

Abstract
Inappropriate activation of CDK5 due to proteolytic release of the activator fragment p25 from the membrane contributes to the formation of neurofibrillary tangles, β-amyloid aggregation and chronic neurodegeneration. At 18 months of age, 3xTg-AD mice were sacrificed after either three weeks (short-term) or one year (long-term) of CDK5 knockdown. In short-term-treated animals, CDK5 knockdown reversed β-amyloid aggregation in the hippocampi via inhibitory phosphorylation of GSK3β Ser 9 and activation of phosphatase PP2A. In long-term-treated animals, CDK5 knockdown induced a persistent reduction in CDK5 and prevented β-amyloid aggregation, but the effect on APP processing was reduced, suggesting that yearly booster therapy would be necessary. These findings further validate CDK5 as a target for preventing or blocking amyloidosis in older transgenic mice.
Keywords: β-amyloid plaques, GSK3 β-Ser 9, PP2A, Alzheimer’s disease, CDK5 RNAi
Introduction
Alzheimer’s disease (AD) is the most common cause of senile dementia, with 24 million people affected around the world. AD patients show progressive declines in cognitive functions, such as memory and learning (Reitz et al. 2011; Reitz and Mayeux 2014). AD brains lose cerebral mass and synaptic plasticity (Sheng et al. 2012). β-amyloid (βA) accumulation and neurofibrillary tangles (NFT) are the main histopathological hallmarks of AD (Bancher et al. 1989). Tau is a microtubule-associated protein that promotes microtubule (MT) assembly and stabilization (Weingarten et al. 1975). Irregular processing of amyloid precursor protein (APP) by β-secretase induces βA accumulation (De Strooper et al. 2010), (Querfurth and LaFerla 2010). Additionally, aberrant regulation of kinases and phosphatases induces tau hyperphosphorylation and NFT formation in AD patients (Giannakopoulos et al. 2003; Iqbal et al. 2010).
In contrast, cyclin-dependent kinase (CDK5) is an important proline-directed serine/threonine kinase involved in several neuronal processes, such as neuronal migration, cytoskeleton remodeling, cortical cytoarchitecture, and synaptic plasticity (Angelo et al. 2006; Ip and Tsai 2008; Lalioti et al. 2010). The major CDK5 activator, p35, is cleaved to p25 by calpain in AD patients, inducing CDK5 overactivation (Kusakawa et al. 2000; Patrick et al. 1999). Increased CDK5 activity is closely related to tau hyperphosphorylation and NFT formation (Baumann et al. 1993). Our previous reports have shown that hippocampal CDK5 silencing induces a decrease in NFTs and improves memory and learning in an aged 3xtgAD mouse model (Castro-Alvarez et al. 2014a; Piedrahita et al. 2010).
CDK5 regulates several types of proteins, including glycogen synthase kinase 3β (GSK3β), and phosphatases, such as protein phosphatase 1 and 2A (PP1 and PP2A) (Castro-Alvarez et al. 2014b; Hou et al. 2013; Li et al. 2006). These proteins are important for the tau phosphorylation rate because GSKβ activity is increased and the activities of phosphatases are decreased in AD (Lee et al. 2011; Martin et al. 2013). However, few details are known regarding the regulation of β-amyloid by CDK5, which is relevant during AD development (Wen et al. 2008a; Wen et al. 2008b). Therefore, in the present study, we evaluated when and how the short- and long-term silencing of CDK5 modifies β-amyloidosis, GSK and phosphatases in 3xTgAD model mice.
Materials and Methods
RNAi design
RNAi (short hairpin microRNA (sh-miR)) sequences used to silence CDK5 (shCDK5miR), and scrambled RNA sequences were used as controls (shSCRmiR) based on previously published sequences (Chang et al. 2006; Piedrahita et al. 2010). These sequences were cloned into human miR-30-based stem loops by polymerase extension of overlapping DNA oligonucleotides. To clone RNAi vectors for AAV production, the following primers were used for polymerase extension: shCDK5miR, forward primer 5′-AAAACTCGAGTGAGCGCTGACCAAGCTGCCAGACTATACTGTAAAGCCACAG ATGGG-3′, and shCDK5miR, reverse primer, 5′-AAAAACTAGTAGGCGTTGAC CAAGCTGCCAGACTATACCCATCTGT-GGCTTTACAG-3′, or shSCRmiR, forward primer, 5′-AAAACTCGAGTGAGCGCA-CCATCGAACCGTCAGAGTTAC TGTAAAGCCACAGATGGG-3′, and shSCRmiR reverse primer, 5′-AAAAACTAGTAGGCGTACCATCGAACCGTCAGAGTTACCCATCTGTGGCTTT ACAG-3′. These extension products were digested with XhoI and SpeI for directional cloning into a U6 expression plasmid cut with XhoI and XbaI (Boudreau et al. 2008).
