfected mouse livers. Standards and samples were run in triplicate with primers to detect recA for quantification of B. burgdorferi or mouse β‐actin for quantification of mouse genomic DNA on a BioRad CFX96 cycler. After generation of a standard curve with BioRad CFX Manager 3.0 software, the experimental value for each sample was extrapolated from the average Ct value for each triplicate and all values were increased by 1 to facilitate plotting on a log scale. Results are presented as the number of B. burgdorferi genomes per 104 mouse genomes for each sample. The limit of detection for Borrelia genomes was six genomes per reaction. The limit of detection for mouse genomes was three haploid genomes per reaction.
Construction and expression of MBP‐P66 fusion proteins
The parental MBP‐P66M fusion was previously described (Coburn et al., 1999) and includes the region of P66 required for integrin binding. Briefly, the pMalC2 vector (New England Biolabs) was utilized to generate a MBP fusion with the middle portion of P66, containing a.a. 170–404. To test the roles of specific amino acids in integrin binding of P66, mutations were generated within the coding sequence of the hypothesized integrin‐binding region (Defoe and Coburn, 2001) of P66 using the Stratagene QuikChange II site‐directed mutagenesis kit (Agilent Technologies) per manufacturer instructions. Primers were designed to generate two single amino acid changes (a.a. D205A and D207A) or to delete a portion of P66 (a.a. 202–208). Primer sequences are described in Supporting Information Table S3. Mutagenized vectors were transformed into E. coli BLR and transformants were plated on LB plates with 0.2% dextrose and 100 μg ml−1 ampicillin for selection. Six clones from each intended mutation were purified by restreaking. Clones confirmed by sequencing to contain the targeted mutations were transformed into E. coli KS330 for optimal protein expression, and are referred to as P66M‐D205A,D207A or P66M‐Del202–208. Protein expression was induced with IPTG (isopropyl β‐D‐1‐thiogalactopyranoside). Cells were lysed by French Press and proteins were purified by amylose affinity chromatography as described previously (Leong et al., 1990; Coburn et al., 1999). MBP‐β‐gal (encoded by the vector) was used as a control for background binding of MBP‐fusion proteins. Protein concentration was quantified using the Bradford Assay (Bio‐Rad) before use in assays.
Construction of B . burgdorferi strains
Construction of HB19 Δp66 (HB19/K04) was previously described (Coburn and Cugini, 2003). Complementation of this strain was performed using the previously described construct, pBSV2Gp66 (Ristow et al., 2012). Successful transformation of this construct into B. burgdorferi results in P66 expression from the autonomously maintained shuttle vector. Targeted mutations of P66 at a.a. 205 and 207 or deletion of a.a. 202–208 were generated as described for the MBP‐P66 fusions using pBSV2Gp66 as a template. Clones were isolated after transformation of these constructs into HB19 Δp66 and screened by PCR for the presence of appropriate drug resistance markers. Restoration of P66 expression was observed by immunoblot using anti‐P66 rabbit antiserum (Coburn and Cugini, 2003) as the primary antibody. Anti‐rabbit IgG‐alkaline phosphatase conjugate (Promega) was used as the secondary antibody for colorimetric visualization of P66 expression. Retention of plasmids found in the parental strain was monitored by PCR using two different primer sets for each clone as previously described (Elias et al., 2002; Bunikis et al., 2011). Clones with restoration of P66 expression, mutations confirmed by sequencing, and retention of the parental genomic content were used in this study and are referred to as p66cp (complement on the plasmid), HD205A,D207A or HDel202–208. These strains were indistinguishable from the WT strain in in vitro growth and were used to quantify binding to mammalian cells.
Construction of B31 A3 Δp66 and p66cc (complement on the chromosome) strains was previously described (Ristow et al., 2012). Targeted mutations within P66 were generated to be expressed in the infectious strain background, B31 A3 (Elias et al., 2002), in the same manner as MBP‐P66 described above, using pGTEp66 (Ristow et al., 2012) as a template. Upon transformation into B. burgdorferi, pGTEp66 allows homologous recombination of the p66 coding region and flanking sequences into the endogenous locus on the chromosome to restore P66 expression to a Δp66 strain. Transformation with the linearized construct resulted in candidate B. burgdorferi clones that were screened as described above for HB19 strains. Clones with restoration of P66 expression, mutations confirmed by sequencing and retention of the complete parental genome were used in the work reported here and are referred to as BD205A,D207A or BDel202–208. These strains were indistinguishable from the WT strain in in vitro growth and were used in transmigration assays, ID50 determinations and quantification of bacterial burdens in different tissue sites.
