

Abstract
Glial cells play a critical role in shaping neuronal development, structure, and function. In a screen for Caenorhabditis elegans mutants that display dopamine (DA)-dependent, Swimming-Induced Paralysis (Swip), we identified a novel gene, swip-10, the expression of which in glia is required to support normal swimming behavior. swip-10 mutants display reduced locomotion rates on plates, consistent with our findings of elevated rates of presynaptic DA vesicle fusion using fluorescence recovery after photobleaching. In addition, swip-10 mutants exhibit elevated DA neuron excitability upon contact with food, as detected by in vivo Ca2+ monitoring, that can be rescued by glial expression of swip-10. Mammalian glia exert powerful control of neuronal excitability via transporter-dependent buffering of extracellular glutamate (Glu). Consistent with this idea, swip-10 paralysis was blunted in mutants deficient in either vesicular Glu release or Glu receptor expression and could be phenocopied by mutations that disrupt the function of plasma membrane Glu transporters, most noticeably glt-1, the ortholog of mammalian astrocytic GLT1 (EAAT2). swip-10 encodes a protein containing a highly conserved metallo-β-lactamase domain, within which our swip-10 mutations are located and where engineered mutations disrupt Swip rescue. Sequence alignments identify the CNS-expressed gene MBLAC1 as a putative mammalian ortholog. Together, our studies provide evidence of a novel pathway in glial cells regulated by swip-10 that limits DA neuron excitability, DA secretion, and DA-dependent behaviors through modulation of Glu signaling.
Keywords: C. elegans, dopamine, glia, glutamate, transporters
Introduction
Dopamine (DA) signaling is a key feature of the neuromodulatory repertoire of animals across phylogeny. In vertebrates, DA regulates cognitive, reward, and motor behaviors, with dysfunction of these processes underlying addiction, attention-deficit hyperactivity disorder, schizophrenia, and Parkinson's disease (Wise, 2004; Mazei-Robinson and Blakely, 2006; Sulzer and Surmeier, 2013; Laruelle, 2014). Through its actions at both D1- and D2-type G-protein coupled receptors, DA regulates the inherent excitability of cells that receive inputs from fast-acting neurotransmitters such as glutamate (Glu), gamma-aminobutyric acid GABA, and acetylcholine (ACh) (Surmeier et al., 2007). Conversely, Glu, GABA, and ACh regulate the excitability of DA neurons, with perturbations of DA neuron excitability and DA secretion contributing to neuropsychiatric and neurodegenerative disorders (Nestler, 2005). Glial mechanisms also factor prominently in the actions of DA; for example, significant contributions of glial Glu transporters (GLTs) have been implicated in the actions of drugs that act through DA signaling, such as cocaine (Kalivas, 2009).
With its short life cycle, simple neural anatomy, genetic tractability, extensive behavioral repertoire, and use of conserved signaling molecules, including DA and Glu, the nematode Caenorhabditis elegans has been used extensively in unbiased screens to discovery novel genetic pathways that control nervous system development and function (Bargmann, 1993; Schafer, 2005). Key to such efforts has been the identification of phenotypes associated with a specific circuit or neurotransmitter (Waggoner et al., 1998; Richmond and Jorgensen, 1999; Nass et al., 2002; Chase et al., 2004; Chase, 2007; Rand, 2007). Previously, we identified a robust locomotory phenotype known as Swimming Induced Paralysis (Swip) that is evident in animals lacking the presynaptic DA transporter (DAT) dat-1 (McDonald et al., 2007). We found that the Swip of dat-1 animals is reversed by pharmacological antagonism of vesicular DA uptake, as well as by mutations in genes that encode the ortholog of tyrosine hydroxylase, cat-2, or the D2-like DA receptor dop-3. We also showed that Swip could be used to identify residues in DAT-1 protein that are critical for transporter synaptic localization and ability to accumulate the neurotoxin 6-OHDA. More recently, we described the use of Swip to support a forward genetic approach to identify novel regulators of DA signaling (Hardaway et al., 2012). This effort led to the identification of multiple, new dat-1 alleles, as well as mutants at other loci that displayed distinct behavioral and neurochemical profiles.
Here, we report that two of the previously identified Swip mutant alleles, vt29 and vt33, harbor independent mutations in a previously unstudied gene, F53B1.6, that we here designate swip-10. Notably, swip-10 contributions to swimming behavior arise from its expression in nematode glial cells, with the effects of mutations consistent with an increase in DA neuron excitability, excess vesicular fusion at DA synapses, and unconstrained DA-mediated inhibition of movement. swip-10 encodes a conserved, metallo-β-lactamase domain (MBD)-containing protein, which our genetic and behavioral studies suggest exerts powerful control over extracellular Glu availability and signaling.
Materials and Methods
C. elegans strains. Strains were maintained as described previously (Brenner, 1974) We thank J. Rand (Oklahoma Medical Research Foundation); the Caenhorhabditis Genetics Center (funded by the National Institutes of Health's Office of Research Infrastructure Programs by Grant P40 OD010440); Shohei Mitani of the National Bioresource Project at Tokyo Women's Medical University; and Shai Shaham, Oliver Hobert, and Leon Avery for providing the strains used in this work. N2 (Bristol) served as our wild-type strain. Strains used in the generation of data that appear in the figures are enumerated as such in Table 1. Additional strains used in this study in the general order of their contributions to the text are as follows: BY1067: vtEx191 (swip-10b::GFP), vprIs128(hlh-17::dsRed2); VPR128 vprIs128 (phlh-17:dsRed2); BY1104-BY1106: swip-10(vt29) vtEx212-214 (hlh-17:: swip-10 cDNA); BY1107-1109: swip-10(vt29) vtEx215-217 (swip-10b:: swip-10 cDNA); TQ800: lite-1(Xu7); BY1056: swip-10(tm5915) lite-1(Xu7); KP4: glr-1(n2461); RB1808: glr-2(ok2342); TM669: glr-2(tm669); TM3506: glr-5(tm3506); TM2877: glr-7(tm2877); VM487: nmr-1(ak4); TM3285: nmr-2(tm3285); TM355: mgl-2(tm355); DA2250: mgl-2(tm355), mgl-1(tm1811); TM1766: mgl-3(tm1766); BY1103: glr-1(n2461), swip-10(vt29); BY1102: glr-2(ok2342), swip-10(vt29); BY926: glr-5(tm3506), swip-10(vt29); BY942: mgl-2(tm355), swip-10(vt29); BY943: mgl-3(tm1766), swip-10(vt29); BY914: nmr-1(ak4), swip-10(vt29); BY923: nmr-2(tm3285), swip-10(vt29); ZB1106: glt-3(bz34); glt-1(ok206); RB2185: glt-4(ok2961); RB1615: glt-5(ok1987); BY1058: glt-3(bz34); swip-10(tm5915); OS2649: hlh-17(ns204); OS2659: hlh-17(ns204) hlh-31(ns217) hlh-32(ns223); OS1914: nsIs105(hlh-17::GFP).
Table 1.
C. elegans strains used in the generation of data that appear in the figures
| Figure | Strain no. | Background alleles | Transgene nos. | Transgene |
|---|---|---|---|---|
| 1 | TM2261 | cat-2(tm2261) | ||
| LX703 | dop-3(vs106) | |||
| BY810 | swip-10(vt29) | |||
| BY828 | swip-10(vt33) | |||
| BY1010/TM5915 | swip-10(tm5915) | |||
| BY957 | cat-2(tm2261);swip-10(vt29) | |||
| BY873 | cat-2(tm2261);swip-10(vt33) | |||
| BY1034 | cat-2(tm2261);swip-10(tm5915) | |||
| BY1030 | swip-10(tm5915) dop-3(vs106) | |||
| 2 | BY1139-BY1141 | N2 | vtEx83-vtEx85 | swip-10a:CFP |
| BY1049-BY1050 | N2 | vtEx191-vtEx192 | swip-10b:GFP | |
| BY859-BY862 | N2 | vtEx94-97 | swip-10c:GFP | |
| BY1037-BY1039 | N2 | vtEx182-vtEx184 | swip-10b:GFP, dat-1:mCherry | |
| OS1917 | N2 | nsIs108 | ptr-10:mRFP | |
| BY1051 | N2 | nsIs108;vtEx191 | ptr-10:mRFP;swip-10b:GFP | |
| 3 | BY837-838 | swip-10(vt29) | vtEx74-75 | swip-10 genomic fragment |
| BY844-846 | swip-10(vt33) | vtEx76-78 | swip-10 genomic fragment | |
| BY1004-1006 | swip-10(vt29) | vtEx159-161 | dat-1:swip-10 cDNA | |
| BY1076-1078 | swip-10(vt29) | vtEx200-202 | ptr-10:swip-10 cDNA | |
| BY1079-1081 | swip-10(tm5915) | vtEx203-205 | ptr-10:swip-10 cDNA | |
| BY1090-1092 | swip-10(vt33) | vtEx206-208 | ptr-10:swip-10 cDNA | |
| BY1093-1095 | swip-10(vt29) | vtEx209-211 | ptr-10:swip-10 cDNA lactamase dead | |
| 4 | IR724 | N2 | uvEx724 | asic-1:SNB-1::SEpHlourin, pRF4 |
| BY1114 | swip-10(tm5915) | uvEx724 | asic-1:SNB-1::SEpHlourin, pRF4 | |
| N2 | ||||
| TM2261 | cat-2(tm2261) | |||
| RM2702 | dat-1(ok157) | |||
| BY810 | swip-10(vt29) | |||
| BY957 | cat-2(tm2261);swip-10(vt29) | |||
| 5 | TQ733 | N2 | XuIs14 | dat-1:GCaMP1.3, dat-1:dsRed2 |
| BY1059 | swip-10(tm5915) lite-1(Xu7) | XuIs14 | dat-1:GCaMP1.3, dat-1:dsRed2 | |
| BY1122 | swip-10(tm5915) lite-1(Xu7) | XuIs14;vtEx227 | dat-1:GCaMP1.3, dat-1:dsRed2, ptr-10:swip-10 cDNA | |
| BY1123 | swip-10(tm5915) lite-1(Xu7) | XuIs14;vtEx228 | dat-1:GCaMP1.3, dat-1:dsRed2, ptr-10:swip-10 cDNA | |
| 6 | MT6308 | eat-4(ky5) | ||
| BY810 | swip-10(vt29) | |||
| BY828 | swip-10(vt33) | |||
| BY890 | eat-4(ky5), swip-10(vt29) | |||
| BY891 | eat-4(ky5), swip-10(vt33) | |||
| RM2702 | dat-1(ok157) | |||
| BY1010/TM5915 | swip-10(tm5915) | |||
| TM3239 | glr-4(tm3239) | |||
| TM2729 | glr-6(tm2729) | |||
| TM1811 | mgl-1(tm1811) | |||
| BY919 | glr-4(tm3239), swip-10(vt29) | |||
| BY1032 | swip-10(tm5915) glr-6(tm2729) | |||
| BY1031 | swip-10(tm5915) mgl-1(tm1811) | |||
| BY1052 | swip-10(tm5915) glr-6(tm2729) mgl-1(tm1811) | |||
| BY1053 | glr-4(tm3239), swip-10(tm5915) mgl-1(tm1811) | |||
| BY1054 | glr-4(tm3239), swip-10(tm5915) glr-6(tm2729) | |||
| BY1097 | glr-4(tm3239), swip-10(tm5915) glr-6(tm2729) mgl-1(tm1811) | |||
| BY1101 | dat-1(ok157), glr-4(tm3239) | |||
| BY1088 | dat-1(ok157), glr-6(tm2729) | |||
| BY1089 | dat-1(ok157), mgl-1(tm1811) | |||
| 7 | ZB1098 | glt-4(bz69) | ||
| ZB1099 | glt-5(bz70) | |||
| TM1316 | glt-6(tm1316) | |||
| TM1641 | glt-7(tm1641) | |||
| BY995 | glt-1(ok206) | |||
| ZB1096 | glt-3(bz34) | |||
| BY920 | cat-2(tm2261), glt-1(ok206) | |||
| BY931 | cat-2(tm2261), glt-3(bz34) | |||
| BY927 | cat-2(tm2261), glt-4(bz69) | |||
| BY1033 | swip-10(tm5915) glt-1(ok206) | |||
| BY1057 | glt-3(bz34), swip-10(vt29) | |||
| BY1058 | glt-3(bz34);swip-10(tm5915) |
Lines are organized by order of figure appearance as noted. Strains used for studies noted only in the text are listed in the Materials and Methods.
