

Abstract
Extracellular signal-regulated kinases 1 and 2 (ERK1/2) are highly homologous yet distinct components of signal transduction pathways known to regulate cell survival and function. Recent evidence indicates an isoform-specific role for ERK2 in pain processing and peripheral sensitization. However, the function of ERK2 in primary sensory neurons has not been directly tested. To dissect the isoform-specific function of ERK2 in sensory neurons, we used mice with Cre-loxP-mediated deletion of ERK2 in Nav1.8+ sensory neurons that are predominantly nociceptors. We find that ERK2, unlike ERK1, is required for peripheral sensitization and cold sensation. We also demonstrate that ERK2, but not ERK1, is required to preserve epidermal innervation in a subset of peptidergic neurons. Additionally, deletion of both ERK isoforms in Nav1.8+ sensory neurons leads to neuron loss not observed with deletion of either isoform alone, demonstrating functional redundancy in the maintenance of sensory neuron survival. Thus, ERK1 and ERK2 exhibit both functionally distinct and redundant roles in sensory neurons.
SIGNIFICANCE STATEMENT ERK1/2 signaling affects sensory neuron function and survival. However, it was not clear whether ERK isoform-specific roles exist in these processes postnatally. Previous work from our laboratory suggested either functional redundancy of ERK isoforms or a predominant role for ERK2 in pain; however, the tools to discriminate between these possibilities were not available at the time. In the present study, we use new genetic knock-out lines to demonstrate that ERK2 in sensory neurons is necessary for development of inflammatory pain and for postnatal maintenance of peptidergic epidermal innervation. Interestingly, postnatal loss of both ERK isoforms leads to a profound loss of sensory neurons. Therefore, ERK1 and ERK2 display both functionally distinct and redundant roles in sensory neurons.
Keywords: ERK, MAP kinase, MEK, nociception, pain, sensory neuron
Introduction
Extracellular signal-regulated kinases 1/2 (ERK1/2) are mitogen-activated protein kinases that integrate signals from growth factor receptors, ion channels, and G-protein coupled receptors. Activated ERK1/2 phosphorylate and activate downstream effectors that support cell survival and alter cell function (Roskoski, 2012). Previously, ERK1 and ERK2 were presumed to be functionally redundant given their 84% sequence homology, shared upstream activators, and similar substrate specificity (Boulton and Cobb, 1991; Boulton et al., 1991). However, genetic deletion of either ERK1 or ERK2 leads to distinct phenotypes, suggesting isoform-specific functions (Pagès et al., 1999; Mazzucchelli et al., 2002; Saba-El-Leil et al., 2003; Nekrasova et al., 2005; Satoh et al., 2007, 2011a; Newbern et al., 2008; Samuels et al., 2008; Alter et al., 2010; Fyffe-Maricich et al., 2011). Still, tissue-specific loss of both ERK1 and ERK2 leads to novel phenotypes that are not observed in single knock-out animals, implying other redundant functions (Newbern et al., 2011; Satoh et al., 2011b; Yasuda et al., 2011). Additional studies must address whether ERK1 and ERK2 are functionally redundant in ERK1/2-dependent processes.
Behavioral hypersensitivity after inflammation is mediated by ERK1/2. Following noxious stimulation or inflammation, phosphorylated (activated) ERK1/2 (pERK1/2) is observed in primary afferents, spinal cord dorsal horn, and brain regions involved in pain processing (Ji et al., 1999; Dai et al., 2002; Obata et al., 2003, 2004; Carrasquillo and Gereau, 2007). Inhibition of the upstream kinase, mitogen-activated protein kinase kinase (MEK), can both reduce and reverse inflammation-induced hypersensitivity (Ji et al., 2002; Obata et al., 2003; Zhuang et al., 2004; Karim et al., 2006). However, these experimental approaches cannot identify isoform-specific or cell type-specific roles of ERK1/2 in pain.
Recent studies have tried to address these questions. Although ERK1 is phosphorylated after injury, global ERK1 expression is dispensable for inflammatory and neuropathic pain (Alter et al., 2010). This suggests either functional redundancy of ERK isoforms or a predominant role of ERK2 in pain signaling. Recent studies support the latter hypothesis that, in the CNS, ERK2 plays a predominant role in behavioral sensitization following injury. Intrathecal delivery of neuron-targeted ERK2 siRNA mitigated inflammation-induced hypersensitivity (Xu et al., 2008). Moreover, conditional deletion of ERK2 in CNS neurons and astrocytes attenuated the development of inflammatory and neuropathic pain (Otsubo et al., 2012). However, neither of these studies addresses the role of ERK2 in sensory neurons or the functionally redundant roles of ERK1 and ERK2 in pain pathways.