Analysis of surface expression of P66
Bacteria from 3 ml of growing cultures (1–5 × 107 bacteria ml−1) of HB19 or B31 A3 strains and derivatives were subjected to centrifugation for 30 min at 1500 × g at ambient temperature (room temperature, RT). After removal of supernatant fluid, bacteria were suspended in 1 ml of PBS and subjected to centrifugation for 8 min at 11 200 × g at RT. After removal of supernatant fluid, bacteria were suspended in 1 ml of PBS and split into four tubes. Bacteria were incubated with varying concentrations of proteinase K (0, 1, 10 and 100 μg ml−1) for 1 h at 33°C. After proteinase K treatment, phenylmethylsulfonyl fluoride was added to a final concentration of 0.2 mM for 15 min at RT. Cultures were subjected to centrifugation for 8 min at 11 200 × g and pelleted bacteria were suspended in PBS to a final concentration of 5 × 109 bacteria per ml. An equal volume of 2 × sample buffer with β‐mercaptoethanol was added before boiling samples at 100°C for 5 min. Ten microlitres of each sample was fractionated by SDS‐PAGE (10% acrylamide), transferred to Immobilon, and probed with rabbit anti‐P66 (1:5000 dilution) and mouse anti‐flagellin H9724 (1:500 dilution) (provided by Dr Tom Schwan) as a loading control and for assessment of outer membrane integrity. Goat anti‐rabbit IgG‐alkaline phosphatase conjugate (1:10 000 dilution) and goat anti‐mouse IgG‐alkaline phosphatase conjugate (1:10 000 dilution) (Promega) were used as secondary antibodies and colorimetric methods were used to visualize bands.
Mammalian cell culture
Human embryonic kidney cell line 293 (HEK293) stably expressing human integrin αvβ3 (293 + αvβ3) (LaFrance et al., 2011) was cultured in Dulbecco's modified Eagle medium plus Ham's F12 nutrient mix (base medium; Gibco) with 5% heat‐inactivated fetal bovine serum (FBS) (Gibco) supplemented with 400 μg ml−1 of G418 (Gibco). Human microvascular endothelial cells (HMEC‐1) (Ades et al., 1992) were cultured in MCDB 131 (base medium; Gibco) with 15% heat‐inactivated Hyclone FBS (Hyclone Laboratories), 1 μg ml−1 of hydrocortisone (Sigma‐Aldrich), 10 ng ml−1 of epidermal growth factor (Gibco) and 25 mM HEPES (Gibco). The mouse lung epithelial cell line LA‐4 was cultured in Kaighn's modification of Ham's F12 medium (F12K) (base medium; Gibco) with 10% heat‐inactivated FBS (Gibco) (Stoner et al., 1975). All cell lines were cultured in the presence of additional L‐glutamine (Gibco) to a final concentration of 2 mM and for general maintenance of cell lines, penicillin at a concentration of 100 U ml−1 and streptomycin to a final concentration of 100 μg ml−1 (Gibco). For bacterial infections, antibiotic containing medium was removed and cells were washed before being lifted with Trypsin‐EDTA (Gibco) and suspended in antibiotic free medium prior to being plated in the absence of antibiotics. Cells were incubated at 37°C under 5% CO2.
Assessment of P66 binding to purified integrin αvβ3
Purified recombinant mouse and human integrin αvβ3 (carrier free) were purchased from R&D Systems. Purified MBP fusions to P66M (WT), D205A,D207A, Del202–208 and β‐galactosidase (β‐gal) were diluted to concentrations indicated in HEPES‐buffered saline with cations (HBSC; 25 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 0.25 mM CaCl2). Interactions of P66 with mouse or human integrin αvβ3 were analysed by SPR using a Biacore 3000 (GE Healthcare). Ten μg of MBP‐P66M, MBP‐D205A,D207A, or MBP‐Del202–208, or MBP‐βgal (negative control) was conjugated to an CM5 chip (GE Healthcare). A control flow cell was injected with HBSC buffer without mouse or human integrin αvβ3. For quantitative SPR experiments to determine integrin αvβ3 binding, 20 μl of increasing concentrations (0, 31.25, 62.5, 125, 250, 500 nM) of a mouse or human integrin αvβ3 were injected into the control cell and flow cells with immobilized MBP‐P66M, MBP‐D205A,D207A, MBP‐Del202–208, or MBP‐βgal at 10 μl min−1, 25°C. To obtain the kinetic parameters of the interaction, sensogram data were fitted by means of BIAevaluation software version 3.0 (GE Healthcare) using the one‐step biomolecular association reaction model (1:1 Langmuir model), resulting in optimum mathematical fit with the lowest chi values.