Plasmid construction and transgenic manipulations.
For promoter fusions, cloning of putative swip-10 promoter elements was performed using overlap PCR fusion (Hobert, 2002). Three different swip-10 genomic fragments were assayed for their ability to drive the expression of fluorescent reporter proteins: (1) the minimal 5′ promoter, a fragment spanning −738 to the ATG start site was fused to CFP coding sequences and injected at 50 ng/μl onto a lin-15(n765ts) background with a lin-15(+) rescue plasmid (pJM23) to generate vtEx83, vtEx84, and vtEx85; (2) the fifth intron (internal promoter), a fragment spanning 1013–2504 downstream of the ATG start site was fused to GFP and injected at 50 ng/μl onto a lin-15(n765ts) background with pJM23 to generate vtEx191 and vtEx192. This fragment was also coinjected with pdat-1:mCherry (10 ng/μl) and pJM23 (10 ng/μl) onto a lin-15(n765ts) background to generate vtEx182–184; and (3) the full-length promoter (containing all putative cis-elements), a fragment spanning −738 upstream to 2504 downstream of the ATG start site was fused to CFP and injected at 50 ng/μl onto a lin-15(n765ts) background with pdat-1:mCherry (20 ng/μl) to generate vtEx94, vtEx95, vtEx96, and vtEx97. For rescue constructs, a full-length 4.3 kb genomic fragment from −738 to 3569 downstream of the ATG start site was amplified and injected at 20 ng/μl onto swip-10(vt29) and swip-10(vt33) with punc-122:GFP (20 ng/μl) to generate vtEx74-75 and vtEx76-78, respectively. For cell autonomy experiments, we used swip-10 cDNA constructs exclusively because of the presence of a large internal promoter in the swip-10 gene. We harvested total RNA from asynchronous N2 animals (TRIzol) and performed first-strand cDNA synthesis using the Superscript III First-Strand Synthesis System (Life Technologies). We amplified a 1.6 kb PCR product using the following primers: ATGCTTTTTCATTTTCTAATCGCTA(S) and TTAACATTTCAAAGCTTTCTT(A). This fragment was TA cloned into PCR8/GW/TOPO for further manipulations.
Rescue constructs were generated in one of four backbones pRB1106-pRB1109, which contain unique restriction sites to allow for subcloning of promoters, open reading frames (ORFs), or 3′-UTRs. In all cases, insertion of the fragment and fidelity of the cassette was confirmed by sequencing. All constructs resulted in C-terminal cDNA fusion to an unc-54 3′UTR. We fused GFP to the C terminus of swip-10 cDNA and subcloned this PCR fragment into pRB1106 (700 bp dat-1 promoter) using AscI and KpnI as 5′ and 3′ restriction sites, respectively, to generate pRB1157. To express swip-10 in glia, we amplified a 315 bp fragment of the ptr-10 promoter from OS1917 worm lysate and subcloned it into pRB1157 using SphI and AscI as 5′ and 3′ sites, respectively, to generate pRB1158. To express swip-10 in cephalic sheath (CEPsh) glia, we amplified a 2.6 kb product from phlh-17:GFP (gift from Shai Shaham) and subcloned it into pRB1106 using FseI and AscI as 5′ and 3′ sites, respectively, generating pRB1159. A 1.5 kb fragment of the swip-10 internal promoter was subcloned into pRB1106 using FseI and AscI as 5′ and 3′ sites, respectively, generating pRB1160. To disrupt the predicted metal-binding capacity and catalytic function of the swip-10 MBD, His residues at amino acid positions 351, 353, 356, 405, and 470 were mutated to Ser via site-directed mutagenesis (QuikChange II XL; Agilent Technologies) in the pRB1158 backbone to generate pRB1162.
Genetic crosses.
Crosses were performed using publically available integrated fluorescent reporter strains to mark chromosomes in trans. Single worm PCR was performed to confirm the presence of the indicated mutation. For all deletions, we used a three primer multiplex strategy that produces PCR amplicons with a 100–200 bp difference between N2 and mutant. This was highly effective in eliminating preferential amplification of a lower-molecular-weight species. In all cases, a synthetic heterozygous control was used to ensure that heterozygous clones could be identified. We identified recombinant lines by PCR genotyping single worm genomic DNA lysates. All PCRs were performed with Platinum PCR Supermix (Life Technologies). In some cases, alleles were sequenced with sequence-specific primers to verify mutation homozygosity.
Whole-genome sequencing and sequence analysis.
Genomic DNA was isolated as described previously (Sarin et al., 2008). Briefly, BY200 (parental strain), vt29, and vt33 worms were harvested from a 10 cM 8p/NA22 plate by rinsing with M9. After a brief preclearing wash with M9, worms were rocked in M9 for 2–3 h to clear ingested bacteria and then washed with M9 and pelleted for DNA extraction. Before extraction, the worm pellet was incubated at −80°C for 1 h to overnight. Genomic DNA was extracted from the worm pellet using a Qiagen Gentra Puregene kit as described by the manufacturer and a post hoc phenol/chloroform extraction, RNase A digestion, and additional phenol/chloroform extraction. Quality of the gDNA was confirmed on a 2% agarose gel before submitting the samples for Illumina sequencing (Vanderbilt Genome Technology Core). Sequencing libraries were generated from gDNA as described previously (Sarin et al., 2008). Each sample was assigned a unique barcode so that samples could be pooled onto several flow cells of an Illumina Genome Analyzer Iix for sequencing as single-end 76mers. Sequence reads were filtered for quality and offloaded in Fastq format for subsequent analysis. Sequence data were analyzed in MaqGene (Bigelow et al., 2009) and a text file containing mutations against the reference sequence (WS180) was extracted. We then compared these mutant strain lists against the parental strain (BY200) to identify mutagen-induced single nucleotide polymorphisms (SNPs) and compared these with the previously mapped interval on LGX (Hardaway et al., 2012) to identify the mutant sites in F53B1.6.
Confocal imaging.
Confocal microscopy of transgenic worm–promoter fusions was performed using either a Zeiss LSM 510 or 710 confocal microscope and LSM or Zen capture software. Worms were immobilized using 20 mm levamisole with 0.1% Tricaine in M9 on a fresh 2% agarose pad and coverslipped with a 1 mm cover glass before sealing with paraffin wax (Smith et al., 2010).
Swip assays.
Manual Swip assays were conducted as described previously (Hardaway et al., 2014). In both manual and single worm analyses, we generated synchronous populations of strains by hypochlorite treatment and the harvesting of L1 arrested animals. Early- to middle-stage L4 animals were identified subsequently by characteristic morphology and used in behavior tests because N2 animals display occasional Swip and quiescent bouts during the last larval molt. For automated analysis, single L4 hermaphrodites were placed in 20 μl of water or drug in a single well of a Pyrex Spot Plate (catalog #13-748B; Fisher), and 10 min movies (uncompressed AVI format) of their swimming behavior were created and analyzed as described previously using SwimR (Hardaway et al., 2014). For this study, we used default SwimR settings without outlier exclusion. For manual assays, data shown for each strain represents the average of at least eight assays performed with three different experimental preps by at least two experimenters for a total of 400–500 worms. N2 and dat-1 were always included in every experiment to assess and control for stochastic environmental or experimental factors that might induce or alter Swip. For assays of transgenic strains, transgenic and nontransgenic progeny were picked to separate plates by virtue of selection markers under epifluorescence, allowed to recover for at least 3 h to avoid pick-transfer suppression of Swip, and assayed blinded to genotype in parallel. At least 100 transgenic and nontransgenic progeny were assayed in this way to generate average responses.
SynaptopHlourin-based imaging of DA vesicle fusion kinetics.