In this study, we evaluated the role of ERK1 and ERK2 in Nav1.8+ sensory neurons that are predominantly nociceptors. Using postnatal, conditional ERK2 deletion in Nav1.8+ sensory neurons (NsERK2 KO) (Agarwal et al., 2004), we find that sensory neuron ERK2 is necessary for development of inflammation-induced hypersensitivity, cold sensation, and for postnatal maintenance of a subset of peptidergic epidermal innervation. Unexpectedly, postnatal loss of both ERK isoforms in Nav1.8+ neurons leads to a loss of sensory neurons that is not observed in either single isoform knock-out animal. Therefore, sensory neuron ERK2 drives peripheral sensitization, whereas expression of at least one ERK isoform is required for postnatal sensory neuron survival.
Materials and Methods
Animals.
All experiments were performed in accordance with National Institute of Health guidelines and were approved by the Animal Care and Use Committee of Washington University School of Medicine. Mice were housed in cages maintained on a 12 h light/dark cycle with ad libitum access to food and water. ERK1-targeted deletion was accomplished by using homologous recombination to replace exons 1–6 with a Neo cassette as previously described (ERK1−/−) (Nekrasova et al., 2005). These mice were backcrossed >10 times to a C57BL/6 background. Mice heterozygous for the ERK1 mutation were crossed to obtain ERK1−/− (ERK1 KO) and ERK1+/+ (WT) littermates. ERK2 conditional deletion in Nav1.8+ sensory neurons (NsERK2 KO) was accomplished using a Cre-LoxP system. The mapk1 gene encoding ERK2 was flanked with loxP sites (ERK2f/f) around exon 2, which, upon Cre-mediated excision, produces a null allele (Samuels et al., 2008). These mice were crossed with a BAC transgenic line expressing Cre recombinase under the Nav1.8 promoter (Nav1.8-cre) (Agarwal et al., 2004). Male ERK2f/f; Nav1.8-cre mice were crossed with ERK2f/f female mice. Through this cross, we obtained ERK2f/f;Nav1.8-cre (NsERK2 KO) and ERK2f/f (control) littermates. NsERK2 KO mice were backcrossed <10 times with C57BL/6 mice to create a mixed Bl6/C57;129 background. ERK1 KO mice were crossed with NsERK2 KO mice to create a mixed Bl6/C57;129 line of sensory-neuron-specific double knock-out mice wherein ERK1 is deleted globally and ERK2 is deleted specifically in Nav1.8+ sensory neurons. This sensory neuron-specific ERK1/2 double knock-out mouse (ERK1−/−;ERK2f/f; Nav1.8-cre) is referred throughout the paper as ERK dKO mice for simplicity of nomenclature. Male ERK1+/−;ERK2f/f;Nav1.8-cre mice were crossed with female ERK1+/−;ERK2f/f mice to obtain the following experimental mice as littermates: ERK1−/−;ERK2f/f (ERK1 KO), ERK1+/+;ERK2f/f (WT), ERK1+/+;ERK2f/f; Nav1.8-cre (NsERK2 KO), and ERK1−/−;ERK2f/f; Nav1.8-cre (ERK dKO). For all studies, littermate controls were used and experimenters were blinded to genotype.
Western blotting.
Sample preparation and immunoblotting were done as previously described (Alter et al., 2010). Mice were acclimated for 2–3 h in behavior chambers. Mice were killed using a guillotine, and spinal cord was extracted via hydraulic extrusion. The lumbar enlargement of the spinal cord was separated and flash frozen on dry ice. Thoracic and lumbar DRGs were isolated, pooled, and flash frozen. Frozen spinal cord and DRGs were homogenized using a Dounce homogenizer in ice-cold buffer containing 20 mm Tris, pH 7.5, 1 mm EDTA, 1 mm Na4P2O7, 25 μg/ml aprotinin (Sigma-Aldrich), 25 μg/ml leupeptin (Sigma-Aldrich), 100 μm PMSF (Roche Applied Science), 1 μg/ml microcystin LR (Enzo Life Science), and 1 mm Na3VO4 (Sigma-Aldrich) in Milli-Q distilled water. A BCA assay (Pierce Biotechnology) was used to measure protein concentration with a SmartSpec 3000 spectrophotometer (Bio-Rad).