Mammalian cell‐binding assays
Confluent monolayers of 293 + αvβ3 cells were used in binding assays. Cells were cultured in sterile 96‐well tissue culture plates 2 days prior to probing with purified recombinant protein. On the day of the assay, cell monolayers were washed once with PBS before the addition of base cell medium supplemented with 1% bovine serum albumin (BSA) (blocking buffer). Plates were blocked at 37°C for 1 h. Proteins were diluted in blocking buffer to 0.1 μM. Blocking buffer was removed and replaced with the test proteins and incubated at 37°C for 1 h. Wells were washed twice with PBS to remove unbound protein before fixing bound protein with 3% paraformaldehyde for 30 min or overnight. The plates were washed three times with Tris‐buffered saline (TBS) to remove fixative, then blocked with TBS + 3% normal goat serum (NGS) + 1% BSA for 1 h. An ELISA was performed to assess bound protein using rabbit anti‐MBP (1:10 000 dilution) in TBS + 3% NGS + 1% BSA as primary antibody, and goat anti‐rabbit horseradish peroxidase (HRP) conjugate (1:10 000 dilution) in TBS + 3% NGS + 1% BSA as secondary antibody. Plates were developed using tetramethylbenzidine substrate tablets (Sigma‐Aldrich) with hydrogen peroxide in a 1:10 mixture of dimethyl sulfoxide (DMSO) and 0.05 M phosphate‐citrate buffer, pH 5.0. Colorimetric development was read by absorbance at 655 nm. After washing away developer, cell retention was measured with crystal violet, read at 595 nm. The ELISA signal was normalized to cell retention for individual wells, with quadruplicate measurements on each plate. Binding of mutant proteins was normalized to binding of MBP‐P66M to allow incorporation of data from multiple experiments into the statistical analyses and figures, as raw optical density (OD) values were not always identical in different experiments. Statistical analyses were performed using one‐way ANOVA with Dunn's multiple comparison post‐test in GraphPad Prism version 5.1.
For assessment of binding of B. burgdorferi strain HB19‐derived bacteria, the protocol was similar. On the day of the assay, after blocking, cell monolayers were washed and probed with 50 μl of bacteria at 2.5 × 107 bacteria per ml for 1 h at 37°C. Unbound bacteria were washed away and bound bacteria were fixed in 3% paraformaldehyde for 30 min or overnight. The plates were washed three times with TBS to remove fixative, then blocked with TBS + 3% NGS + 1% BSA for 1 h. An ELISA was performed using rabbit anti‐Lyme spirochete serum (1:10 000 dilution) (provided by Dr Allen Steere) in TBS + 3% NGS + 1% BSA as primary antibody, followed by goat anti‐rabbit HRP conjugate (1:10 000 dilution) in TBS + 3% NGS + 1% BSA as secondary antibody. Plates were developed and ELISA signal was normalized to cell retention as described for MBP‐fusion proteins. Binding of bacteria expressing mutant P66 proteins was normalized to binding of WT bacteria to allow incorporation of data from multiple experiments into the statistical analyses and figures, as raw OD values were not always identical in different experiments. Statistical analyses were performed using one‐way ANOVA with Dunn's multiple comparison post‐test in GraphPad Prism version 5.1.
In vitro transmigration
HMEC‐1 cells were plated onto 3 μm pore size Transwell inserts (Costar #3472 polyester membrane Transwell inserts) in growth medium without antibiotics, as previously described (Martinez‐Lopez et al., 2010). Endothelial cell monolayers were allowed to reach confluence over 72 h before infection with B. burgdorferi at an MOI of 20. The percentage of bacteria crossing into the chamber below was monitored over 72 h by removing aliquots from both top and bottom chambers and enumerating the bacteria in a Petroff‐Hausser counting chamber using dark‐field microscopy. Raw numbers of spirochetes were comparable between strains throughout the time course, with all strains replicating through the 48 h time point. Beyond 48 h, bacterial health and numbers began to diminish and data were not reproducible. In parallel, FITC‐Dextran 40 000 Da was added to an insert to assess confluence of the plated monolayers. Inserts without cells plated were used as controls for crossing of the membrane alone. Assays were performed in triplicate. Statistical analyses were performed using two‐way ANOVA with Bonferroni's post‐test in GraphPad Prism version 5.1.