Young adult animals were staged by picking L4 animals and growing at 20°C overnight. The day of the experiment, animals were mounted on 2% agarose pads and immobilized using 0.05% levamisole. Fluorescence recovery after photobleaching (FRAP) experiments were performed using a Zeiss LSM 510 inverted confocal microscope. PDE synapses were chosen for these analyses due to the more 2D layout and lack of overlap of the PDE processes compared with the cephalic (CEP) and anterior deirid (ADE) projections. Synapses were identified by Synaptobrevin::SEpHluorin fluorescence in the PDE processes and were bleached with a laser at 488 nm, 15 mW for 5–10 s to an intensity 20–30% that of the original fluorescence value. Fluorescence was then monitored every 10 s for 2 min and analyzed using Zeiss LSM 510 software. Percentage recovery was then calculated as the fluorescence at each time point divided by the initial fluorescence value after bleaching. We analyzed 20–25 PDE synapses per genotype, with an average of 3–5 synapses per animal. Recovery plots were fit by nonlinear regression methods to a one-phase exponential model in Prism 5.0 (GraphPad). Average K values (the inverse rate constant) were determined for each experiment, averaged, and subjected to a two-tailed Student's t test with p < 0.05 taken as significant.
In vivo imaging of DA neuron Ca2+ signaling.
Calcium imaging of freely moving worms was performed using a Calcium Ratiometric Imaging of Behaving Nematodes (CARIBN) system as described previously (Piggott et al., 2011). The CARIBN system allows for the imaging of freely behaving worms on the surface of a nematode growth medium (NGM) plate in an open environment without any physical constraint. Because it has been predicted that the mechanical stimulation of DA neuron dendrites produces an increase in DA neuron excitability, we recorded calcium transients from CEP soma during the basal slowing response (Sawin et al., 2000). Briefly, 4 spots of freshly-grown food (OP50 bacteria) were spotted on the surface of an NGM plate, with each containing 2 μl of OP50 culture spaced ∼0.5–0.7 mm apart. Single young adult animals were picked into a food-free area of the plate. Images before and after food contact were acquired at 22 Hz with 15 ms of exposure time per frame for 4 min. G-CaMP/DsRed ratio was calculated as described previously (Piggott et al., 2011).
Locomotor speed analysis on solid substrate.
L4 worms were picked to a fresh plate and incubated at 12–15°C overnight until worms reached the young adult stage (12–16 h). We avoided animals that were actively laying eggs in these analyses, preferring the 3–4 h before active egg-laying bouts. Food-free NGM plates were prepared fresh and used within a week. Plates containing precipitate were not used. Immediately before recording, plates containing young adults were placed at room temperature and allowed to acclimate to the ambient environment for 20 min. For recordings, individual young adults were picked using an eyelash pick, rinsed briefly in a well containing 200 μl of M9 (not water) to remove food from the animal, and then placed on a food-free NGM plate. Using this rinse method, each worm swims for only <5 s in the M9 before being transferred to the NGM assay plate. Assays using rinses from an entire plate or plate area yielded more inconsistent results. We picked as many animals in this way for 2 min (normally 10 worms) and then began a recording that lasted for 7 min. For the recordings, the plate was illuminated with an LED illuminator passed through a housing unit containing a mirror and a polarizing filter beneath a base plate (MBF Biosciences). The illuminator was placed on a camera stand beneath video recording components consisting of an AVT Stingray F-504B CCD Monochrome Camera and adjustable Nikon Zoom lens with a C-mount adapter (MBF Biosciences). Videos were recorded using the WormLab Image Capture module (MBF Biosciences) using the native resolution 2456 × 2058 at 7.5 frames/s. On average, the field size for recording was 1.6 × 1.2 cm and the resolution <10 μm/pixel. We analyzed videos using WormLab tracking and analytical plugins. Proper head/tail and track assignment was validated by eye using manual tracking features in WormLab to correct the assignment or track when necessary.
Swip-10/MBLAC1 sequence evaluation.
Putative paralogs and orthologs of swip-10 were identified using online databases at the National Center for Biotechnology Information website, most notably AceView. Multiple sequence alignments were performed and illustrated using the MegAlign program of DNAStar and Clustal V-based protein alignments.
Evaluation of mouse brain MBLAC1 mRNA expression.
Procedures using mice (14- to 16-week-old C57BL/6 males) were performed under an animal care protocol approved by the Vanderbilt Institutional Animal Care and Use Committee. After rapid decapitation, we isolated brain and peripheral tissues on ice followed by total RNA extraction in TRIzol (Sigma-Aldrich) according to manufacturer's instructions. TRIzol extraction was repeated to reduce genomic DNA contamination. To generate first-strand cDNA, we subjected 500 ng of total RNA to reverse transcriptase reactions using Superscript III Reverse Transcriptase (Life Technologies), with reactions lacking RT used to control for genomic contamination. cDNAs were diluted to 100 ng/μl and used as a template for PCR. PCRs were performed using Platinum PCR Supermix (Life Technologies) according to the manufacturer's instructions. PCR products were sequenced with the forward (sense) primer to validate amplicon identity. The primers used were as follows: Mblac1: RB4442(For), CCTGGGGCTGTTTCCCGAGGCAG; RB4393(Rev), TGCCTTCACATATCAAATCCTC; Mblac2: RB4394(For), ACCTCTACTCCTCTGGCCTCTT; RB4443(Rev), GCCCAGGAAGCACCTTCTCCACAAG; Gapdh: RB4495(For), GCACAGTCAAGGCCGAGAAT; RB4496(For), GGCCTTCTCCATGGTGGTGAA.
Statistical analyses.
Raw thrashing data and heat maps of time-dependent thrashing frequency were generated in SwimR as described previously (Hardaway et al., 2014). All statistical comparisons and graphs were generated in GraphPad Prism version 6.0 and figures were assembled using Adobe Illustrator version 5.1.
Results
Swip-causing mutations vt29 and vt33 are located in the F53B1.6 gene
In our previous study (Hardaway et al., 2012), we identified two reserpine-sensitive mutants, vt29 and vt33, that induce DA-dependent Swip and established that neither carry mutations in dat-1 from which the Swip phenotype was first identified (McDonald et al., 2007). Linkage analysis revealed that the Swip behavior of both vt29 and vt33 segregates with markers located at the left end of LGX (Fig. 1A; Hardaway et al., 2012 and data not shown). In addition, the results of complementation studies provided evidence that these mutations reside in the same gene (data not shown). To identify the site of the molecular lesions in these lines, we performed whole-genome sequencing on vt29, vt33, and the parental strain used for mutagenesis (Sarin et al., 2008). After filtering datasets for SNPs present in the parental strain, we identified a single locus, F53B1.6, that harbored potentially disruptive mutations in both vt29 and vt33 (Fig. 1A and see Materials and Methods). The F53B1.6 gene is predicted to include 9 exons with a large intron that separates a group of 4 exons at the 5′ end of the gene from a group of 5 exons at the 3′ end. The F53B1.6 transcript is predicted to encode an ORF of 421 amino acids (www.wormbase.org); however, our studies demonstrated that the full-length protein contains 486 aa (see below). The mutation identified in vt33 produces a nonconservative coding substitution (427 Gly→Glu), whereas the mutation in vt29 generates a premature stop codon at aa 442 (Fig. 1A).
Figure 1.

DA- and dop-3-dependent Swip mutants vt29 and vt33 harbor mutations in F53B1.6. A, Genomic localization of mutations in vt29 and vt33 and gene diagram of F53B1.6. Lines vt29 and vt33 harbor, respectively, nonsense and a nonconservative missense mutation in exon 10 of FB3B1.6. The tm5915 allele deletes 339 bp spanning F53B1.6 exons 2, 3, and 4 along with a 2 bp insertion. B, Manual Swip analyses of vt29, vt33, and tm5915 demonstrated that these lines exhibit DA synthesis (cat-2)- and DA receptor (dop-3)-dependent Swip. Assays were performed as described in the Materials and Methods. Data were analyzed using one-way ANOVA with multiple Bonferroni posttests where ****p < 0.0001 and error bars represent SEM. Asterisks indicate a comparison of double mutants back to the single mutant vt29, vt33, and tm5915, respectively. Loss of cat-2 and dop-3 alone does not result in a significant change in swimming.
Three additional findings support the conclusion that the identified sequence variants in F53B1.6 are causal for Swip. First, a predicted null allele of F53B1.6 (tm5915) displays a highly penetrant Swip phenotype, comparable to vt29 and vt33 (Fig. 1B). In addition, we determined that the Swip phenotype of tm5915, as for vt29 and vt33, depends on DA synthesis and signaling because double mutants produced with cat-2/TH or the D2 type DA receptor dop-3(vs106), swim at wild-type rates (Fig. 1B). Finally, we found that transgenic expression of a genomic fragment spanning F53B1.6 rescues the Swip defect of both vt29 and vt33 (see Fig. 3).
Figure 3.

Expression of F53B1.6 in glial cells is sufficient to rescue Swip. Expression of a swip-10 genomic fragment significantly restores swimming behavior in vt29 (second bar) and vt33 (seventh bar) mutants. Similarly, expression of swip-10 cDNA under the control of a pan glial promoter ptr-10, and not in DA neurons, significantly rescues vt29, vt33, and tm5915. Mutation of histidine residues within the conserved canonical β-lactamase motif significantly reduces the ability of the pptr-10: swip-10 cDNA to rescue vt29. For all experiments, transgenic lines were generated and tested as described in the Materials and Methods. The promoter used to drive expression of the swip-10 gene, and swip-10 coding sequences (CDS; either genomic or cDNA) are noted beneath the bars. The basal Swip values from nontransgenic animals shown for swip-10(vt29), swip-10(vt33), and swip-10(tm5915) are representative averages obtained during these experiments and consistent with values presented in Figure 1. Values from at least three independent transgenic lines were pooled to generate the averages shown. Data were analyzed using an unpaired Student's t test comparing nontransgenic and transgenic progeny assayed in parallel. **p < 0.01, ***p < 0.001, ****p < 0.0001, ####p < 0.0001 from a t test between averages of transgenic progeny.