Immunoblotting experiments were performed by loading 10 μg of protein homogenates into an SDS-PAGE gel (Bio-rad) for protein size separation. Protein homogenates were loaded into precast Tris-HCl 4% stacking and 10% separating polyacrylamide midi-gels (Bio-Rad). Following size separation on a SDS-PAGE gel, protein was transferred from the gel to a nitrocellulose membrane using a wet transfer system (Bio-Rad). Odyssey blocking buffer (LI-COR Biosciences) was used to block the membrane for 1 h at room temperature. The membrane was then incubated in primary antibodies diluted in Odyssey blocking buffer for 1 h at room temperature. The following primary antibodies were diluted in Odyssey blocking buffer (LI-COR Biosciences) and used for immunoblotting experiments: anti-pERK1/2 (mouse monoclonal, 1:1000; Cell Signaling Technology), anti-ERK1/2 (rabbit polyclonal, 1:1000; Cell Signaling Technology), anti-β-tubulin (mouse monoclonal, 1:10,000, Sigma), and mouse anti-actin (mouse monoclonal, 1:2000; Sigma-Aldrich). Following several washes, membranes were incubated for 1 h in the fluorescently tagged secondary antibodies, goat anti-rabbit IRDye800 (1:20,000; LI-COR Biosciences) and goat anti-mouse AlexaFluor-680 (1:20,000; Invitrogen) diluted in Odyssey blocking buffer (LI-COR Biosciences). Odyssey Infrared Fluorescence Imaging System (LI-COR Biosciences) was used to detect antibody binding at 700 nm (pERK1/2, β-tubulin, and actin) and 800 nm (ERK1/2) wavelength fluorescent emissions. Densitometry was used quantify band intensity using the Odyssey Infrared Fluorescence Imaging System software (LI-COR Biosciences).
Behavioral studies.
Male (see Figs. 1, 2, 3, 4) or female (see Figs. 6, 7) littermate mice were tested between 7 and 10 weeks of age during their light cycle. Behavioral experiments were performed in isolated rooms held at room temperature (∼25°C) with white noise on. Before behavioral testing, mice were acclimated to the testing area for at least 2 h. During thermal gradient experiments, mice were placed in individual lanes separated to prevent mice from seeing each other. For responses to thermal and mechanical stimuli, mice were placed into individual, Plexiglas behavioral chambers (width, 10 cm; length, 10 cm; height, 15 cm) separated by opaque dividers to prevent mice from seeing each other during testing.
Figure 1.

Conditional deletion of ERK2 is DRG specific. A, Schematic illustrates two constructs inserted in the mouse genome: a BAC-transgene with Cre-recombinase driven by a Nav1.8 promotor and the endogenous ERK2 allele with loxP sites (illustrated by triangles) inserted flanking exon 2. In ERK2f/f; SNS-Cre (called NsERK2 KO) expressing both constructs, Nav1.8 promotor-driven Cre expression led to excision of exon 2 at loxP site to create a null allele in Nav1.8+ sensory neurons. B, Western blot and quantification of isolated DRG lysates demonstrate ERK1 and ERK2 expression in NsERK2 KO (CKO) and ERK2f/f (flox) control mice with anti-ERK1/2 and anti-βIII tubulin as a loading control. C, Western blots and quantifications were performed to assess ERK1 and ERK2 expression in SC, Cx, CB, or Hippo using an anti-actin as a loading control. For quantification of ERK bands in B and C, integrated intensities of each isoform were divided by integrated intensity of total βIII tubulin (B) or actin (C) expression and then normalized to ERK2f/f controls (flox) (n = 7/genotype). ***p < 0.001.
Figure 2.

Conditional deletion of ERK2 impairs cold sensation without affecting heat or mechanical thresholds. A, D, NsERK2 KO (A; n = 7 or 8/group) and ERK1 KO (D; n = 5 or 6/group) mice were assessed for thermotaxis behavior during a 2 h trial on temperature gradient ranging from 49°C to 13°C. Anymaze software (Stoelting) was used to measure time that the mouse's center of mass spent in each zone during the entire test (mean ± SEM). ***p < 0.001. B, E, Using the extended cold plantar assay, paw withdrawal latency (seconds) to a cold stimulus in NsERK2 KO (B; n = 8/group) and ERK1 KO (E; n = 9/group) mice was measured and compared with control littermates on glass held at room temperature (22°C) or cooled to either 17°C or 12°C. *p < 0.05. C, NsERK2 KO (n = 14) and ERK2f/f (n = 14) control littermates were assessed for paw withdrawal latency (seconds) to a radiant heat source using the Hargreaves test. F, NsERK2 KO (n = 9) and ERK2f/f (n = 9) control littermates were assessed for paw withdrawal thresholds (in grams) using calibrated von Frey filaments. Data for paw withdrawal latencies and thresholds are expressed as mean ± SEM. G, Open field locomotor behavior was assessed over 1 h in NsERK2 KO (n = 7) and ERK2f/f (n = 7) control littermates. Data are binned into 5 min intervals and expressed as mean ± SEM. H, Time to fall off of an accelerating Rotarod was measured in both NsERK2 KO (n = 7) and ERK2f/f (n = 7) control littermates. Data are graphed for each of the five trials and expressed as mean ± SEM.
Figure 3.