Pore formation assay
Cultures of HB19 strains were grown until mid‐late logarithmic phase and outer membrane proteins were purified as previously described (Magnarelli et al., 1989). Further purification of P66 was performed by anion exchange chromatography using a HiPrep Q XL 16/10 column (GE Healthcare). The method for black lipid bilayer experiments has been described previously (Benz et al., 1978). The bilayer equipment consists of a Teflon chamber with two compartments, separated by a thin wall with a small circular hole (0.4 mm2) that connects the two compartments. Over the small hole, an artificial bilayer membrane can be painted, into which proteins with channel‐forming capabilities can insert. Insertion of pore‐forming proteins causes a change in the membrane current that can be measured and used to calculate the size of the channel formed. The membrane solution was formed from 1% (w v−1) diphytanoyl phosphatidylcholine (Avanti Polar Lipids) in n‐decane. The protein containing solution was diluted 1:1 in 1% Genapol and added to the electrolyte on both sides when the membrane turned black. The two compartments contained 1 M KCl salt solution and the voltage applied was 20 mV. Membrane current was measured with a pair of Ag/AgCl electrodes with salt bridges switched in series with a voltage source and a current amplifier (Keithley 427). All experiments were performed at RT.
Supporting information
Fig. S1. Wild‐type and Δp66 Borrelia burgdorferi bacterial burdens are unaltered in β3 −/− mice compared with β3 +/+ mice. β3 +/+ (filled symbols) or β3 −/− (open symbols) mice were inoculated with B31‐A3 (wild‐type) (squares) or Δp66 (triangles) B. burgdorferi at a range of doses (103–109). Tissues were harvested 4 weeks post‐inoculation and total DNA was purified and analyzed by qPCR with primers to detect Borrelia genomes or mouse genomes. Bacterial loads in mice inoculated at 105 are shown. Mean bacterial load is indicated with error bars representing standard deviation. Statistical analyses were performed using a two‐way ANOVA with Bonferroni's post‐test. *P < 0.05, **P < 0.01, ***P < 0.001. n = 5 mice per group.
Fig. S2. Mutation of p66 does not increase sensitivity to complement. (A) B. burgdorferi strains in the B31‐A3 background were incubated with 40% complete or 40% heat‐inactivated (HI) human serum in triplicate. Aliquots from each condition were plated in solid BSK II at 1, 2, 4 and 16 h, incubated at 33°C for 2 weeks and colonies were enumerated. The mean concentration of viable bacteria per millilitre is indicated with error bars representing standard error. n = 3. (B) E. coli MC1061 bacteria were incubated with no serum, 40% complete or 40% heat‐inactivated (HI) human serum in triplicate. A dilution series from each condition was plated on LB agar at 1, 2, 4 and 16 h, incubated at 37°C for 16 h and colonies were enumerated. The mean concentration of viable bacteria per millilitre is indicated with error bars representing standard error. n = 3.
Fig. S3. Depletion of macrophages or dendritic cells does not affect infection by either wild‐type or Δp66 B. burgdorferi. (A and B) The site of inoculation was harvested at days 1, 2, 3, 4, 5, 6 and 8 from transgenic mice expressing diphtheria toxin receptor (DTxR) on macrophages inoculated with either B31‐A3 (wild‐type) or Δp66 bacteria plus diphtheria toxin (DTx) or the enzymatically inactive cross‐reactive mutant form of the toxin (CRM) (A), or from C57Bl/6 mice inoculated with wild‐type or Δp66 bacteria with no toxin (B). Sham‐inoculated mice were used as controls on days 1 and 8 (B). Note that DTx‐treated mice were euthanized and tissues harvested as the mice began to show signs of illness, so CRM‐treated mice and C57Bl/6 mice were not always euthanized and harvested for the same time points. (C and D) The site of inoculation was harvested at days 1, 2, 4 and 5 from transgenic mice expressing DTxR on dendritic cells inoculated with either B31‐A3 (wild‐type) or Δp66 bacteria plus DTx or CRM (C), or from C57Bl/6 mice inoculated with wild‐type or Δp66 bacteria with no toxin (D). Sham‐inoculated mice were used as controls on days 1 and 5 (D). DNA was purified from the tissue and analysed by qPCR with primers to detect Borrelia DNA or mouse DNA. Statistical analyses were performed between groups of mice on each day of harvest using one‐way ANOVA Kruskal–Wallis test with Dunn's multiple comparison post‐test. *P < 0.05, **P < 0.01. Mean bacterial load is indicated with error bars representing standard deviation. n = 5 mice per condition.