Viral particle production
In the protocol used to produce the AAV particles, we accomplished the large-scale production of heterologous proteins using an Sf9 insect cell culture to co-infect recombinant baculovirus derived from the Autographa californica nuclear polyhedrosis virus (Urabe et al. 2002). shCDK5miR and shSCRmiR expression cassettes (driven by the mouse U6 promoter) were cloned into pAAV.CMV.hrGFP, which contains AAV serotype 2/5 inverted terminal repeats and a CMV-humanized Renilla GFP (hrGFP)-simian virus 40 poly(A) reporter cassette (Boudreau et al. 2009; Urabe et al. 2002). AAV titers were determined by quantitative PCR and/or DNA slot blot analysis. The AAV particles were dialyzed before use.
Animal procedures
A total of 47 18-month-old 3xTg-AD mice (Oddo et al. 2003) and 6 18-month-old control mice from a previous study were used. The mice were trained on a water maze for 10 days before being sacrificed (Castro-Alvarez et al. 2014a). The animals were obtained from our in-house, specific pathogen-free (SPF) colony at the vivarium at SIU, University of Antioquia, Medellin, Colombia, maintained on a 12:12-h dark:light cycle with food and water ad libitum. The animals were handled according to Colombian standards (law 84/1989 and resolution 8430/1993) and the NIH animal welfare care guidelines (Public Law 99-158, November 20, 1985, "Animals in Research"). Special care was taken to minimize animal suffering and to reduce the number of animals used.
Animals were anesthetized (ketamine (5%) and xylazine (2%) with a 50:5 mg/kg dose) and bilaterally injected with 1 μl of AAV2-shSCRmiR (shSCRmiR) or AAV2-shCDK5miR (shCDK5miR) into both hippocampi (bregma coordinates were −1.7 anteroposterior, 0.8 (right) and −0.8 (left) lateral, and 4 mm dorsoventral from the top of the skull). The injections were performed with a 10 μL Hamilton syringe at a rate of 0.1 μL/min, and 5 min elapsed after infusion before withdrawal of the syringe. The experimental groups consisted of the following animals: aged control mice that did not undergo injection, aged 3xTg-AD short-term-treated mice (ST-3xTg-AD) (18-month-old mice evaluated 3 weeks after injection) and 3xTg-AD long-term-treated mice (LT-3xTg-AD) (6-month-old mice evaluated one year after injection).
Western blotting
After behavioral testing, the animals were sacrificed, and the hippocampi were dissected, immediately frozen in liquid nitrogen and stored at −80°C until use. The samples were lysed in 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% NP40, 1 nM orthovanadate, 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Sigma-Aldrich) (Cardona-Gomez et al. 2004). The proteins were loaded on 8%, 10% and 10–20% Tricine gels and transferred to nitrocellulose membranes (GE Healthcare) at 250 mA for 2 h using an electrophoretic transfer system. The membranes were incubated overnight at 4°C with anti-CDK5 (C-8), anti-Akt, anti-I1PP2A, anti-I2PP2A, anti-PP1 (Santa Cruz Biotechnology), anti-PP2A, anti-GSK3β, anti-GSK3β pSer9, anti-Akt pSer473 (Cell Signaling), anti-I2PP1 (R&D Systems), anti-GSK3β pTyr216 (Sigma), anti-β-amyloid 1-16 (6E10) (SIGNET), anti-APP A4 (Millipore) anti-APP C-terminal antibody (Sigma) and mouse anti-βIII tubulin (Promega). IRDye 800CW goat anti-mouse or rabbit antibodies (LI-COR) and anti-mouse IgG or anti-rabbit IgG peroxidase conjugated antibodies (Pierce Biotechnology) were used as secondary probes. The blots were developed using the Odyssey Infrared Imaging System or the chemiluminescence method (ECL Western blotting system; GE Healthcare) followed by exposure to a radiographic film (ECL Hyperfilm; GE Healthcare). The films were analyzed using Quantity One, version 4.3.0 (Bio-Rad).
Immunoprecipitation
Animals (n=5 for each experimental group) were sacrificed by decapitation, and their brains were quickly removed. The hippocampi were dissected out and frozen at −80°C until analysis. The samples were homogenized in lysis buffer containing 150 mM NaCl, 20 mM Tris, pH 7.4, 10% glycerol, 1 mM EDTA, 1% NP40, 100 μM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin and leupeptin (Sigma) and 100 μM orthovanadate. The lysates were clarified by spinning at 14,000 rpm for 5 min. A protein assay was performed, and 180 μg of extract was incubated overnight at 4°C in the presence of anti-I2PP1 (R&D Systems) (1:250). Protein G-sepharose beads were added, and the incubation was continued for an additional two hours at room temperature. The immune complexes were washed three times with an immunoprecipitation lysis buffer prior to analysis by SDS-PAGE and immunoblotting. The proteins were separated with 10% SDS-PAGE, transferred to nitrocellulose membranes (Amersham), and probed with anti-PP1 (Santa Cruz Biotechnology) (1:1000). The lysates were used as positive controls, and IgG peptide incubation was used as a negative control for immunoprecipitation.