To establish the coding potential of F53B1.6 mRNA, we first searched for cDNAs matching F53B1.6 in a genomewide ORF cloning project (WORFDB; http://worfdb.dfci.harvard.edu/) that included three ORF sequence tags (OSTs). One OST (A) derived from the 5′-most 4 exons of the predicted full-length mRNA, including a predicted start site for translation and an encoded peptide of 249 aa and containing a predicted stop codon. A second OST (B) spanned the 3′ group of exons and contains a translation start site predicting a 180 aa protein and included an exon that was not predicted by the genomic sequence. A third OST (C) was 1266 bp and spanned the fully predicted F53B1.6 ORF with a predicted size of 421 aa that initiated at the same ATG as noted for cDNA (A). We verified the presence of mRNA encoding the (C) ORF by RT-PCR, although our clone revealed an additional exon that is absent from the (C) OST but present in the (B) OST, overall yielding a total ORF length of 1478 bp and a predicted protein of 486 aa. In addition to evidence from cDNA cloning, three pieces of evidence suggest that both 5′ and 3′ sets of exons are required to encode functional F53B1.6 protein. First, we found that transgenic expression of a genomic PCR fragment containing the large intron and only the 3′ group of exons failed to rescue the Swip of vt29 (data not shown). Second, the F53B1.6 tm5915 allele deletes 3 exons located within the 5′ group. Last, the mutations present in vt29 and vt33 reside in the 3′ group of exons. Together, these findings indicate that functional protein derived from the F53B1.6 gene arises from the combined coding potential of the 5′ and 3′ clusters of exons. Hereafter, we refer to F53B1.6 as swip-10.
swip-10 is expressed in glial cells
In an effort to determine the cells in which swip-10 is expressed, we generated a series of GFP reporter fusions with potential regulatory regions from the swip-10 gene region. Analysis of conserved nucleotides (UCSC Genome Browser; http://genome.ucsc.edu/) indicated the presence of putative cis-transcriptional regulatory elements in the region just 5′ of the translation start site in exon 1 as well as within the large fifth intron. We therefore generated a series of transgenic strains expressing PCR fusion fragments spanning either or both of these regions (Fig. 2A). The swip-10a construct, consisting of 738 bp of sequence immediately 5′ of the predicted swip-10 translational start site, showed GFP expression in head and tail epidermis (data not shown), uterine muscle-like cells (data not shown), and in an unidentified single cell in the nerve ring (Fig. 2B). The swip-10b construct, generated with a 1.4 kb region spanning the swip-10 fifth intron, drove expression in the head epidermis (data not shown), as well as cells that envelop the nerve ring or that send processes along the length of anterior sensory neurons (Fig. 2C–E). The swip-10c construct, produced from a 3.2 kb genomic fragment that includes the upstream regions of swip-10a fragment and the swip-10b fragment produced an similar expression pattern as the swip-10b construct (Fig. 2I–K). To identify sites of swip-10:GFP expression, we compared its expression with other known cellular markers. swip-10:GFP expression was either weak (swip-10c:GFP; Fig. 2I–K) or undetectable (swip-10b:GFP) in dat-1:mCherry-labeled dopaminergic neurons (Fig. 2F–H). We observed that both the swip-10b:GFP and swip-10c:GFP fusions labeled cells that send cellular processes to the anterior sensilla adjacent to the CEP dendrites (Fig. 2C–E), resembling the morphology of glial-like neuronal support cells (Oikonomou and Shaham, 2011). Indeed, a line expressing both the swip-10b promoter fusion and a pan glial reporter (ptr-10:mRFP; Yoshimura et al., 2008) demonstrated significant reporter colocalization, confirming the glial nature of swip-10 expression (Fig. 2L–N).
Figure 2.

Expression pattern of swip-10. A, Summary diagram of constructs used for swip-10 promoter GFP fusion experiments. Data derive from L4 animals. PCR products were generated via overlap PCR as described in the Materials and Methods. B, GFP expression under the control of the swip-10a promoter in hypodermal cells. C–E, GFP expression driven by swip-10b is visible along processes (arrowheads) that run parallel to DA neuron dendrites as revealed by coexpression with dat-1: mCherry. F–H, swip-10b:GFP is expressed in a number of cells located in the head that do not overlap with DA neurons. Arrowheads in F denote processes similar to C–E. I–K, swip-10c:GFP is expressed in multiple head cells, include low expression in DA neurons (arrowheads). L–N, swip-10b:GFP expression (L) is colocalized in multiple cells with reporter driven by the pan-glial promoter ptr-10 (M). Scale bars, 10 μm in all images. Dotted lines denote the outline of the worm head as revealed by DIC imaging.
swip-10 acts in glial cells to support DA-dependent swimming behavior
To test the hypothesis that swip-10 function is required in glial cells to support normal swimming behavior, we expressed the full-length 486 aa swip-10 cDNA fused to GFP (swip-10::GFP) under the control of ptr-10 and dat-1 promoters, respectively. As shown in Figure 3, ptr-10 driven expression of swip-10 produced a robust rescue of Swip in all three alleles of swip-10 to a level comparable to that seen with transgenic expression of a genomic fragment spanning the swip-10 gene. In contrast, dat-1-driven expression of swip-10 cDNA (Fig. 3) failed to restore normal swimming behavior (Fig. 3). Together, these findings support the idea that swip-10 functions in glia in support of mechanisms required to prevent excess DA signaling and Swip.
swip-10 regulates basal and context-triggered changes in DA neuron excitability
Uptake of the neurotoxic agent 6-OHDA depends on DAT-1 function and results in the death of DA neurons (Nass et al., 2002). In our previous study, we demonstrated that swip-10 mutation does not impair the DA neuron-specific toxicity of 6-OHDA (Hardaway et al., 2012). We therefore reasoned that swip-10 might function in a pathway that acts in parallel to DAT-1 to influence the release of DA. Given the expression of swip-10 in glia juxtaposed to DA neurons, we reasoned that swip-10 could act non-cell autonomously to influence DA neuron excitability and DA release, overwhelming endogenous DAT-1 activity. To test this hypothesis, we examined the rate of vesicular release at DA-releasing synapses using FRAP. We examined DA synapses in the ventral nerve cord of the PDE neuron from animals expressing a pH-sensitive GFP (pHlourin) fused to the C terminus of the synaptic vesicle protein SNB-1 (Fig. 4A). Because pHlourin only produces a fluorescent signal in the neutral pH of the extracellular space, the rate of FRAP can be used as an indirect readout of the rate of synaptic vesicle fusion. Using these animals, we first validated prior findings (Voglis and Tavernarakis, 2008) showing that the presynaptic ion channel asic-1 enhances fusion rates of DA synaptic vesicles (data not shown). In our experiments, we observed that loss of swip-10 resulted in a significant, ∼2-fold elevation in the rate of SNB-1::GFP fluorescence recovery relative to wild-type animals (Fig. 4B). These findings indicate that the basal excitation of DA neurons may be elevated in swip-10 animals. Consistent with this idea, when we quantified movement of crawling animals off of food using an automated movement assessment platform (WormLab), we observed that swip-10 animals exhibited a significant decrease in crawling speed on solid substrate relative to the N2 strain, a reduction that was rescued by loss of cat-2 (Fig. 4C). Interestingly, only a small, nonsignificant decrease in crawling speed was evident with dat-1 animals, suggesting that, under basal conditions, swip-10 has a stronger influence on DA signaling than dat-1.
Figure 4.
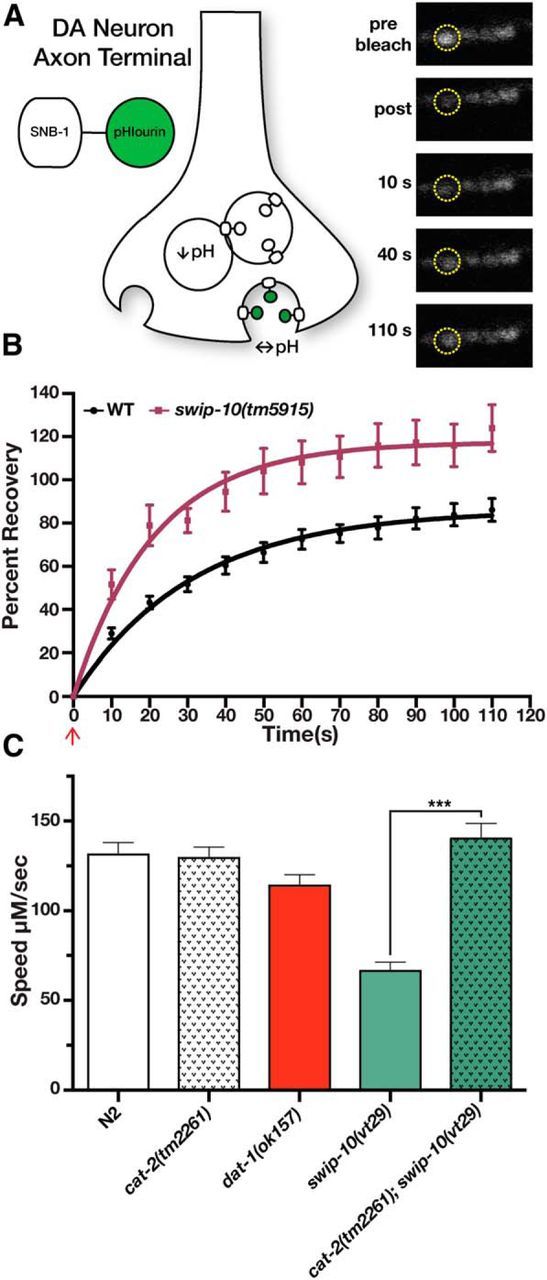
Loss of swip-10 results in elevated vesicular release in DA terminals and DA-dependent reductions in crawling speed. A, Schematic illustration of the experimental transgenes used in B. The pH-sensitive GFP fluorophore pHlourin fused in frame to the C terminus of the synaptic vesicle protein SNB-1 was expressed solely in DA neurons via the asic-1 promoter as in Voglis and Tavernarakis (2008). Vesicular fusion rates at DA neuron synapses was then monitored using FRAP. Images illustrate a wild-type PDE neuron synapse before bleaching and during different points of recovery. B, Loss of swip-10 results in a significantly elevated rate of fluorescence recovery, indicative of a more rapid vesicular fusion rate. Data were fit to a one phase exponential using nonlinear regression. K (rate constant) = 0.032 ± 0.002 s−1 for N2 vs 0.063 ± 0.01 s−1. p < 0.05, Student's two-tailed t test. Red arrow denotes the time point immediately after photobleaching. Confocal images and FRAP were performed as described in the Materials and Methods. C, Loss of swip-10 results in a DA-dependent decrease in crawling on solid substrate. Crawling videos and tracking were performed using WormLab as described in the Materials and Methods. Data were analyzed using one-way ANOVA with multiple Tukey posttests. Unlike dat-1, swip-10(vt29) demonstrates a significantly reduced speed relative to N2 that is fully restored by loss of cat-2. ***p < 0.001.