Fig. S4. Depletion of macrophages or dendritic cells in DTxR transgenic mice treated with DTx. Top row: immunohistochemical staining of anti‐F4/80‐reactive cells in inoculation site skin of mice treated with CRM (left panel) or DTx (right panel) at day 1 post‐inoculation and intoxication. Scale bars = 20 μm; images acquired at 100× magnification. Bottom row: fluorescence imaging of the site of inoculation at day 1 post‐inoculation and intoxication with B31‐A3 (wild‐type) B. burgdorferi expressing tdTomato (red) and CRM (left panel) or DTx (right panel). Dendritic cells in this transgenic mouse model express DTxR and eGFP (green). Spirochetes are indicated with an arrow. Scale bars = 20 μm; images acquired at 200× magnification.
Table S1. Subcutaneous ID50 determinations of B31‐A3 (wild‐type) and Δp66 Borrelia burgdorferi in β3 +/+ or β3 −/− mice 4 weeks post‐inoculation.
Table S2. Bacterial strains and plasmids used in this study.
Table S3. Oligonucleotide primers, 5′ → 3′.
Video S1. Motility of wild‐type and Δp66 B. burgdorferi is similar at the site of inoculation in vivo. RFP expressing B31 A3 and GFP expressing Δp66 B. burgdorferi were imaged at the site of inoculation 1 h post‐intradermal inoculation. A time‐lapse video was acquired at a magnification of 20×, exposure time of 2 s for each colour (green and red), time lapse set at 2 s interval for 5 min. For movie conversion, output timing was set at the interval of 200 ms.
Appendix S1. Supplemental experimental procedures.
Appendix S2. Supplemental experiments.
Acknowledgements
We thank Dr Dara Frank for providing access to her fluorescence microscope, Drs Yi‐Guang Chen, Calvin Williams and Dipica Haribhai for their expertise and training in the diphtheria toxin receptor mouse model, Tarrant Csida and Jennifer Ritchie for performing intravenous mouse inoculations, Dr Allen Steere and Dr Tom Schwan for providing antibodies, the Ragon Institute Imaging Core Facility for providing access to SPR instrumentation, Michael Waring of the Imaging Core for assistance with SPR, and the staff in the Children's Hospital histology core for sectioning and staining mouse skin samples. This work was supported by NIH grants (R01‐AI084873 and R01‐AI093104), by the Swedish Research Council grant (07922), and by generous gifts from the MACC Fund and John B. and Judith A. Gardetto.