PP2A activity measurement
PP2A phosphatase activity from the hippocampi of 3xTg-AD mice (18-month-old) treated for three weeks or for 12 months with shCDK5miR or shSCRmiR respectively, was analyzed with a PP2A Immunoprecipitation Phosphatase Assay Kit (Millipore) as described in the manufacturer’s instructions.
Immunofluorescence
Mouse brains were fixed with 4% paraformaldehyde in PBS, cryopreserved with 30% sucrose and stored at 20°C. The brains were cut into 50 μm coronal sections with a vibratome (Leica 1000) and treated with 50 mM NH4Cl for 10 min at room temperature. The slices were preincubated for 1 h in 1% BSA with 0.3% Triton X-100 in 0.1 M PB. The primary antibodies, including anti-β-amyloid 1-16 (6E10) (SIGNET), were incubated overnight at 4°C. The secondary antibodies were conjugated to the fluorophore Alexa 594 (Molecular Probes). The slices were observed under an Olympus IX-81 confocal-DSU microscope. Intracellular β-amyloid immunofluorescence in pyramidal neurons from the CA1 area was analyzed for fluorescence intensity using Image Scope-Pro software (Media Cybernetics).
Immunohistochemistry
Aged control mouse ST-3xTg-AD and LT-3xTg-AD brain sections (50 μm) were pre-treated with formic acid (20%) for 20 min for β-amyloid detection. Then, they were treated for 20 minutes in 0.1 M PB (methanol (1:1) and 1% hydrogen peroxide) and incubated for 1 h in 0.1 M PB with 1% BSA and 0.3% Triton X-100. The slices were then incubated with the primary antibody, anti-β-amyloid 1-16 (6E10) (SIGNET), overnight at 4°C in 0.1 M PB with 0.3% BSA and 0.3% Triton X-100. The slices were incubated with biotinylated mouse secondary antibody and then with the ABC-HRP complex (Pierce Biotechnology). Diaminobenzidine (DAB) was used for detection. The tissue was dehydrated, covered with mounting solution and observed under an Eclipse E200 optical microscope (Nikon). The number of extracellular β-amyloid plaques was quantified in the CA1 area of the hippocampus.
Aβ40/Aβ42 ELISA
The Aβ40 and Aβ42 protein levels from the hippocampi of ST-3xTg-AD and LT-3xTg-AD mice were measured by ELISA as described in the manufacturer’s protocol (Convance BetaMark x-42 ELISA kit and BetaMark x-40 ELISA kit).
Statistics
At least n=3–5 mice were used for the histological and biochemical studies. Parametric data were compared using Student’s t-test for independent samples. Nonparametric data were evaluated using the Mann-Whitney U nonparametric test. SPSS software was used for the statistical analysis, and results were considered significant at p≤0.05. The values were expressed as means ± SEM.
Results
Reduction of βA aggregation in 3xTg-AD mice following short-term treatment with shCDK5miR
The role of CDK5 in AD pathogenesis has been widely studied for tauopathies but less so for β-amyloid aggregation. We evaluated the number of βA plaques in the hippocampus of ST-3xTg-AD mice via immunohistochemical staining with the 6E10 antibody and found reduced staining in the region of the CA1 injection site with shCDK5miR treatment. This finding was not observed with the shSCRmiR control and was comparable to observations of aged littermates (Fig. 1a). This result was supported by a significant reduction of β-amyloid immunofluorescence (70% vs control, p=0.05) (Fig. 1b). βA was also diminished in hippocampal lysates from ST-3xTg-AD mice treated with shCDK5miR. βA 40 and 42 fragments evaluated using ELISA showed a significant decrease in concentration (βA 40 (635.7 pg/mL ±251.9 (shCDK5miR) vs 1567 pg/mL ±247.2 (shSCRmiR); p>0.001) and βA 42 (1530 pg/mL ±183.5 (shCDK5miR) vs 2136 pg/mL ±150.2 (shSCRmiR); p=0.012)) (Fig. 1c). Biochemical analysis of APP and its cleavage fragments, soluble APP β (sAPPβ) and C-terminal fragment β (CTFβ), showed a significant decrease in shCDK5miR-treated animals (APP (68.8% ±7.1 vs control; p=0.048), sAPPβ (75.63% ±2.7 vs control; p=0.0014), and CTFβ (81.7% ±4.2 vs control; p=0.024)) (Fig. 1d).
Figure 1.

shCDK5miR treatment reduced amyloid deposition in the hippocampus at 3 weeks after administration. a) βA immunohistochemistry revealed a reduction in extracellular β-amyloid plaques in animals injected (short-term) with shCDK5miR compared to scrambled (shSCRmiR) injection in 18-month-old 3xTgAD mouse hippocampi and aged control mice. Magnification, 10X and 40X, scale bar, 100 μm and 20 μm. n=3–4. b) The fluorescence intensity of intracellular βA was diminished by short-term therapy with shCDK5miR in the hippocampal CA1 area of 3xTg-AD mice. Representative images of green fluorescence from pAAV.CMV.hrGFP for the shSCRmiR and shCDK5miR conditions are shown. c) These results were confirmed by a decrease in βA 1-40 and βA 1-42 levels measured using ELISA in the hippocampus of 18-month-old 3xTgAD mice injected with shCDK5miR or shSCRmiR. d) Western blots of APP, sAPPβ and CTFβ revealed a reduction in protein levels 3 weeks after the injection of shCDK5miR in 18-month-old 3xTgAD mice. Tubulin was used as a loading control, and densitometry quantification was performed. n= 5, *=p<0.05, **=p<0.01, ***=p<0.001.