Sensory neuron ERK2 plays a complex role in formalin- and CFA-induced hypersensitivity. A, Following intraplantar injection of 10 μl of 5% formalin, time spent in spontaneous behavior (e.g., licking, lifting, or flicking the injured paw) was assessed for ERK2f/f (n = 5) and NsERK2 KO (n = 8) littermates and plotted in 5 min bins for 1 h. B, Time spent in spontaneous behavior following 5% formalin intraplantar injection was parsed into first (0–10 min) and second (10–60 min) phase of spontaneous behavior. *p < 0.05. **p < 0.01. C, Paw withdrawal thresholds (in grams) in ERK2f/f (n = 8) and NsERK2 KO (n = 9) mice were measured at baseline and after intraplantar injection of 10 μl of 1 mg/ml CFA. *p < 0.05. **p < 0.01. D, Paw withdrawal latency to a radiant heat source was measured in ERK2f/f (n = 9) and NsERK2 KO (n = 10) mice and expressed as a percentage of baseline for 2 d following intraplantar injection of 10 μl CFA. E, F, CFA-induced paw edema was assessed by measuring paw thickness (in millimeters) in NsERK2 KO (E) and ERK1 KO mice (F) compared with littermate controls (n = 4–6) both at baseline and after CFA.
Figure 4.

ERK1 and sensory neuron ERK2 differentially modulate NGF-induced heat, but not mechanical, hypersensitivity. A, Paw withdrawal threshold (in grams) was measured at baseline and after an intraplantar injection of 0.2 μg/10 μl NGF using calibrated filaments in ERK2f/f (n = 13), NsERK2 KO (n = 12), ERK1 WT (n = 13), and ERK1 KO (n = 13) mice. Data are expressed as a paw withdrawal threshold in grams. B, Paw withdrawal threshold data were averaged from the first 3 h following intraplantar injection of 0.2 μg/10 μl NGF for each ERK knock-out and their control littermate (n = 12 or 13/group). Data are expressed as a percentage of baseline ± SEM. C, Following intraplantar injection of 0.2 μg/10 μl NGF, paw withdrawal latency to a radiant heat source was assessed in ERK2f/f (n = 9), NsERK2 KO (n = 11), ERK1 WT (n = 12), and ERK1 KO (n = 12) mice. Each time point was divided by the baseline withdrawal latency to express data as a percentage of the baseline latency. *p < 0.05. D, Percentage of baseline data was averaged from the first 3 h following intraplantar injection of 0.2 μg/10 μl NGF for each ERK knock-out and their control littermate (n = 9–12/group). **p < 0.05. Data are expressed as a mean ± SEM. E, Paw withdrawal latency to radiant heat source was measured in ERK2f/f (n = 8) and NsERK2 KO (n = 10) and expressed as a percentage of baseline averaging data from 1 to 3 h following intraplantar injection of 10 μl of 5% formalin. F, G, Mustard oil-induced heat hypersensitivity in NsERK2 KO (F; n = 4 or 5/group) and ERK1 KO (G; n = 5/group) mice was measured and expressed as a percentage of baseline averaging data from 1 to 3 h after 10 μl of 0.75% mustard oil. Data are mean ± SEM.
Figure 6.

Sensory neuron-specific ERK1/2 double knock-out mice exhibit altered baseline sensitivity to noxious stimuli. A, B, Paw withdrawal latency (in seconds) to a radiant heat source directed at either the hindpaw (A) or tail (B) was measured in female ERK dKO mice compared with control and single knock-out littermates (n = 5–8/genotype; one-way ANOVAs). **p < 0.01. C, Paw withdrawal threshold to an innocuous, calibrated von Frey mechanical stimulus was measured in female ERK dKO mice and compared with control and single knock-out littermates (n = 5–8/genotype, one-way ANOVA). D, Randall-Selitto tail pinch assay was used to measure tail withdrawal thresholds in female ERK dKO mice compared with control and single knock-out littermates (n = 5–8/genotype). ***p < 0.001 (one-way ANOVA). E, Data from Randall-Selitto assay in D were separated into distal (1 mm from tail end), medial (2 mm from tail end), and proximal (3 mm from tail end) bins (n = 5–8/genotype). ***p < 0.001 (two-way RM ANOVA with Bonferroni post test).
Figure 7.