References
- Ades, E.W. , Candal, F.J. , Swerlick, R.A. , George, V.G. , Summers, S. , Bosse, D.C. , and Lawley, T.J. (1992) HMEC‐1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol 99: 683–690. [DOI] [PubMed] [Google Scholar]
- Bárcena‐Uribarri, I. , Thein, M. , Sacher, A. , Bunikis, I. , Bonde, M. , Bergström, S. , and Benz, R. (2010) P66 porins are present in both Lyme disease and relapsing fever spirochetes: a comparison of the biophysical properties of P66 porins from six Borrelia species. Biochim Biophys Acta 1798: 1197–1203. [DOI] [PubMed] [Google Scholar]
- Bagos, P.G. , Liakopoulos, T.D. , Spyropoulos, I.C. , and Hamodrakas, S.J. (2004) PRED‐TMBB: a web server for predicting the topology of beta‐barrel outer membrane proteins. Nucleic Acids Res 32: W400–W404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour, A.G. , Burgdorfer, W. , Grunwaldt, E. , and Steere, A.C. (1983) Antibodies of patients with Lyme disease to components of the Ixodes dammini spirochete. J Clin Invest 72: 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczyk, M. , Carracedo, S. , and Gullberg, D. (2010) Integrins. Cell Tissue Res 339: 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera, A.K. , Durand, E. , Cugini, C. , Antonara, S. , Bourassa, L. , Hildebrand, E. , et al (2008) Borrelia burgdorferi BBB07 interaction with integrin alpha3beta1 stimulates production of pro‐inflammatory mediators in primary human chondrocytes. Cell Microbiol 10: 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz, R. , Janko, K. , Boos, W. , and Läuger, P. (1978) Formation of large, ion‐permeable membrane channels by the matrix protein (porin) of Escherichia coli . Biochim Biophys Acta 511: 305–319. [DOI] [PubMed] [Google Scholar]
- Boyden, S. (1962) The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med 115: 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunikis, I. , Kutschan‐Bunikis, S. , Bonde, M. , and Bergström, S. (2011) Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi . J Microbiol Methods 86: 243–247. [DOI] [PubMed] [Google Scholar]
- Coburn, J. , and Cugini, C. (2003) Targeted mutation of the outer membrane protein P66 disrupts attachment of the Lyme disease agent, Borrelia burgdorferi, to integrin alphavbeta3. Proc Natl Acad Sci USA 100: 7301–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn, J. , Leong, J.M. , and Erban, J.K. (1993) Integrin alpha IIb beta 3 mediates binding of the Lyme disease agent Borrelia burgdorferi to human platelets. Proc Natl Acad Sci USA 90: 7059–7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn, J. , Barthold, S.W. , and Leong, J.M. (1994) Diverse Lyme disease spirochetes bind integrin alpha IIb beta 3 on human platelets. Infect Immun 62: 5559–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn, J. , Magoun, L. , Bodary, S.C. , and Leong, J.M. (1998) Integrins alpha(v)beta3 and alpha5beta1 mediate attachment of lyme disease spirochetes to human cells. Infect Immun 66: 1946–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn, J. , Chege, W. , Magoun, L. , Bodary, S.C. , and Leong, J.M. (1999) Characterization of a candidate Borrelia burgdorferi beta3‐chain integrin ligand identified using a phage display library. Mol Microbiol 34: 926–940. [DOI] [PubMed] [Google Scholar]
- Cugini, C. , Medrano, M. , Schwan, T.G. , and Coburn, J. (2003) Regulation of expression of the Borrelia burgdorferi beta(3)‐chain integrin ligand, P66, in ticks and in culture. Infect Immun 71: 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defoe, G. , and Coburn, J. (2001) Delineation of Borrelia burgdorferi p66 sequences required for integrin alpha(IIb)beta(3) recognition. Infect Immun 69: 3455–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias, A.F. , Stewart, P.E. , Grimm, D. , Caimano, M.J. , Eggers, C.H. , Tilly, K. , et al (2002) Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun 70: 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herter, J. , and Zarbock, A. (2013) Integrin regulation during leukocyte recruitment. J Immunol 190: 4451–4457. [DOI] [PubMed] [Google Scholar]
- Hodivala‐Dilke, K.M. , McHugh, K.P. , Tsakiris, D.A. , Rayburn, H. , Crowley, D. , Ullman‐Culleré, M. , et al (1999) Beta3‐integrin‐deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy, M.R. , Luthra, A. , Anand, A. , Dunn, J.P. , Radolf, J.D. , and Akins, D.R. (2014) Structural modeling and physicochemical characterization provide evidence that P66 forms a beta‐barrel in the Borrelia burgdorferi outer membrane. J Bacteriol 196: 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFrance, M.E. , Pierce, J.V. , Antonara, S. , and Coburn, J. (2011) The Borrelia burgdorferi integrin ligand P66 affects gene expression by human cells in culture. Infect Immun 79: 3249–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong, J.M. , Fournier, R.S. , and Isberg, R.R. (1990) Identification of the integrin binding domain of the Yersinia pseudotuberculosis invasin protein. EMBO J 9: 1979–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong, J.M. , Morrissey, P.E. , Marra, A. , and Isberg, R.R. (1995) An aspartate residue of the Yersinia pseudotuberculosis invasin protein that is critical for integrin binding. EMBO J 14: 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnarelli, L.A. , Anderson, J.F. , and Barbour, A.G. (1989) Enzyme‐linked immunosorbent assays for Lyme disease: reactivity of subunits of Borrelia burgdorferi . J Infect Dis 159: 43–49. [DOI] [PubMed] [Google Scholar]