Under the same short-term experimental conditions, the CDK5 protein level and its activity were reduced (Castro-Alvarez et al. 2014a). We evaluated the GSK3/AKT pathway, PP1, PP2A and their activators. We found a significant decrease in total GSK3β (90.1% ±2.7 vs control; p=0.037); however, there was an insignificant trend of increased GSK3β pSer9 and Akt pSer473 (GSK3β pSer9 158.7% ±46.1 vs control; p=0.293; Akt pSer473 135.3% ±47.8 vs control; p=0.513) (Fig. 2a). PP1 (90.3% ±1.8 vs control; p=0.012) and its inhibitor I2PP1 (94.8% ±0.9 vs control; p=0.028) were significantly reduced (Fig. 1a), but when we evaluated the complex of PP1/I2PP1, the data showed that shCDK5miR treatment induced a significant increase in PP1 associated with I2PP1 (50% vs control, p=0.04) (Fig. 2b). Additionally, PP2A was reduced (57.6% ±6.5 vs control; p=0.007); its inhibitor, I1PP2A, was unmodified, but another inhibitor, I2PP2A (30% vs control, p=0.05) (Fig. 2a), and PP2A (20% vs control, p=0.04) activity were significantly increased (Fig. 2b). Hence, CDK5 interference can not only diminish tau hyperphosphorylation but also reduce β-amyloidosis and modulate other proteins related to both proteinopathies, mainly by increasing PP2A activity during short-term treatment.
Figure 2.
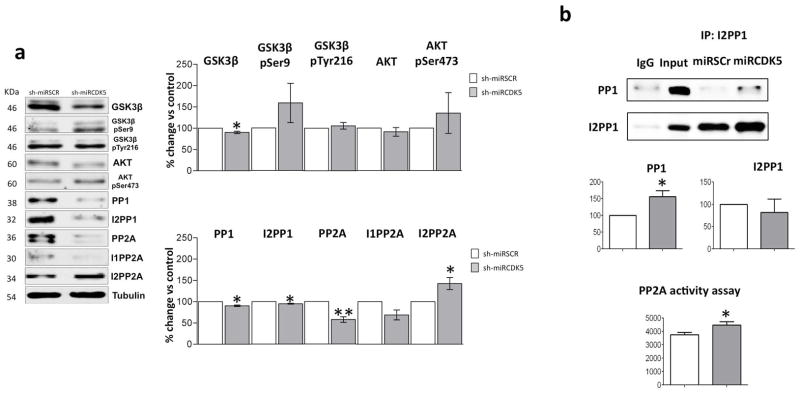
Reduced CDK5 activity during short-term treatment with shCDK5miR indirectly modulates other kinases and phosphatases. a) Changes in the GSK3β/AKT pathway, PP1 and PP2A phosphatases and their inhibitor levels were evaluated by Western blotting. Representative blots are shown. Tubulin was used as a loading control, and densitometry quantification was performed. n=3, *=p<0.05, **=p<0.01, ***=p<0.001. b) The I2PP1 and PP1 association was evaluated using immunoprecipitation. An elevated association of PP1 and I2PP1 and a trend of increased GSK3β-p Ser 9 suggests PP1 inactivation. Additionally, increased PP2A activity was directly detected using a phosphatase assay kit. Quantification and representative blots are shown. Densitometry quantification was performed. n=5, *=p<0.05.
Persistent effect of CDK5 knockdown on β-amyloid aggregation one year after delivery
Additionally, long-term CDK5 interference therapy continued to prevent β-amyloidosis, although the number of extracellular βA plaques were unmodified (Fig. 3a), the intracellular βA immunofluorescence presented a significant reduction (90% vs control, p=0.05) (Fig. 3b). However, it did not continue to have an effect on APP processing in the hippocampus of 3xTg-AD mice, since the protein levels of βA 40 and 42 did not significantly change but showed a tendency to increase (βA 40 (1245 pg/mL ±238.2 (shCDK5miR) vs 731.3 pg/mL ±49.7 (shSCRmiR); p=0.202) and βA 42 (1983 pg/mL ±118.6 (shCDK5miR) vs 1154 pg/mL ±317.5 (shSCRmiR); p=0.066)) (Fig. 3c). Additionally, the cleavage fragment CTFβ was significantly increased after one year of shCDK5miR treatment (358.2 ±74.7 vs control; p=0.02) (Fig. 3c). APP and sAPPβ showed a tendency to increase, but these changes were not significant (APP 165.2% ±25.6 vs control; p=0.084; sAPPβ 298.3% ±59.7 vs control; p=0.08) (Fig. 3d).