Due to the use of immobilized animals for FRAP recording, we sought a more direct measure that could interrogate DA neuron excitability under physiological conditions. For this effort, we used an optical approach to assess DA neuron excitability via in vivo Ca2+ imaging while animals executed locomotory responses to contact with food (i.e., a bacterial lawn). Therefore, we monitored Ca2+ levels in CEP neurons from animals expressing a Ca2+-sensitive version of GFP (GCaMP) while the freely moving animals approached and made contact with food, a stimulus known to induce a DA-dependent basal slowing response (Sawin et al., 2000). To control for body movements, we normalized recordings using a Ca2+-independent fluorophore (dsRed2) expressed in the same cells (Fig. 5A). As hypothesized by Sawin et al. (2000), we observed that DA neurons become activated upon entering a lawn of bacteria, demonstrating a robust elevation in normalized Ca2+ signal upon food touch (Fig. 5A). Consistent with our hypothesis that increased DA neuron excitability may underlie Swip behavior, swip-10(tm5915) animals displayed an enhanced Ca2+ response to food. Importantly, the augmented Ca2+ response to food contact could be normalized by glial restoration of swip-10 (Fig. 5B,C). Together, these observations support the hypothesis that swip-10 functions in glia to limit DA neuron excitability and vesicular fusion at DA neuron synapses that, in excess, produces DA-dependent Swip.
Figure 5.
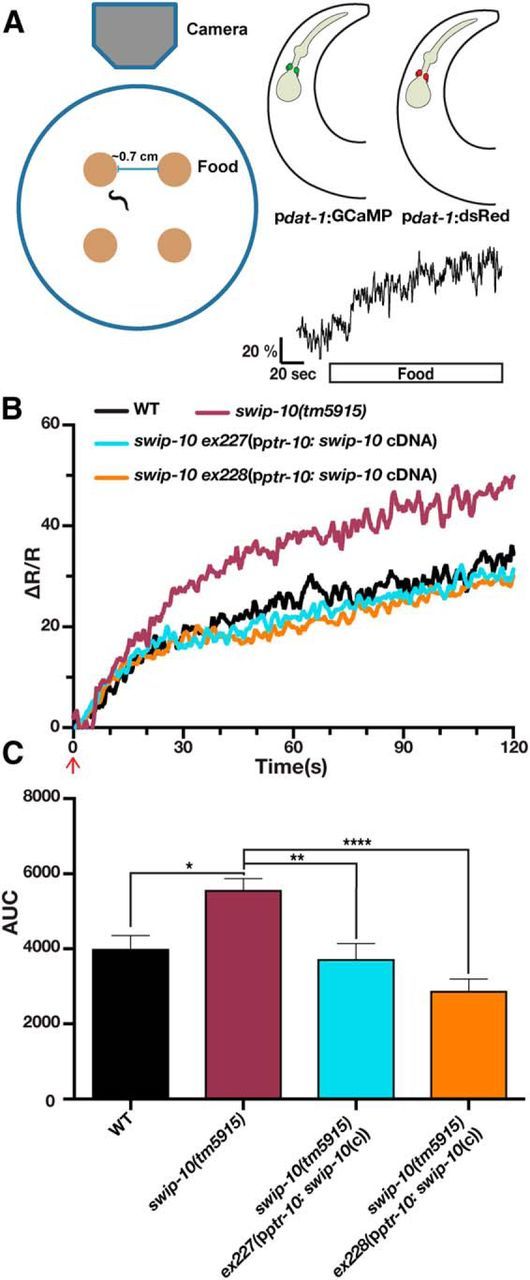
Loss of swip-10 results in elevated DA neuron excitability that is restored by expression of swip-10 in glia. A, Left, Schematic of recording apparatus and assay conditions. Top right, Illustration of background transgenic strain used for CARIBN experiments. Bottom right, Representative trace of wild-type background strain before entering a lawn of food (white bar). Scale bar, ΔR/R of 20% and 20 s. B, Food triggers an increase in DA neuron activity that is greater in worms lacking swip-10. Expression of swip-10 under a ptr-10 promoter restores the magnitude of the GCaMP/dsRed ratio to wild-type levels. Data represent average traces of WT, swip-10, and rescued strains after encountering food (red arrow). n = at least 17 worms per group. The WT strain is XuIs14(pdat-1: GCaMP, pdat-1:dsRed); lite-1(Xu7). C, Area under the curve quantification for B. swip-10(tm5915) displays a significantly elevated AUC from WT levels that is significantly restored by expression of swip-10 expressed with a ptr-10 promoter. Data were analyzed by one-way ANOVA with multiple Bonferroni posttests where *, **, and *** indicate a *p < 0.05, **p < 0.01, ***p < 0.001, respectively. No significant differences were found between WT and strains containing ex227 and ex228.
Contribution of Glu signaling to swip-10
In mammals, Glu is responsible for a major fraction of excitatory signaling, including aspects of the excitatory drive on DA neurons (Karreman et al., 1996; Dobi et al., 2010; Qi et al., 2014). In C. elegans, Glu and DA regulate each other reciprocally (Lee et al., 1999; Hills et al., 2004; Kindt et al., 2007; Hukema et al., 2008; Baidya et al., 2014). Our evidence of enhanced DA neuron excitability in swip-10 animals led us to consider whether swip-10 DA phenotypes might be driven by an excess of Glu signaling either directly onto DA neurons themselves or onto upstream neurons that control DA neuron activity. Synaptic Glu packaging and release is dependent on the activity of vesicular glutamate transporters (vGLUTs) (Liu and Edwards, 1997; Lee et al., 1999). In C. elegans, the most well characterized vGLUT is eat-4 (Lee et al., 1999), although other putative vGLUT proteins (vglu-2 and vglu-3) are present in the genome. Consistent with the hypothesis that Glu signaling is involved in the elevated excitability of DA neurons in swip-10 animals, the eat-4 mutation suppressed the Swip exhibited by both vt29 and vt33 (Fig. 6A), which was more pronounced in the phenotypically weaker vt33 allele.
Figure 6.

swip-10 paralysis is dependent on vesicular Glu release and GluRs. A, Automated thrashing analysis reveals that swip-10 paralysis suppressed by loss of the vesicular GLT eat-4. Curves represent the average thrashing frequency of at least 25 independent animals recorded over the course of several sessions. eat-4;swip-10(vt29) is significantly suppressed from swip-10(vt29) progeny during minutes 1–3 and eat-4;swip-10(vt33) is significantly suppressed from swip-10(vt33) from minutes 2–10. We did not observe any significant difference between eat-4 and N2 (data not shown). B, Loss of glr-4, glr-6, and mgl-1 additively suppress swip-10 paralysis. Individual mutations of these GluRs have no impact on swimming behavior. C, Loss of glr-4 significantly suppresses swip-10(vt29). glr-4;swip-10 is significantly elevated from swip-10(vt29) starting in the first minute. D, Loss of glr-6 significantly suppresses swip-10(tm5915) in minutes 4–10. E, Loss of mgl-1 significantly suppresses swip-10(tm5915) in minutes 3–10. F, Loss of mgl-1 significantly restores dat-1 Swip, whereas loss of glr-4 or glr-6 does not affect dat-1 paralysis. For all panels, double and triple mutant strains were constructed and assays were performed as described in the Materials and Methods. For B and F, data were analyzed by one-way ANOVA with multiple Bonferroni posttests. ***p < 0.001, ****p < 0.0001. For A, B, D, and E, single worm recordings were performed as described in the Materials and Methods and analyzed with SwimR. Data were analyzed by two-way ANOVA with multiple Bonferroni posttests at each time point.
Glu actions in the worm, as in vertebrates, are mediated by a combination of AMPA-type and NMDA-type ionotropic Glu receptors (GluRs), as well as by metabotropic GluRs (Brockie and Maricq, 2006; Dillon et al., 2006). We screened mutant alleles of these receptors for their ability to suppress Swip behavior in swip-10 animals. As with loss of vesicular Glu packaging, we did not observe significant paralysis with loss of GluR function (Fig. 6B and data not shown). In swip-10 double mutants, however, three GluR mutant alleles disrupting function of glr-4, glr-6, and mgl-1 individually suppressed Swip, as measured by manual or automated swimming analysis (Fig. 6B–E). Combinations of these alleles enhanced suppression, with the strongest effect conferred by the simultaneous loss of glr-4, glr-6, and mgl-1 (Fig. 6B). These observations are consistent with the hypothesis that the DA-dependent Swip of swip-10 mutants requires Glu signaling and involves contributions from multiple GluRs, including both ionotropic (glr-4 and glr-6) and metabotropic (mgl-1) receptors.
Until this point, our evidence was consistent with a loss of swip-10 function producing Swip via Glu-dependent mechanisms that result in excessive DA signaling, whereas the Swip of dat-1 mutants is generated by excess extrasynaptic DA arising from a loss of DAT-1-mediated DA clearance capacity. To test this idea, we investigated whether the GluR mutants shown to suppress the Swip of swip-10 animals also affect paralysis of dat-1 animals (Fig. 6F). Notably, we found that glr-4 and glr-6 mutations had no impact on dat-1 Swip, whereas loss of mgl-1 nearly completely restored normal swimming behavior, with an effect exceeding that seen with the swip-10; mgl-1 double mutant (Fig. 6F). These data suggest that mechanisms engaged by loss of SWIP-10 exerts a broad effect on GluR signaling mechanisms, engaging both ionotropic and metabotropic receptors to influence DA signaling, with the MGL-1 component possibly tied most intimately to DAT-1 function.