- Martinez‐Lopez, D.G. , Fahey, M. , and Coburn, J. (2010) Responses of human endothelial cells to pathogenic and non‐pathogenic Leptospira species. PLoS Negl Trop Dis 4: e918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, T.J. , Norman, M.U. , Colarusso, P. , Bankhead, T. , Kubes, P. , and Chaconas, G. (2008) Real‐time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog 4: e1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, T.J. , Shi, M. , Lin, Y.P. , Ebady, R. , Zhou, H. , Odisho, T. , et al (2012) Vascular binding of a pathogen under shear force through mechanistically distinct sequential interactions with host macromolecules. Mol Microbiol 86: 1116–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, M.U. , Moriarty, T.J. , Dresser, A.R. , Millen, B. , Kubes, P. , and Chaconas, G. (2008) Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS Pathog 4: e1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando, R.A. , and Cheresh, D.A. (1991) Arginine‐glycine‐aspartic acid binding leading to molecular stabilization between integrin alpha v beta 3 and its ligand. J Biol Chem 266: 19543–19550. [PubMed] [Google Scholar]
- Pinne, M. , Thein, M. , Denker, K. , Benz, R. , Coburn, J. , and Bergström, S. (2007) Elimination of channel‐forming activity by insertional inactivation of the p66 gene in Borrelia burgdorferi . FEMS Microbiol Lett 266: 241–249. [DOI] [PubMed] [Google Scholar]
- Preac‐Mursic, V. , Wilske, B. , and Schierz, G. (1986) European Borrelia burgdorferi isolated from humans and ticks culture conditions and antibiotic susceptibility. Zentralbl Bakteriol Mikrobiol Hyg A 263: 112–118. [DOI] [PubMed] [Google Scholar]
- Reed, L.J. , and Muench, H. (1938) A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27: 493–497. [Google Scholar]
- Ristow, L.C. , Miller, H.E. , Padmore, L.J. , Chettri, R. , Salzman, N. , Caimano, M.J. , et al (2012) The beta(3)‐integrin ligand of Borrelia burgdorferi is critical for infection of mice but not ticks. Mol Microbiol 85: 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels, D.S. (1995) Electrotransformation of the spirochete Borrelia burgdorferi . Methods Mol Biol 47: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skare, J.T. , Mirzabekov, T.A. , Shang, E.S. , Blanco, D.R. , Erdjument‐Bromage, H. , Bunikis, J. , et al (1997) The Oms66 (p66) protein is a Borrelia burgdorferi porin. Infect Immun 65: 3654–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, T.A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76: 301–314. [DOI] [PubMed] [Google Scholar]
- Stephens, L.E. , Sutherland, A.E. , Klimanskaya, I.V. , Andrieux, A. , Meneses, J. , Pedersen, R.A. , and Damsky, C.H. (1995) Deletion of beta 1 integrins in mice results in inner cell mass failure and peri‐implantation lethality. Genes Dev 9: 1883–1895. [DOI] [PubMed] [Google Scholar]
- Stoner, G.D. , Kikkawa, Y. , Kniazeff, A.J. , Miyai, K. , and Wagner, R.M. (1975) Clonal isolation of epithelial cells from mouse lung adenoma. Cancer Res 35: 2177–2185. [PubMed] [Google Scholar]
- Szczepanski, A. , Furie, M.B. , Benach, J.L. , Lane, B.P. , and Fleit, H.B. (1990) Interaction between Borrelia burgdorferi and endothelium in vitro . J Clin Invest 85: 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran van Nhieu, G.T. , and Isberg, R.R. (1991) The Yersinia pseudotuberculosis invasin protein and human fibronectin bind to mutually exclusive sites on the alpha 5 beta 1 integrin receptor. J Biol Chem 266: 24367–24375. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Wild‐type and Δp66 Borrelia burgdorferi bacterial burdens are unaltered in β3 −/− mice compared with β3 +/+ mice. β3 +/+ (filled symbols) or β3 −/− (open symbols) mice were inoculated with B31‐A3 (wild‐type) (squares) or Δp66 (triangles) B. burgdorferi at a range of doses (103–109). Tissues were harvested 4 weeks post‐inoculation and total DNA was purified and analyzed by qPCR with primers to detect Borrelia genomes or mouse genomes. Bacterial loads in mice inoculated at 105 are shown. Mean bacterial load is indicated with error bars representing standard deviation. Statistical analyses were performed using a two‐way ANOVA with Bonferroni's post‐test. *P < 0.05, **P < 0.01, ***P < 0.001. n = 5 mice per group.