Figure 3.

Long-term therapy (one year after injections beginning at 6 months of age) with shCDK5miR prevents βA aggregation in the hippocampus of 3xTgAD mice. a) Immunohistochemistry did not indicate any changes in the extracellular β-amyloid plaques in 18-month-old 3xTgAD mouse hippocampi injected with shCDK5miR compared to shSCRmiR one year after treatment. Magnification, 10X and 40X, scale bars, 100 μm and 20 μm. n=3–4. b) The fluorescence intensity of intracellular βA in the CA1 region of 3xTg-AD mice remained reduced during long-term therapy with shCDK5miR. Representative images of green fluorescence from pAAV.CMV.hrGFP for the shSCR and shCDK5miR treatments are shown. c) The βA40 and βA42 levels were not affected at one year after treatment in the hippocampi of 18-month-old 3xTgAD mice injected with shCDK5miR or shSCRmiR. d) Western blotting of APP, sAPPβ and CTFβ showed an increasing trend during long-term treatment with shCDK5miR in 18-month-old 3xTgAD mice. Tubulin was used as a loading control, and densitometry quantification was performed. n=5, *=p<0.05.
Although CDK5 protein levels remained decreased, CDK5 activity showed no significant changes under the same long-term experimental conditions in 3xtgAD mice (Castro-Alvarez et al. 2014a). However, the long-term effect of CDK5 interference suggested its participation in GSK3β, PP1 and PP2A modulation. shCDK5miR treatment revealed that GSK3β expression (87.3% ±44.1 vs control; p=0.02) remained decreased after one year (Fig. 4a), and inhibitory phosphorylation of GSK3β at Ser 9 was significantly increased in 3xTg-AD mice after one year of therapy (199.2% ±22.2 vs control; p=0.021) (Fig. 4a). PP1 and its inhibitor I2PP1 were unmodified, but PP1 expression remained significantly increased in the complex with I2PP1 (50% vs control, p=0.04). PP2A expression remained reduced (72.2% ±7.4 vs control; p=0.033), I1PP2A achieved a significant reduction (40% vs control, p=0.04), and I2PP2A remained increased (51% vs control, p=0.04); however, PP2A activity was unmodified (p=0.485) (Fig. 4b).
Figure 4.

Inhibition of the association between GSK3β (p-Ser 9) and PP1/I2PP1 was detected during long-term treatment with shCDK5miR. a) Changes in the GSK3β and PP2A/I2PP2A protein levels were maintained after one year of CDK5 knockdown in 3xTgAD mice. Representative blots are shown. Tubulin was used as a loading control, and densitometry quantification was performed. n=3, *=p<0.05, **=p<0.01, ***=p<0.001. b) The I2PP1 and PP1 association was evaluated using Immunoprecipitation. This association suggests persistent PP1 inactivation, and changes in PP2A activity were not detected at one year post-injection. Quantification and representative blots are shown. Densitometry quantification was performed. n=5, *=p<0.05.
Discussion
Our previous research showed that short- and long-term CDK5 silencing reduces the phosphorylation of tau and improves learning and memory in triple transgenic Alzheimer’s mice (Castro-Alvarez et al. 2014a). Using those animals in the present study, we also found that intracellular βA was decreased following both time intervals of CDK5 silencing treatment, in contrast to extracellular βA plaques, APP processing and βA 1-40/1-42 production, which were only decreased with short-term treatment. This result suggests that CDK5 knockdown is relevant in the reversion of βA protein levels and for intra- and extracellular β-amyloidosis in older 3xTgAD mice for a shorter treatment period, considering that soluble βA (βA 1-40) is also involved in the pathogenesis of Alzheimer’s disease (Haass and Selkoe 2007; Benilova et al. 2012, Abelein et al. 2013). This finding also suggests that in an independent mode of this histopathological hallmark, CDK5 reduction down-regulates insoluble tau and improves learning and memory with long-term treatment, despite the recovery of basal enzymatic activity at one year post-injection. These observations are supported by similar results showing a reduction of p35 cleavage to p25 by calpain inhibition, which prevented cognitive alterations in transgenic mouse models (Liang et al. 2010; Medeiros et al. 2012). Furthermore, Crews et al. have shown the rescue of altered neurogenesis after pharmacological and genetic inhibition of CDK5 in APP transgenic mice (Crews et al. 2011).
The involvement of kinases and phosphatases in the process of phosphorylation/dephosphorylation of tau is essential for the development of tauopathy (Wang et al. 2007). GSK3β and CDK5 are the most important kinases in the hyperphosphorylation of tau and are crucial to the development of diseases associated with tau hyperphosphorylation (Plattner et al. 2006). However, it is difficult to determine the effect of CDK5 on GSK3β, especially in a chronic model of tauopathy, such as aged 3xTgAD mice (Noble et al. 2003; Wen et al. 2008a). In our work, we found that GSK3β may be modulated by CDK5 knockdown by reducing the basal protein levels and inducing a clear trend or increase in inhibitory phosphorylation at Ser 9 of GSK3β, which may be correlated with the short- and long-term decrease in hyperphosphorylated tau and PHF-1 formation. This study is the first report of this association in a model of AD.