Loss of Glu transport capacity induces Swip
In mammals, extracellular Glu levels are tightly controlled by a family of Na+-dependent plasma membrane GLTs (Sheldon and Robinson, 2007). GLT proteins are highly conserved across phylogeny, including in C. elegans, in which six family members can be identified by sequence conservation (Mano et al., 2007). If hyperexcitation of DA neurons in swip-10 animals is driven by elevated extracellular Glu levels, we hypothesized that the loss of one or more worm GLTs should also confer Swip. As shown in Figure 7A, glt-1, glt-3, and glt-4 animals displayed significant Swip when assessed in endpoint assays (Fig. 8A), whereas glt-6 displayed weaker but significant Swip and glt-5 or glt-7 animals failed to paralyze (Fig. 8A). Real-time analyses (Fig. 7B–D) of glt-1, glt-3, and glt-4 mutants revealed a significantly faster and more penetrant Swip effect for glt-1. Indeed, glt-1 animals demonstrated substantial paralysis within the first minute of swimming (Fig. 8E), as much or more than swip-10 animals. To determine whether loss of glt-1, glt-3, or glt-4 induces Swip through mechanisms shared by swip-10 mutation, we assessed time-dependent swimming behavior in swip-10; glt double mutants (Fig. 7B–D). These studies demonstrated a lack of additivity after the first few minutes of recording, consistent with the function of swip-10 and glts in a common pathway. The significant deviation from nonadditivity in the first few minutes of recording, particularly with glt-1, suggests possible DA independence, whereas, at later times, all effects involve DA signaling. In support of this idea, loss of DA synthesis capacity (cat-2) restored normal swimming behavior of glt-1, glt-3, and glt-4 animals in endpoint assays (Fig. 7F). Together, these experiments demonstrate that an inability to clear extracellular Glu can result in DA-dependent Swip and that SWIP-10 and GLTs may act in a common pathway.
Figure 7.

Loss of GLT genes produces DA-dependent Swip. A, Manual Swip assays of glutamate transporter alleles. Loss of glt-1, glt-3, glt-4, or glt-6 produces a significant Swip phenotype relative to N2 when assessed at a single 10 min time point. B, Automated thrashing analysis reveals that loss of glt-1 produces a significant Swip phenotype relative to N2 (significant reduction at all time points). Multiple comparisons of swip-10 versus swip-10 glt-1 mutants show that swip-10 glt-1 mutants are significantly reduced from swip-10 mutants from time points 0–48 s. C, Automated thrashing analysis reveals that loss of glt-3 produces a significant Swip phenotype relative to N2 (significant reduction at scattered time points during minutes 6–9 from 9:13–10:00). Multiple comparisons of swip-10 versus glt-3;swip-10 mutants show that glt-3;swip-10 mutants are significantly reduced from swip-10 mutants from time points 1:12–1:18, 1:29–1:32, and 1:34–1:39. D, Automated thrashing analysis reveals that loss of glt-4 produces a significant Swip phenotype relative to N2 (significant reduction at scattered time points during minutes 2–6). Multiple comparisons of swip-10 versus swip-10 glt-4 mutants show that swip-10 glt-4 mutants are significantly reduced from swip-10 mutants from time points 0:55–1:31, 1:37–2:01, and 2:07–2:19. E, Heat maps of the first minute of automated recordings for N2, swip-10, glt-1, and swip-10 glt-1 strains. Colors represent normalized thrashing values within that strain where red is the highest thrashing value and green is no movement. F, Swip in glt-1, glt-3, and glt-4 is DA dependent because loss of cat-2 restores swimming behavior in glt-1, glt-3, and glt-4 to wild-type swimming levels. For A and F, data were analyzed using one-way ANOVA with multiple Bonferroni posttests where ****p < 0.0001 and error bars represent SEM. For B–D, single worm recordings were performed as described in the Materials and Methods and analyzed with SwimR. Data were analyzed by two-way ANOVA with multiple Bonferroni posttests at each time point. n ≥ 33 for each strain.
Figure 8.

Phylogenetic conservation of swip-10 and CNS expression of the swip-10 mouse homolog Mblac1. A, The SWIP-10 protein exhibits significant conservation with its nematode homolog CBG14084 across phylogeny, concentrated at the protein's C-terminal MBD. The vertebrate homologs have much shorter protein sequences and only begin to align at 260 aa into the SWIP-10 protein sequence. Shaded residues indicate amino acids that match the SWIP-10 sequence. Red bar indicates the span of the MBD that begins with a canonical “ILVDTG” motif. Blue boxes indicate histidine residues that are predicted to be critical for metal binding, including a canonical HxHxDH motif that is typical of the entire metallo β-lactamase superfamily. Yellow boxes indicate amino acids that are altered in the strains isolated from the Swip-based mutagenesis screen. swip-10(vt29) harbors a SNP that results in the conversion of a conserved tryptophan at position 377 to a stop codon, resulting in a truncated MBD. The mutation in swip-10(vt33) converts a conserved glycine at position 362 to glutamic acid. Sequences were aligned using ClustalW (DNAStar). B, Dendrogram generated from multiple sequence alignment of swip-10 with Mblac1, MBLAC1, and another MBD containing protein in mouse and human named Mblac2 and MBLAC2, respectively. swip-10 exhibits greater identity with Mblac1 and MBLAC1. C, RT-PCR of Mblac1, Mblac2, and Gapdh from different areas of the mouse brain, heart, lung, and liver. We observed the presence of both RNAs in tissue harvested from all tissues assayed, but not in whole-brain lysates lacking reverse transcriptase.
Structural basis of swip-10 function and conservation
To determine the structural basis for SWIP-10 protein function, we searched protein databases for conserved sequence motifs that could identify functional domains. This effort identified a single conserved structural element, a metallo β-lactamase (MBD) (Bebrone, 2007) residing in the SWIP-10 C terminus (Fig. 9A). MBDs derive their name from the conserved domain organizing the active site of bacterial enzymes that hydrolyze β-lactam antibiotics such as penicillin and the cyclosporins. The MBD of SWIP-10 possesses the “HxHxDH” motif that supports binding of one or more metal (e.g., Zn2+) ions. In prokaryotic MBDs, metal binding is used to coordinate water molecules used for hydrolytic attack on specific substrates (Bebrone, 2007). Sequence alignments further revealed the presence of two additional, conserved His residues (His405 and His470) that may also contribute to metal ion binding. We mutated these five His residues to Ser and then tested the ability of mutant constructs to rescue swip-10(vt29) paralysis. We found the His mutated swip-10 to be significantly impaired in its ability to rescue Swip (Fig. 3), supporting a hypothesized role for metal coordination in swip-10 function and a likely enzymatic function of SWIP-10 protein.
Figure 9.

Extrasynaptic glutamate model for swip-10-dependent hyperdopaminergia. Schematic rendition of the hypothesis that, whereas Swip of dat-1 animals arises from an inability to clear DA at normal rates of Glu-dependent DA neuron excitation, paralysis of swip-10 animals arises from an increase in extrasynaptic Glu that results in changes in DA neuron excitability and excess DA release, overwhelming DA clearance mechanisms.
Analysis of inferred C. elegans proteins revealed multiple, candidate swip-10 paralogs encoded by the genes C03F11.2, C23H3.9, and B0432.9. SWIP-10 identities with these proteins are largely confined to their MBDs (MBD identity: C03F11.2, 36.2%; C23H3.9, 29.4%; B0432.9, 25.3%). In contrast, the C. briggsae gene CBG14084, also uncharacterized, bears identity throughout the protein, suggesting its identity as a swip-10 ortholog. Both SWIP-10 and the CBG14084 protein contain an ∼300 aa sequence that precedes their single MBD domain (Fig. 8A). We identified related swip-10 proteins in searches of the fly, zebrafish, mouse, rat, and human genomes. In each case, these swip-10 family members possess a highly conserved MBD, but lack the N-terminal amino acid sequences preceding the MBD of SWIP-10 in C. elegans. The most promising candidate ortholog in humans is the uncharacterized gene MBLAC1 (MBD containing protein 1). The MBD of SWIP-10 and MBLAC1 exhibit ∼40% identity, whereas the MBDs of putative C. elegans swip-10 paralogs show lower identity with MBLAC1 (29–33%). Importantly, the loss-of-function mutations that we identified in vt29 and vt33 are both positioned at sites conserved in the MBD domain of MBLAC1 (Fig. 9A). Mouse and human genomes also contain an additional, more distantly related MBD-containing protein annotated in humans as MBLAC2 (Fig. 9B). MBLAC2 also lacks the N-terminal extension found preceding the MDB in SWIP-10. The MBD of MBLAC2 exhibits less identity with the MBD of SWIP-10 (23.5%) than seen for the MBLAC1 MBD and even less compared with the MBD of MBLAC1 (21%). These findings suggest that MBLAC2 diverged early from the common precursor of MBLAC1/SWIP-10. To validate expression of Mblac1/Mblac2 in vivo, we used RT-PCR of mouse mRNA and found both genes to be widely expressed, including in various brain regions (Fig. 8C).
Discussion
The findings of the present study support the contribution of a novel, glial-expressed protein to constraining excitability and synaptic vesicle fusion in DA neurons. We show that loss of swip-10 results in an increase in evoked intracellular Ca2+ signaling (an indirect measurement of excitability) and elevated rates of vesicular release and DA-dependent behaviors engaging both the ionotropic and metabotropic Glu signaling pathways. We provide evidence that SWIP-10 and GLTs may act in the same pathway, consistent with a critical role for SWIP-10 in glial-dependent control of extracellular Glu homeostasis. Finally, we identify putative mammalian SWIP-10 orthologs that share a highly conserved MBD.
We previously reported the identification of novel mutant lines that exhibit DA-dependent Swip (Hardaway et al., 2012). Among these lines, vt29 demonstrated a normal sensitivity to the DA-specific neurotoxin 6-OHDA and an additive behavioral genetic interaction with dat-1. These observations led us to hypothesize that vt29 (and the noncomplementing vt33 allele) harbors a mutation in a gene that functions in a pathway parallel to DA clearance. Our identification of swip-10, and characterization of its impact on DA neuron excitability and vesicular release, validates this idea.