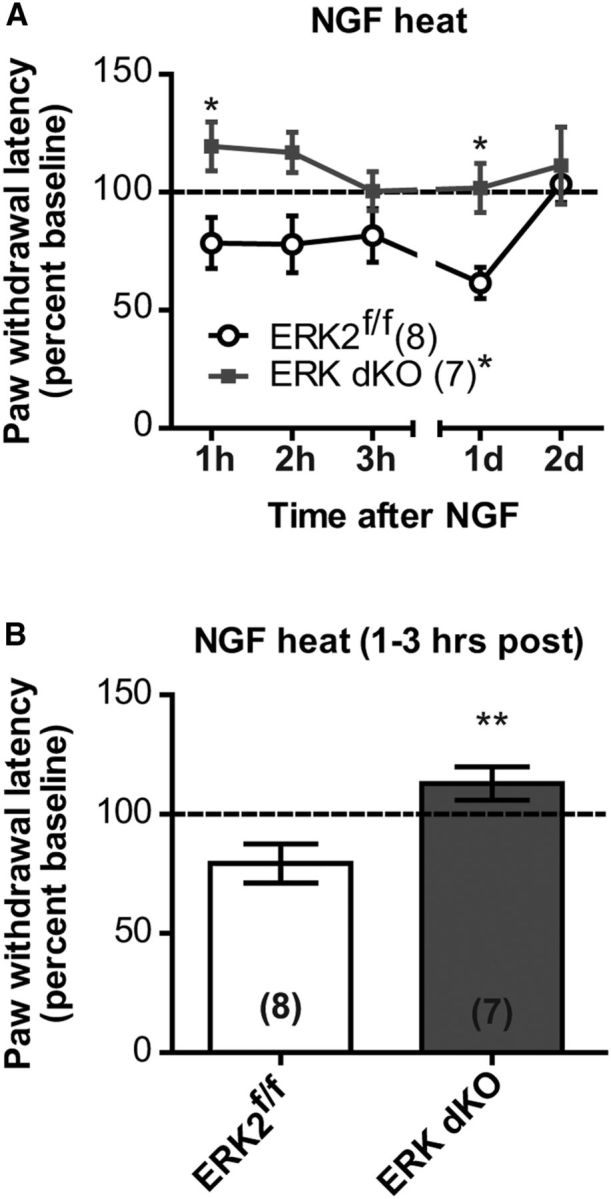
Sensory neuron-specific ERK1/2 double knock-out mice do not develop NGF-induced heat hypersensitivity. A, Following intraplantar injection of 0.2 μg/10 μl NGF, paw withdrawal latency to a radiant heat source was assessed in ERK dKO (n = 8) and ERK2f/f (n = 7) control littermates, and each time point was divided by the baseline paw withdrawal latency to express data as a percentage of the baseline latency. *p < 0.05 (two-way RM ANOVA with Bonferroni post test). B, Percentage of baseline data was averaged from the first 3 h following intraplantar injection of 0.2 μg/10 μl NGF for ERK dKO (n = 8) and their ERK2f/f control littermates (n = 7; unpaired t test). **p < 0.01. A, B, Data are expressed as a mean of the percentage baseline ± SEM.
Thermal gradient.
Naive mice were assessed for thermotaxis behavior on a thermal gradient ranging from 13°C to 49°C as described previously (Dhaka et al., 2007). Mice were acclimated to the room in their home cages before each day of testing. On the first day, mice were allowed to move freely for 2 h on the apparatus with no temperature gradient. On the day of testing, the gradient was turned on during acclimation of the mice in their home cage. Before testing, the temperatures of 20 equally sized zones were determined by averaging the temperature at the zone's edges. Mice were then allowed to freely move on the gradient over the next 2 h. On both days, Anymaze software (Stoelting) continuously tracked the center of each mouse to measure the time spent in each zone during the 2 h trial. Data were averaged for each genotype and graphed as mean ± SEM of time spent in each zone over the entire 2 h test.
Extended cold plantar assay.
Naive mice were tested for their response to a cold stimulus (dry ice) applied to the glass plate directly underneath the plantar area of their hindpaw as done previously (Brenner et al., 2012, 2014b). For all experiments, mice were placed in behavioral chambers on ¼-inch thick glass purchased from Stemmerich. A cold stimulus was applied to the glass underneath the hindpaw using powdered dry ice packed into a modified 3 ml syringe (BD Biosciences). Mirrors underneath the glass were used to accurately target the stimulus. Mice were assessed for their paw withdrawal latency to this cold stimulus every 7 min alternating between paws for each mouse. Only awake, still mice were tested. A 20 s cutoff was used to avoid tissue damage. Three to five measurements were taken using a stopwatch to determine average paw withdrawal latencies for each mouse.
The glass plate was either maintained at room temperature or cooled to either 17°C or 12°C. To uniformly cool the glass, either wet (17°C) or dry ice (12°C) was placed in two custom-built aluminum boxes (4.5 inches wide and 3 inches tall with a lid) that stretched the length of the glass on either side of the behavioral chambers (Brenner et al., 2014b). To cool the plate to 17°C, wet ice was added to these aluminum boxes positioned ∼0.25 inches away from the behavioral chambers on either side. To cool the plate to 12°C, dry ice pellets were added to the aluminum boxes that were positioned ∼1.25 inches away from the behavioral chambers on either side. Once the plate reached the desired temperature (within ∼1 h), the mice were placed in their behavioral chambers and acclimated there for at least 2 h. The temperature was continuously monitored at the middle of the plate using a 24P T-type filament thermocouple probe (Physitemp Instruments) and logged by a EA15 Data-Logging Dual Input Thermometer (Extech Instruments). The glass temperature was modified by either adding more ice or adjusting the position of the aluminum containers with respect to the behavioral chambers. Mice were only tested when the plate temperature was within ±1.0°C of the target temperature. Mice were tested on room temperature glass on the first day, and 17°C and 12°C glass on subsequent days.