Contrary to our observations, it has been reported that CDK5 can phosphorylate and promote the activation of the ErbB receptor (epidermal growth factor receptor), which, when activated by the binding of neuregulin (a member of the family of epidermal growth factor proteins), can induce the phosphorylation and activation of AKT. AKT is a serine/threonine kinase involved in cell survival pathways (Li et al. 2003) and may inactivate GSK3 through phosphorylation at Ser 9, preventing β-amyloidosis but inducing phosphorylated tau in older mice (Cross et al. 1995, Wen et al. 2008). However, both findings indicate that CDK5 has different direct or indirect means of regulating GSK3 activity, and its inhibition could be relevant for functions other than protein aggregation.
Other proteins involved in AD pathology include phosphatases, which have the ability to regulate the degree of phosphorylation of various proteins via dephosphorylation (Braithwaite et al. 2012). The most important phosphatases associated with the AD are PP1 and PP2A, which are reported to have the highest tau dephosphorylation activity (Liu et al. 2005). These phosphatases, in turn, have control over other substrates involved in the signaling pathway of AD (Braithwaite et al. 2012; Liu et al. 2013; Ducruet et al. 2005). PP1 can activate GSK3 via dephosphorylation at Ser 9 (Hernandez et al. 2010; Bennecib et al. 2000), and PP2A can dephosphorylate and regulate AKT, thereby inhibiting its action on GSK3 (Resjo et al. 2002; Mora et. al. 2002) Thus, phosphatases can influence the pathological conditions of AD through various pathways in which phosphatase activity can be altered to induce tau dysregulation (Liu et al. 2005). However, an imbalance in GSK3/PP1 is implicated in βA aggregation (Lopez-Menendez et al. 2013; Vintem et al. 2009). Because activation of GSK3β induces phosphorylation of the inhibitor I2PP1, the activation of PP1 in the GSK/I2PP1/PP1 molecular complex (Sakashita et al. 2003) may be a key factor in disease development.
Nevertheless, CDK5 is involved in the regulation of PP1 through the phosphorylation of inhibitor 1 (I1PP1) and inhibitor 2 (I2PP1), which both regulate activity by binding to PP1 (Oliver et al. 1998). The dephosphorylated form of I2PP1 can bind to and inhibit PP1, CDK5 and GSK3 can phosphorylate and prevent the action of I2PP1 on PP1 (Agarwal-Mawal and Paudel 2001). Unlike I2PP1, the dephosphorylated form of I1PP1 remains inactive and does not perform any function on PP1, but when I1PP1 is phosphorylated by PKA, CDK5 can bind to and inhibit PP1 (Huang and Paudel 2000; Nguyen et al. 2007). Interestingly, the 3xTg-AD mice treated with shCDK5miR for both evaluated time intervals showed co-immunoprecipitation of I2PP1 and PP1, which is associated with the inhibition of phosphatase PP1 and most likely associated with the trend toward increased phosphorylation of GSK3 at Ser 9 during the short- and long-term treatments. This process could be related to the significant reduction of intracellular β-amyloid in the present study, as well as the reversion and prevention of hyperphosphorylated tau and the consequent improvement of cognitive function during short- and long-term treatments in older 3xTgAD mice (Castro-Alvarez et. al. 2014a).
PP2A is the most important phosphatase in the process of tau phosphorylation; its inhibition under normal conditions is associated with the hyperphosphorylation of tau (Liu et al. 2005; Louis et al. 2011; Planel et al. 2001). Additionally, its inhibitor, I2PP2A, is directly involved in the abnormal hyperphosphorylation of tau and the inhibition of PP2A activity (Chohan et al. 2006, Arif et al. 2014). Interestingly, our data demonstrated that CDK5 interference significantly increased PP2A activation despite the reduction in PP2A and increase in I2PP2A total protein levels. These findings could be supported by a recent report in which the inhibitory phosphorylation of GSK3 at Ser 9 constrained I2PP2A to the cytoplasm, avoiding an inactivation with PP2A (Yu et al. 2013). Furthermore, GSK3β phosphorylated at Ser 9 induces PP2A activity, thereby preventing amyloidosis (Noh et al. 2013).
However, our data suggest that when shCDK5miR loses its efficiency for reducing CDK5 activity at one year post-injection (Castro-Alvarez et al. 2014a), there is a concomitant loss of PP2A activity, loss of efficiency on the reversion of APP processing and the presence of extracellular plaques, suggesting a close relationship between CDK5 and PP2A in the control of βA aggregation, possibly through the regulation of GSK3β. Which could be supported by previous studies indicating that PP2A and GSK3β are involved in βA accumulation (Liu et al. 2013; Ryder et al. 2003; Wen et al. 2008a).