In a series of transgenic promoter fusion experiments, we observed expression of swip-10 in cells of the epidermis, in uterine muscle cells, and in multiple cells of the head. Prominently among the head cells are glial-like support cells, as identified by ptr-10-driven reporter expression, which includes CEP and amphid sheath and socket cells (www.wormatlas.org; Perens and Shaham, 2005; Yoshimura et al., 2008). Elements affording glial expression of swip-10 were mapped to the large fifth intron. Although we also observed a weak swip-10:GFP signal in DA neurons, exclusive expression of the wild-type SWIP-10 protein in glial cells rescued the swip-10 locomotory defect, whereas expression in DA neurons did not. On the basis of these results, we conclude that SWIP-10 exerts a non-cell-autonomous effect on DA neuron excitability that depends on SWIP-10 function in glial cells. Despite a report that CEPsh glia contribute to DA-dependent behaviors (Felton and Johnson, 2014), we were unable to reverse Swip in swip-10 mutants using the CEPsh-specific promoter hlh-17 (data not shown and Yoshimura et al., 2008), so further studies are needed to further subdivide contributions from glial subtypes. Our reporter studies indicate that swip-10 expression may also be tightly coupled to the developmental stage of the animal, with high levels of expression in the epidermis of early larval stage animals (data not shown), whereas expression in head cells and putative glia dominates during the L3 and L4 stage, coinciding with the emergence of highly penetrant Swip during the L4 stage of dat-1 and swip-10 animals.
Through in vivo monitoring of DA vesicle fusion, we obtained evidence to support the idea that mutation of swip-10 enhances the fusion rate of DA synaptic vesicles. Lateral diffusion of unbleached, surface-associated SNB-1:pHluorin molecules could confound our interpretation, but we are unaware of a mechanism by which such a process would be accelerated by loss of glial swip-10 expression. Although the FRAP approach we have used does not measure extracellular DA per se, our findings, in the context of the swip-10 hyperdopaminergic phenotype, strongly suggest that Swip derives from excess fusion of DA synaptic vesicles, leading to elevated extracellular DA. This finding is consistent with behavioral assays that detected DA-dependent reduction in crawling speed and of reduced DA content (Hardaway et al., 2012), which we hypothesize derives from excess tonic DA release. Direct monitoring of in vivo DA neuron Ca2+ dynamics in freely moving animals provided evidence of increased DA neuron excitability. We have demonstrated here for the first time that DA neurons are activated upon encountering a lawn of food through a mechanism that is likely to depend on physical contact and TRP-4 channel function (Sawin et al., 2000; Chase et al., 2004; Li et al., 2006; Kindt et al., 2007; Kang et al., 2010). Loss of swip-10 results in an increase in the magnitude of the evoked Ca2+ responses in this behavioral assay. Importantly, elevated Ca2+ responses were normalized by glial restoration of wild-type swip-10 expression. To our knowledge, these studies are the first to demonstrate that a glial-expressed gene regulates the in vivo function of DA neurons.
The importance of mammalian glia in regulating neuronal excitability via control of extracellular Glu homeostasis (Anderson and Swanson, 2000; Kalivas, 2009) suggested to us the hypothesis that SWIP-10 protein might function in a glial pathway that normally constrains Glu excitation of DA neurons. The best characterized presynaptic determinant of Glu signaling is the vesicular GLT EAT-4 (Lee et al., 1999; Serrano-Saiz et al., 2013). Consistent with the hypothesis that excess Glu signaling drives the swip-10 Swip phenotype, a loss-of-function eat-4 mutation significantly suppressed swip-10 paralysis. The incomplete suppression of Swip by eat-4 may indicate a Glu-independent component of swip-10 action or may arise from Glu signaling supported by the uncharacterized eat-4 homologs vglu-2 and vglu-3. Alternatively, nonvesicular Glu release mechanisms may regulate DA neuron signaling. In mammals and flies, extrasynaptic Glu availability is regulated by a process of glial cystine/glutamate exchange (system Xc−; Augustin et al., 2007; Bridges et al., 2012). Multiple transporter proteins exist that could support XC-like activity in C. elegans (Veljkovic et al., 2004), but none has yet been linked to Glu signaling. Further studies are needed to explore this possibility.
Because we observed significant Swip suppression in swip-10;eat-4 double mutants, we sought further evidence for Glu involvement via genetic experiments with GluR mutants. The C. elegans genome encodes multiple families of ionotropic and metabotropic GluRs (Hart et al., 1995; Maricq et al., 1995; Brockie et al., 2001; Brockie and Maricq, 2006; Dillon et al., 2006; Greer et al., 2008). Mutations in glr-4, glr-6, and mgl-1 led to a significant, additive suppression of swip-10 Swip, which is consistent with a model whereby excess Glu signaling to DA neurons supports paralysis behavior (Fig. 9). Interestingly, GLR-4 and GLR-6 appear to make a unique contribution to the Swip defect of swip-10 mutants versus that of dat-1 animals. Conversely, MGL-1 appears largely involved in the dat-1 pathway. The mechanistic basis for the separate roles of ionotropic versus metabotropic glutamate receptors is unknown at present, but may reflect a greater role of ionotropic receptors in neuronal excitability as reported for swip-10 versus metabotropic receptor control of DA release or response. Further studies that clarify the sites of expression where different GluRs suppress Swip are needed to address this issue. Interestingly, tiling array experiments demonstrate mRNA expression of both glr-6 and mgl-1 in DA neurons (WormViz; Spencer et al., 2011).
Tight control of mammalian Glu signaling is achieved through the activity of multiple members of the SLC1 family of Na+-dependent, plasma membrane GLTs (EAATs in humans), which are expressed by both glia and neurons (Sheldon and Robinson, 2007). GLT dysfunction has also been suggested to contribute to DA-linked disorders including addiction (Knackstedt et al., 2010; Trantham-Davidson et al., 2012) and Parkinson's disease (Assous et al., 2014). We screened all viable alleles of the C. elegans family of sodium-dependent GLTs for Swip. DA-dependent Swip was evident for glt-1, glt-3, and glt-4, although the most penetrant effects were seen with glt-1 mutants. Additivity experiments suggest that the GLTs and SWIP-10 act through a common pathway, the disruption of which generates Swip. The greater penetrance of the glt-1 mutation may reflect a greater role of this transporter in the control of Glu-dependent DA neuron excitability. On the basis of reporter studies, C. elegans GLTs have been reported to exhibit a broad expression profile, including body wall and pharyngeal muscle, neurons, and specific single cell types such as the excretory canal cell (Mano et al., 2007), leading to a hypothesis that they may collaborate to set basal extracellular Glu levels; however, this does not rule out more selective contributions to specific circuits.
Analysis of the predicted SWIP-10 protein sequence identified a single conserved protein domain with similarity to the MBD sequence. MBDs are ancient structural elements found in hydrolytic enyzmes (Aravind, 1999; Daiyasu et al., 2001) in which 3–5 conserved His residues (core motif = HxHxDH) coordinate metal ions (e.g., Zn2+) to support water polarization and substrate hydrolysis. Mutation of this canonical motif in SWIP-10 resulted in a loss of its ability to rescue Swip in the vt29 strain, although we cannot exclude the possibility that mutation of His residues in the MBD resulted in protein destabilization because imaging of the swip-10::GFP protein has proved unsuccessful thus far. This may reflect secretion of SWIP-10 protein (although it is not conserved in C. briggsae, a sequence that meets criteria for a secretory signal is present in the SWIP-10 N terminus) or that protein levels sufficient for protein visualization are not achieved in vivo due to tight, posttranslational regulation of SWIP-10 protein.
Best known of the β-lactamase substrates are the fungal β-lactam antibiotics, typified by penicillin, ampicillin, and cephalosporin, although eukaryotic MBD-containing enzymes hydrolyze a wide variety of substrates, from s-lactoyl glutathione and cAMP to RNA and DNA (Moshous et al., 2001; Mandel et al., 2006; Bebrone, 2007). Interestingly, treatment of rodents with β-lactam antibiotics such as ceftriaxone upregulates the expression of the astrocytic glutamate transporter EAAT2/GLT-1, as well of the cystine/Glu exchanger (xCT) (Rothstein et al., 2005; Lewerenz et al., 2009; Knackstedt et al., 2010). GLT-1 is thought to maintain low levels of synaptic Glu, whereas astrocytic xCT proteins export Glu extrasynaptically. Studies indicate that β-lactam antibiotic treatments lead to an alteration in extracellular Glu levels and multiple Glu-linked physiological/behavioral changes, including neuroprotection in neurodegenerative disease models (Rothstein et al., 2005; Chu et al., 2007; Leung et al., 2012), suppression of reinstatement after cocaine withdrawal (Knackstedt et al., 2010), and reduction of depressive behavior (Mineur et al., 2007). Our studies suggest that, in C. elegans, an MBD-containing protein plays a critical role in extracellular Glu homeostasis, possibly coordinated through GLT activities. Our ongoing studies therefore seek to determine whether MBLAC1 represents a key determinant of the CNS actions of β-lactam antibiotics.
Footnotes
J.A.H., C.L.S., and D.P.B. were supported by the Vanderbilt Brain Institute and the Graduate Neuroscience Training Program through the National Institutes of Health (NIH Grant MH064913). P.O. was supported by the Silvio O. Conte Center for Neuroscience Research (NIH Grant MH078028 to R.D.B.). Additional support for project efforts was derived from the NIH (Grants MH093102 to J.A.H., DA035559 to D.P.B., MH095044 to R.D.B., HG004263 and NS26115 to W.C.S. and D.M.M., and GM083241 to Z.L. and X.Z.S.X.). L.C. was supported by the NIH-funded Centers of Biomedical Research Excellence (COBRE Grant P20 GM104360-01). Experiments were performed through the use of the VUMC Cell Imaging Shared Resource, which is supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY08126. We thank Sarah B. Robinson, Cassandra L. Retzlaff, Hussain Jinnah, and Dan Chase for helpful discussions during the course of this work and Chris Svitek, Jane Wright, Qiao Han, Tracy Jarrett-Moore, and Angela Steele for expert research support.
The authors declare no competing financial interests.