Hargreaves' test.
Mice were evaluated for their response to a radiant heat source (Hargreaves apparatus) applied to the plantar surface of their hindpaw as previously described (Alter et al., 2010). Mice were placed in behavioral chambers on a prewarmed (∼30°C) glass plate of 390G Plantar Test Apparatus (IITC Life Sciences). Following acclimation on the apparatus, paw withdrawal latencies were determined using a radiant heat source stimulus set at a 14%–17% active intensity and 3%–5% inactive intensity. A 20 s cutoff was used to avoid tissue damage. For baseline paw withdrawal latencies, five independent measurements on each paw were performed separated by at least 30 min on the same paw. Following injection of inflammatory or algogenic compounds, two independent measurements per hour were taken for each paw and averaged for hourly time points. On each subsequent day, three independent measurements were acquired for each paw and averaged for the day's mean value. Data were then converted to and graphed as a percentage of baseline paw withdrawal latency.
von Frey.
Mice were tested for their 50% paw withdrawal threshold using calibrated von Frey filaments (North Coast Medical) as previously described (Alter et al., 2010). Mice were acclimated in behavioral chambers on a wire mesh for at least 2 h. During testing, filaments were pressed against the area between the anterior and posterior footpads of the hindpaw for ∼1 s. Awake, still mice were tested five times with each filament at a rate of ∼0.5 Hz starting with the lightest filament (0.08 g). Testing progressed to heavier filaments until a filament evoked a withdrawal response in at least three of five applications and was thus considered to be the 50% paw withdrawal threshold. A response was counted when the mouse withdrew its hindpaw from the mesh immediately following the filament application. For baselines, three to five independent measurements were taken for each paw separated by at least 30 min on the same paw. Following NGF-induced and complete Freund's adjuvant (CFA)-induced inflammation, paw withdrawal thresholds were determined hourly after injury from a single measurement. On subsequent days, three independent measurements were acquired and averaged for the day's mean value. Data were then graphed as the paw withdrawal threshold.
Radiant tail flick assay.
Mice were assessed for the tail withdrawal latency to a radiant heat source directed at their tail. Mice were restrained in a 50 ml conical tube equipped with a stopper and air holes. The mouse's tail was placed under radiant heat source of the Tail Flick Meter (IITC) maintained at an inactive intensity of 0%. When mice reached a still, awake state, the radiant heat source was applied to the tail with an active intensity of 50%. Once the mouse flicked its tail away from the radiant heat source, the trial was stopped and the tail flick latency was recorded. Care was taken to avoid visibly injured areas present on mice. Three independent measures were recorded at least 15 min apart.
Randall-Selitto tail pinch assay.
Mice were assessed for the sensitivity to a ramping mechanical force directed on their tail. Mice were restrained in a 50 ml conical tube equipped with a stopper and air holes. The mouse's tail was placed under the pusher on an Analgesia-Meter (Ugo Basile). When mice reached a still, awake state, the instrument was used to apply a force on the tail increasing at a rate of 16 g/s. The mechanical stimulus was stopped manually when the mouse began to struggle, and the cumulative force (in grams) needed to evoke a response was recorded as the tail withdrawal threshold. Three independent trials were performed at least 15 min apart starting at a distal region ∼1 mm from the end of the tail and moving ∼1 mm proximally for subsequent measures. Care was taken to avoid visibly injured areas present on mice.
Formalin-induced spontaneous behaviors.
To assess formalin-induced spontaneous behaviors, mice were given an intraplantar injection of 5% formalin and subsequently assessed for time spent in spontaneous pain behavior, including licking, lifting, and flicking. Mice were first acclimated in behavioral chambers for at least 2 h. Mice were then injected subcutaneously with 10 μl of 5% formalin (Sigma-Aldrich) diluted in 0.9% NaCl into the plantar surface of the hindpaw. Mice were immediately returned to their behavior chamber and the experimenter exited the room. A video was recorded from below using a webcam (Logitech) with resolution set at 960 × 720 for 1 h following injection. Total time spent licking, lifting, and flicking the injured paw was assessed post hoc from the videos by a blinded experimenter using a stopwatch. Total time spent in these spontaneous behaviors was binned every 5 min for the hour following formalin injection. Time spent in spontaneous behavior was also parsed into Phase 1 (0–10 min after injection) and Phase 2 (10–60 min after injection).
Inflammation- and algogen-induced hypersensitivity.