Together, our experimental data suggest that gene therapy based on silencing CDK5 prevents not only tauopathy but also intracellular β-amyloidosis during short- and long-term treatment through regulation of the phosphates PP1 and PP2A and inhibition of GSK3 β at Ser 9. This conclusion suggests that CDK5 can control tauopathy (Castro-Alvarez & Uribe-Arias et al. 2014b) and possibly β-amyloidosis. However, the recovery of CDK5 activity after long-term therapy introduces variation in the control of phosphatases and extracellular β-amyloidosis. Therefore, our findings could indicate that a therapy booster is necessary each year to achieve efficiency in CDK5 silencing.
Acknowledgments
We would like to thank Dr Frank Laferla at the Institute for Brain Aging and Dementia, University of California in Irvine, USA for donating the 3xtg AD mice; Dr Beverly Davidson and Dr Maria Scheel at the Viral Vector Core and Davidson Laboratory, University of Iowa, USA for viral vector expert advice. JFC-A was awarded a PhD Fellowship by Colciencias (AU-A, Young Researcher Program 2012 from Colciencias). Research reported in this publication was supported by Colciencias Projects #111545921503 and 111551928905 (GPC-G), Fogarty International Center and the NIA NIH Institute under Award Number RO1-AG029802-01 (CoPI GPC-G).
Footnotes
Disclosure statement
The authors report no conflicts of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Angelo M, Plattner F, Giese KP. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem. 2006;99(2):353–370. doi: 10.1111/j.1471-4159.2006.04040.x. [DOI] [PubMed] [Google Scholar]
- Agarwal-Mawal A, Paudel HK. Neuronal Cdc2-like protein kinase (Cdk5/p25) is associated with protein phosphatase 1 and phosphorylates inhibitor-2. J Biol Chem. 2001;276(26):23712–8. doi: 10.1074/jbc.M010002200. [DOI] [PubMed] [Google Scholar]
- Arif M, Wei J, Zhang Q, Liu F, Basurto-Islas G, Grundke-Iqbal I, Iqbal K. Cytoplasmic Retention of Protein Phosphatase 2A Inhibitor 2 (I2PP2A) Induces Alzheimer-like Abnormal Hyperphosphorylation of Tau. J Biol Chem. 2014;289(40):27677–91. doi: 10.1074/jbc.M114.565358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989;477(1–2):90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336(3):417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000;485(1):87–93. doi: 10.1016/s0014-5793(00)02203-1. [DOI] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–57. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17(6):1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Monteys AM, Davidson BL. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. Rna. 2008;14(9):1834–1844. doi: 10.1261/rna.1062908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer’s disease. Prog Mol Biol Transl Sci. 2012;106:343–79. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona-Gomez P, Perez M, Avila J, Garcia-Segura LM, Wandosell F. Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci. 2004;25(3):363–373. doi: 10.1016/j.mcn.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Castro-Alvarez JF, Uribe Arias SA, Mejia Raigoza D, Cardona Gomez GP. Cyclin-dependent kinase 5, a node protein in diminished tauopathy: a systems biology approach. Frontiers in Aging Neuroscience. 2014a:6. doi: 10.3389/fnagi.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alvarez JF, Uribe-Arias SA, Kosik KS, Cardona-Gomez GP. Long and short-term CDK5 knockdown prevents spatial memory dysfunction and tau pathology of triple transgenic Alzheimer’s mice. Frontiers in Aging Neuroscience. 2014b:6. doi: 10.3389/fnagi.2014.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Elledge SJ, Hannon GJ. Lessons from Nature: microRNA-based shRNA libraries. Nat Methods. 2006;3(9):707–714. doi: 10.1038/nmeth923. [DOI] [PubMed] [Google Scholar]
- Chohan MO, Khatoon S, Iqbal IG, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006;580(16):3973–3979. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Crews L, Patrick C, Adame A, Rockenstein E, Masliah E. Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer’s disease. Cell Death Dis. 2011;2:e120. doi: 10.1038/cddis.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducruet AP, Vogt A, Wipf P, Lazo JS. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer’s disease. Annu Rev Pharmacol Toxicol. 2005;45:725–50. doi: 10.1146/annurev.pharmtox.45.120403.100040. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Langa E, Cuadros R, Avila J, Villanueva N. Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol Cell Biochem. 2010;344(1–2):211–5. doi: 10.1007/s11010-010-0544-0. [DOI] [PubMed] [Google Scholar]
- Hou H, Sun L, Siddoway BA, Petralia RS, Yang H, Gu H, Nairn AC, Xia H. Synaptic NMDA receptor stimulation activates PP1 by inhibiting its phosphorylation by Cdk5. J Cell Biol. 2013;203(3):521–535. doi: 10.1083/jcb.201303035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KX, Paudel HK. Ser67-phosphorylated inhibitor 1 is a potent protein phosphatase 1 inhibitor. Proc Natl Acad Sci U S A. 2000;97(11):5824–9. doi: 10.1073/pnas.100460897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip N, Tsai LH. Cyclin Dependent Kinase 5 (Cdk5) New York: Springer; 2008. [Google Scholar]