Fig. S2. Mutation of p66 does not increase sensitivity to complement. (A) B. burgdorferi strains in the B31‐A3 background were incubated with 40% complete or 40% heat‐inactivated (HI) human serum in triplicate. Aliquots from each condition were plated in solid BSK II at 1, 2, 4 and 16 h, incubated at 33°C for 2 weeks and colonies were enumerated. The mean concentration of viable bacteria per millilitre is indicated with error bars representing standard error. n = 3. (B) E. coli MC1061 bacteria were incubated with no serum, 40% complete or 40% heat‐inactivated (HI) human serum in triplicate. A dilution series from each condition was plated on LB agar at 1, 2, 4 and 16 h, incubated at 37°C for 16 h and colonies were enumerated. The mean concentration of viable bacteria per millilitre is indicated with error bars representing standard error. n = 3.
Fig. S3. Depletion of macrophages or dendritic cells does not affect infection by either wild‐type or Δp66 B. burgdorferi. (A and B) The site of inoculation was harvested at days 1, 2, 3, 4, 5, 6 and 8 from transgenic mice expressing diphtheria toxin receptor (DTxR) on macrophages inoculated with either B31‐A3 (wild‐type) or Δp66 bacteria plus diphtheria toxin (DTx) or the enzymatically inactive cross‐reactive mutant form of the toxin (CRM) (A), or from C57Bl/6 mice inoculated with wild‐type or Δp66 bacteria with no toxin (B). Sham‐inoculated mice were used as controls on days 1 and 8 (B). Note that DTx‐treated mice were euthanized and tissues harvested as the mice began to show signs of illness, so CRM‐treated mice and C57Bl/6 mice were not always euthanized and harvested for the same time points. (C and D) The site of inoculation was harvested at days 1, 2, 4 and 5 from transgenic mice expressing DTxR on dendritic cells inoculated with either B31‐A3 (wild‐type) or Δp66 bacteria plus DTx or CRM (C), or from C57Bl/6 mice inoculated with wild‐type or Δp66 bacteria with no toxin (D). Sham‐inoculated mice were used as controls on days 1 and 5 (D). DNA was purified from the tissue and analysed by qPCR with primers to detect Borrelia DNA or mouse DNA. Statistical analyses were performed between groups of mice on each day of harvest using one‐way ANOVA Kruskal–Wallis test with Dunn's multiple comparison post‐test. *P < 0.05, **P < 0.01. Mean bacterial load is indicated with error bars representing standard deviation. n = 5 mice per condition.
Fig. S4. Depletion of macrophages or dendritic cells in DTxR transgenic mice treated with DTx. Top row: immunohistochemical staining of anti‐F4/80‐reactive cells in inoculation site skin of mice treated with CRM (left panel) or DTx (right panel) at day 1 post‐inoculation and intoxication. Scale bars = 20 μm; images acquired at 100× magnification. Bottom row: fluorescence imaging of the site of inoculation at day 1 post‐inoculation and intoxication with B31‐A3 (wild‐type) B. burgdorferi expressing tdTomato (red) and CRM (left panel) or DTx (right panel). Dendritic cells in this transgenic mouse model express DTxR and eGFP (green). Spirochetes are indicated with an arrow. Scale bars = 20 μm; images acquired at 200× magnification.
Table S1. Subcutaneous ID50 determinations of B31‐A3 (wild‐type) and Δp66 Borrelia burgdorferi in β3 +/+ or β3 −/− mice 4 weeks post‐inoculation.
Table S2. Bacterial strains and plasmids used in this study.
Table S3. Oligonucleotide primers, 5′ → 3′.
Video S1. Motility of wild‐type and Δp66 B. burgdorferi is similar at the site of inoculation in vivo. RFP expressing B31 A3 and GFP expressing Δp66 B. burgdorferi were imaged at the site of inoculation 1 h post‐intradermal inoculation. A time‐lapse video was acquired at a magnification of 20×, exposure time of 2 s for each colour (green and red), time lapse set at 2 s interval for 5 min. For movie conversion, output timing was set at the interval of 200 ms.
Appendix S1. Supplemental experimental procedures.
Appendix S2. Supplemental experiments.