Inflammatory and algogenic compounds were injected subcutaneously into the plantar surface of the hindpaw. CFA-induced inflammation was caused by a 10 μl injection of 1 mg/ml CFA (Sigma-Aldrich). NGF-induced inflammation was caused by a 10 μl injection of 0.2 μg NGF diluted in 0.9% NaCl. As in spontaneous behavior experiments, 10 μl of 5% formalin (Sigma-Aldrich) diluted in 0.9% NaCl was injected to induce hypersensitivity. Mustard oil-induced hypersensitivity was caused by an injection of 10 μl of 0.75% mustard oil diluted in light mineral oil. Hypersensitivity following each algogen was measured as described above for thermal (Hargreaves' test) and mechanical (von Frey) stimuli.
CFA-induced paw edema.
Paw inflammation following CFA injection was monitored in both ERK1 KO and NsERK2 KO mice compared with control littermates. As in behavioral experiments, mice were habituated for 2 h in individual behavioral chambers before testing. Following habituation, 150 mm stainless dial calipers (Chicago Brand) were used to measure paw thickness (in millimeters) to establish a baseline reading. Mice then received an intraplantar injection of 10 μl injection of 1 mg/ml CFA (Sigma-Aldrich). CFA-induced inflammation was monitored by measuring paw thickness (in millimeters) over the same time course used in the CFA-induced behavioral hypersensitivity assays. Data were graphed as paw thickness (in millimeters).
Accelerating rotarod.
Untrained NsERK2 KO and control (ERK2f/f) littermate mice were assessed for their gross motor function using an accelerating Rotarod (Ugo Basile). Five consecutive trials were performed with at least 5 min between trials. Latency to fall or roll on the Rotarod was measured as the apparatus accelerated from 4 to 40 rpm over 5 min.
Open field.
Locomotion was assessed using an open field equipped with a Versamax Animal Activity Monitoring System (AccuScan Instruments). Before testing, mice were habituated to the room in their home cages for at least 1 h. Naive NsERK2 KO and control (ERK2f/f) littermates were then placed in the open field during individual trials and allowed to freely explore after the experimenter exited the room. The number of photobeam breaks was calculated to measure the horizontal activity of mice in the chamber (42 × 42 × 30 cm, length × width × height) during the 1-h-long trial.
Immunohistochemistry (IHC).
Tissues for IHC experiments were collected as previously described (Golden et al., 2010). Tissue from 7- to 10-week-old male littermates was fixed with either Zamboni's fixative (footpads) or via transcardial perfusion with 4% PFA (all other tissue). Following rinsing with PBS, tissue was cryoprotected in 30% sucrose. For Ret immunostaining, DRGs were fresh frozen. Tissue was sectioned using a cryostat set to cut either 30 μm (footpads and spinal cord) or 18-μm-thick (DRG) sections.
Immunostaining experiments were performed as follows. Following brief postfix in 4% paraformaldehyde in PBS and a couple washes in TBS, sections were blocked using a 1% BSA (Sigma-Aldrich) and 0.2% Difco milk (BD Biosciences) solution dissolved in TBS with 0.1% Triton X-100. Subsequently, sections were incubated overnight at 4°C in the following primary antibodies diluted in Tyramide Signal Amplification (TSA) blocking reagent (PerkinElmer): rabbit anti-Ret (1:50, IBL), rabbit anti-TrkA (1:300, Millipore), goat anti-rat calcitonin gene-related peptide (CGRP, 1:400, Serotec AbD), mouse anti-NF200 (1:200, Sigma), rabbit anti-βIII-tubulin (1:1000, Covance), and mouse anti-βIII-tubulin (1:1000, Millipore). IB4+ labeling was performed overnight at 4°C using an Alexa-conjugated IB4 (1:200, Invitrogen) diluted in TSA blocking reagent (PerkinElmer). The following day, a few washes in TBS with 0.1% Triton X-100 were performed before incubation with fluorescent-conjugated secondary antibodies (Invitrogen) diluted in TSA blocking reagent (PerkinElmer). The following fluorescent-conjugated secondary antibodies were used from Invitrogen: donkey anti-rabbit 555 (1:200), donkey anti-goat 488 (1:200 or 1:350), donkey anti-mouse 555 (1:200), and donkey anti-rabbit 488 (1:200).
TMP histochemistry.
Following several washes in Tris-maleate buffer, spinal cord sections were incubated at 37°C in Tris-maleate buffer containing 0.25% thioamine phosphate (TMP; Sigma-Aldrich) and 0.08% lead nitrate (Sigma-Aldrich) for 30 min. Following another round of washes, the reaction was developed briefly in an aqueous solution of 1% sodium sulfide (Sigma-Aldrich). The reaction was quenched in Tris-maleate buffer. Then, the slides were dehydrated and coverslipped.
Microscopy.