- Iqbal K, Wang X, Blanchard J, Liu F, Gong CX, Grundke-Iqbal I. Alzheimer’s disease neurofibrillary degeneration: pivotal and multifactorial. Biochem Soc Trans. 2010;38(4):962–966. doi: 10.1042/BST0380962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275(22):17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lalioti V, Pulido D, Sandoval IV. Cdk5, the multifunctional surveyor. Cell Cycle. 2010;9(2):284–311. doi: 10.4161/cc.9.2.10466. [DOI] [PubMed] [Google Scholar]
- Lee S, Hall GF, Shea TB. Potentiation of tau aggregation by cdk5 and GSK3beta. J Alzheimers Dis. 2011;26(2):355–364. doi: 10.3233/JAD-2011-102016. [DOI] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278(37):35702–9. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry. 2006;45(10):3134–3145. doi: 10.1021/bi051635j. [DOI] [PubMed] [Google Scholar]
- Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2010;285(36):27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. The European journal of neuroscience. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Liu GP, Wei W, Zhou X, Shi HR, Liu XH, Chai GS, Yao XQ, Zhang JY, Peng CX, Hu J, Li XC, Wang Q, Wang JZ. Silencing PP2A inhibitor by lenti-shRNA interference ameliorates neuropathologies and memory deficits in tg2576 mice. Mol Ther. 2013;21(12):2247–2257. doi: 10.1038/mt.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Menendez C, Gamir-Morralla A, Jurado-Arjona J, Higuero AM, Campanero MR, Ferrer I, Hernandez F, Avila J, Diaz-Guerra M, Iglesias T. Kidins220 accumulates with tau in human Alzheimer’s disease and related models: modulation of its calpain-processing by GSK3beta/PP1 imbalance. Hum Mol Genet. 2013;22(3):466–482. doi: 10.1093/hmg/dds446. [DOI] [PubMed] [Google Scholar]
- Louis JV, Martens E, Borghgraef P, Lambrecht C, Sents W, Longin S, Zwaenepoel K, Pijnenborg R, Landrieu I, Lippens G, Ledermann B, Gotz J, Van Leuven F, Goris J, Janssens V. Mice lacking phosphatase PP2A subunit PR61/B′delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A. 2011;108(17):6957–6962. doi: 10.1073/pnas.1018777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F. Tau protein phosphatases in Alzheimer’s disease: the leading role of PP2A. Ageing Res Rev. 2013;12(1):39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Kitazawa M, Chabrier MA, Cheng D, Baglietto-Vargas D, Kling A, Moeller A, Green KN, LaFerla FM. Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol. 2012;181(2):616–625. doi: 10.1016/j.ajpath.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, Centeno F. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell Signal. 2002;14(6):557–62. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38(4):555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Noh MY, Koh SH, Kim SM, Maurice T, Ku SK, Kim SH. Neuroprotective effects of donepezil against Abeta42-induced neuronal toxicity are mediated through not only enhancing PP2A activity but also regulating GSK-3beta and nAChRs activity. J Neurochem. 2013;127(4):562–574. doi: 10.1111/jnc.12319. [DOI] [PubMed] [Google Scholar]
- Nguyen C, Nishi A, Kansy JW, Fernandez J, Hayashi K, Gillardon F, Hemmings HC, Jr, Nairn AC, Bibb JA. Regulation of protein phosphatase inhibitor-1 by cyclin-dependent kinase 5. J Biol Chem. 2007;282(22):16511–20. doi: 10.1074/jbc.M701046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:D961–72. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla F, Gallego-Gomez JC, Kosik KS, Cardona-Gomez GP. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci. 2010;30(42):13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276(36):34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281(35):25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Mayeux R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol. 2014;88(4):640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resjo S, Göransson O, Härndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14(3):231–8. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312(4):922–929. doi: 10.1016/j.bbrc.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Sakashita G, Shima H, Komatsu M, Urano T, Kikuchi A, Kikuchi K. Regulation of type 1 protein phosphatase/inhibitor-2 complex by glycogen synthase kinase-3beta in intact cells. J Biochem. 2003;133(2):165–171. doi: 10.1093/jb/mvg020. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4(5) doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13(16):1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Vintem AP, Henriques AG, da Cruz ESOA, da Cruz ESEF. PP1 inhibition by Abeta peptide as a potential pathological mechanism in Alzheimer’s disease. Neurotoxicol Teratol. 2009;31(2):85–88. doi: 10.1016/j.ntt.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25(1):59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, Duff KE. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008a;28(10):2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I, Maurin T, Wang L, Figueroa H, Herman M, Krishnamurthy P, Liu L, Planel E, Lau LF, Lahiri DK, Duff K. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008b;57(5):680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Yan T, Feng Y, Liu X, Xia Y, Luo H, Wang JZ, Wang X. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol Aging. 2013;34(7):1748–1758. doi: 10.1016/j.neurobiolaging.2012.12.025. [DOI] [PubMed] [Google Scholar]