References
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Aravind L. An evolutionary classification of the metallo-beta-lactamase fold proteins. In Silico Biol. 1999;1:69–91. [PubMed] [Google Scholar]
- Assous M, Had-Aissouni L, Gubellini P, Melon C, Nafia I, Salin P, Kerkerian-Le-Goff L, Kachidian P. Progressive Parkinsonism by acute dysfunction of excitatory amino acid transporters in the rat substantia nigra. Neurobiol Dis. 2014;65:69–81. doi: 10.1016/j.nbd.2014.01.011. [DOI] [PubMed] [Google Scholar]
- Augustin H, Grosjean Y, Chen K, Sheng Q, Featherstone DE. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J Neurosci. 2007;27:111–123. doi: 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidya M, Genovez M, Torres M, Chao MY. Dopamine modulation of avoidance behavior in Caenorhabditis elegans requires the NMDA receptor NMR-1. PLoS One. 2014;9:e102958. doi: 10.1371/journal.pone.0102958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI. Genetic and cellular analysis of behavior in C. elegans. Annu Rev Neurosci. 1993;16:47–71. doi: 10.1146/annurev.ne.16.030193.000403. [DOI] [PubMed] [Google Scholar]
- Bebrone C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol. 2007;74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Bigelow H, Doitsidou M, Sarin S, Hobert O. MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods. 2009;6:549. doi: 10.1038/nmeth.f.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges RJ, Natale NR, Patel SA. System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Maricq AV. Ionotropic glutamate receptors: genetics, behavior and electrophysiology. WormBook. 2006:1–16. doi: 10.1895/wormbook.1.61.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Madsen DM, Zheng Y, Mellem J, Maricq AV. Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J Neurosci. 2001;21:1510–1522. doi: 10.1523/JNEUROSCI.21-05-01510.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL. Biogenic amine neurotransmitters in C. elegans. WormBook. 2007:1–15. doi: 10.1895/wormbook.1.132.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR. Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci. 2004;7:1096–1103. doi: 10.1038/nn1316. [DOI] [PubMed] [Google Scholar]
- Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, Roh JK. Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke. 2007;38:177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- Daiyasu H, Osaka K, Ishino Y, Toh H. Expansion of the zinc metallo-hydrolase family of the beta-lactamase fold. FEBS Lett. 2001;503:1–6. doi: 10.1016/S0014-5793(01)02686-2. [DOI] [PubMed] [Google Scholar]
- Dillon J, Hopper NA, Holden-Dye L, O'Connor V. Molecular characterization of the metabotropic glutamate receptor family in Caenorhabditis elegans. Biochem Soc Trans. 2006;34:942–948. doi: 10.1042/BST0340942. [DOI] [PubMed] [Google Scholar]
- Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci. 2010;30:218–229. doi: 10.1523/JNEUROSCI.3884-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton CM, Johnson CM. Dopamine signaling in C. elegans Is mediated in part by HLH-17-dependent regulation of extracellular dopamine levels. G3 (Bethesda) 2014;4:1081–1089. doi: 10.1534/g3.114.010819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer ER, Pérez CL, Van Gilst MR, Lee BH, Ashrafi K. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 2008;8:118–131. doi: 10.1016/j.cmet.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Hardie SL, Whitaker SM, Baas S, Zhang B, Bermingham DP, Lichtenstein AJ, Blakely RD. Forward genetic analysis to identify determinants of dopamine signaling in Caenorhabditis elegans using swimming-induced paralysis. G3 (Bethesda) 2012;8:961–975. doi: 10.1534/g3.112.003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Wang J, Fleming PA, Fleming KA, Whitaker SM, Nackenoff A, Snarrenberg CL, Hardie SL, Zhang B, Blakely RD. An open-source analytical platform for analysis of C. elegans swimming-induced paralysis. J Neurosci Methods. 2014;232:58–62. doi: 10.1016/j.jneumeth.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AC, Sims S, Kaplan JM. Synaptic code for sensory modalities revealed by C. elegans GLR-1 glutamate receptor. Nature. 1995;378:82–85. doi: 10.1038/378082a0. [DOI] [PubMed] [Google Scholar]
- Hills T, Brockie PJ, Maricq AV. Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J Neurosci. 2004;24:1217–1225. doi: 10.1523/JNEUROSCI.1569-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. BioTechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- Hukema RK, Rademakers S, Jansen G. Gustatory plasticity in C. elegans involves integration of negative cues and NaCl taste mediated by serotonin, dopamine, and glutamate. Learn Mem. 2008;15:829–836. doi: 10.1101/lm.994408. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kang L, Gao J, Schafer WR, Xie Z, Xu XZ. C. elegans TRP family potein TRP-4 Is a pore-forming subunit of a native mechanotransduction channel. Neuron. 2010;67:381–391. doi: 10.1016/j.neuron.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreman M, Westerink BH, Moghaddam B. Excitatory amino acid receptors in the ventral tegmental area regulate dopamine release in the ventral striatum. J Neurochem. 1996;67:601–607. doi: 10.1046/j.1471-4159.1996.67020601.x. [DOI] [PubMed] [Google Scholar]
- Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, Rankin CH, Schafer WR. Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron. 2007;55:662–676. doi: 10.1016/j.neuron.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M. Schizophrenia: from dopaminergic to glutamatergic interventions. Curr Opin Pharmacol. 2014;14:97–102. doi: 10.1016/j.coph.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci. 1999;19:159–167. doi: 10.1523/JNEUROSCI.19-01-00159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TC, Lui CN, Chen LW, Yung WH, Chan YS, Yung KK. Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem Neurosci. 2012;3:22–30. doi: 10.1021/cn200072h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- Li W, Feng Z, Sternberg PW, Xu XZ. A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature. 2006;440:684–687. doi: 10.1038/nature04538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Edwards RH. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci. 1997;20:125–156. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, Tong L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature. 2006;444:953–956. doi: 10.1038/nature05363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano I, Straud S, Driscoll M. Caenorhabditis elegans glutamate transporters influence synaptic function and behavior at sites distant from the synapse. J Biol Chem. 2007;282:34412–34419. doi: 10.1074/jbc.M704134200. [DOI] [PubMed] [Google Scholar]
- Maricq AV, Peckol E, Driscoll M, Bargmann CI. Mechanosensory signalling in C. elegans mediated by the GLR-1 glutamate receptor. Nature. 1995;378:78–81. doi: 10.1038/378078a0. [DOI] [PubMed] [Google Scholar]
- Mazei-Robinson MS, Blakely RD. ADHD and the dopamine transporter: are there reasons to pay attention? Handb Exp Pharmacol. 2006;175:373–415. doi: 10.1007/3-540-29784-7_17. [DOI] [PubMed] [Google Scholar]
- McDonald PW, Hardie SL, Jessen TN, Carvelli L, Matthies DS, Blakely RD. Vigorous motor activity in Caenorhabditis elegans requires efficient clearance of dopamine mediated by synaptic localization of the dopamine transporter DAT-1. J Neurosci. 2007;27:14216–14227. doi: 10.1523/JNEUROSCI.2992-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/S0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Oikonomou G, Shaham S. The glia of Caenorhabditis elegans. Glia. 2011;59:1253–1263. doi: 10.1002/glia.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perens EA, Shaham S. C. elegans daf-6 encodes a patched-related protein required for lumen formation. Dev Cell. 2005;8:893–906. doi: 10.1016/j.devcel.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Piggott BJ, Liu J, Feng Z, Wescott SA, Xu XZ. The neural circuits and synaptic mechanisms underlying motor initiation in C. elegans. Cell. 2011;147:922–933. doi: 10.1016/j.cell.2011.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Zhang S, Wang HL, Wang H, de Jesus Aceves Buendia J, Hoffman AF, Lupica CR, Seal RP, Morales M. A glutamatergic reward input from the dorsal raphe to ventral tegmental area dopamine neurons. Nat Commun. 2014;5:5390. doi: 10.1038/ncomms6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. Acetylcholine. WormBook. 2007:1–21. doi: 10.1895/wormbook.1.131.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O'meara MM, Pe'er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods. 2008;5:865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/S0896-6273(00)81199-X. [DOI] [PubMed] [Google Scholar]
- Schafer WR. Deciphering the neural and molecular mechanisms of C. elegans behavior. Curr Biol. 2005;15:R723–R729. doi: 10.1016/j.cub.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Serrano-Saiz E, Poole RJ, Felton T, Zhang F, De La Cruz ED, Hobert O. Modular control of glutamatergic neuronal identity in C. elegans by distinct homeodomain proteins. Cell. 2013;155:659–673. doi: 10.1016/j.cell.2013.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, Spencer WC, O'Brien T, Cha B, Albeg A, Treinin M, Miller DM., 3rd Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Dev Biol. 2010;345:18–33. doi: 10.1016/j.ydbio.2010.05.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, Reinke V, Petrella L, Strome S, Stetina Von Stetina SE, Katz M, Shaham S, Rätsch G, Miller DM., 3rd A spatial and temporal map of C. elegans gene expression. Genome Res. 2011;21:325–341. doi: 10.1101/gr.114595.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord. 2013;28:41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, Knackstedt LA. Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci. 2012;32:12406–12410. doi: 10.1523/JNEUROSCI.1976-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veljkovic E, Stasiuk S, Skelly PJ, Shoemaker CB, Verrey F. Functional characterization of Caenorhabditis elegans heteromeric amino acid transporters. J Biol Chem. 2004;279:7655–7662. doi: 10.1074/jbc.M309528200. [DOI] [PubMed] [Google Scholar]
- Voglis G, Tavernarakis N. A synaptic DEG/ENaC ion channel mediates learning in C. elegans by facilitating dopamine signalling. EMBO J. 2008;27:3288–3299. doi: 10.1038/emboj.2008.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner LE, Zhou GT, Schafer RW, Schafer WR. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 1998;21:203–214. doi: 10.1016/S0896-6273(00)80527-9. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Murray JI, Lu Y, Waterston RH, Shaham S. mls-2 and vab-3 control glia development, hlh-17/Olig expression and glia-dependent neurite extension in C. elegans. Development. 2008;135:2263–2275. doi: 10.1242/dev.019547. [DOI] [PubMed] [Google Scholar]