Images were taken using either a Nikon 80i upright epifluorescence microscope with a CoolSnap ES camera or an inverted Leica DMI400 CSQ Confocal Microscope equipped with a spectral detector for imaging at 488 and 543 nm. Images were then processed in National Institutes of Health ImageJ, version 1.48 or Adobe Photoshop software using only global adjustments to brightness and contrast.
Stereological counts.
Stereological counts were done as previously described (Golden et al., 2010). To determine intraepidermal nerve fiber density (IENFD) for total (βIII tubulin+) and peptidergic (CGRP+) fibers, five randomly selected areas of thick, glaborous skin (≥120 μm apart) were assessed for fibers coursing through a measured length of the dermal–epidermal border. To determine the total numbers of βIII-tubulin+, Ret+, TrkA+, CGRP+, and NF200+ neurons, 18 μm sections were collected from L3-L5. Positively immunostained profiles were counted in every fourth section of one DRG (L3, L4, or L5), and final counts were obtained by multiplying counts by 4. To determine percentages of DRG neuron subpopulations, three randomly selected sections (≥70 μm apart) per DRG were counted for the number of labeled neurons as a percentage of the total (βIII tubulin+) neurons. Neuron sizes were determined manually using MetaMorph software. Spinal cord immunohistochemistry experiments were assessed qualitatively by blinded experimenters.
Statistical analyses.
All data were analyzed using Microsoft Excel (Microsoft) and GraphPad Prism (GraphPad Software). Data are expressed as mean ± SEM. The text and figure legends indicate the statistical test that was used for each dataset. The text also indicates the group size and degrees of freedom for each experiment. The following tests were used to analyze data under various circumstances (outlined in parentheses): unpaired t test (two groups), one-way repeated-measures (RM) ANOVA (>2 groups), two-way RM ANOVA (time course with 2 or more groups) or Kologorov–Smirnov test (comparing two distributions to each other). The criterion for significance was set at p < 0.05 a priori.
Results
DRG-specific conditional deletion of ERK2
Despite the known importance of ERK1/2 signaling in peripheral sensitization (Ji et al., 1999; Dai et al., 2002; Obata et al., 2003; Karim et al., 2006), our previous report demonstrated that ERK1 is dispensable for behavioral hypersensitivity following inflammation or peripheral nerve injury (Alter et al., 2010). To test the hypothesis that ERK2 plays a predominant role in peripheral sensitization, we conditionally deleted ERK2 from primary sensory neurons using Nav1.8 promoter-driven Cre-recombinase expression from a BAC transgene (Fig. 1A) (Agarwal et al., 2004; Samuels et al., 2008). Cre expression was driven primarily in small, unmyelinated, nociceptive sensory neurons but was also in approximately one-third of large, myelinated, Aβ sensory neurons (Agarwal et al., 2004; Shields et al., 2012). Compared with control littermates lacking the Cre transgene (ERK2f/f), there was a decrease in ERK2 expression in the DRG of NsERK2 KO mice (Fig. 1B; unpaired t test, ***p < 0.0001). As expected, NsERK2 KO DRG retained some ERK2 likely due to its expression in Nav1.8-negative sensory neurons and non-neuronal cells. Importantly, ERK1 expression in DRG was unaltered by deletion of ERK2 (Fig. 1B; unpaired t test, p = 0.6644). Additionally, NsERK2 KO mice did not exhibit any changes in ERK1 or ERK2 expression in spinal cord (SC), cerebellum (CB), cortex (Cx), and hippocampus (Hippo) compared with control littermates (Fig. 1C; unpaired t tests, for ERK1: p = 0.9153 for SC, p = 0.7644 for CB, p = 0.6534 for Cx, p = 0.5949 for Hippo; and for ERK2: p = 0.9113 for SC, p = 0.4681 for CB, p = 0.6281 for Cx, p = 0.9815 for Hippo). ERK2 expression was not altered in control littermates (ERK2f/f) compared with wild-type mice (data not shown). Overall, these data are consistent with a selective loss of ERK2 in Nav1.8+ sensory neurons.
Conditional deletion of ERK2 impairs cold sensation without affecting heat or mechanical thresholds
Although pharmacological inhibition of ERK1/2 signaling does not alter sensory function in naive mice (Dai et al., 2002; Obata et al., 2003), we tested whether conditional deletion of ERK2 altered sensation using naive NsERK2 KO mice. To assess thermal detection ability, we monitored thermotaxis behavior on a temperature gradient ranging from 49°C to 13°C (Golden et al., 2010). NsERK2 KO mice demonstrated a moderate, but significant, shift in their thermotaxis behavior toward cooler temperatures compared with control littermates (Fig. 2A; two-way RM ANOVA with a Bonferroni post test: significant interaction between zone and genotype, F = 4.42, degrees of freedom numerator (dfn) = 19, degrees of freedom denominator (dfd) = 260, ***p < 0.0001). Importantly, NsERK2 KO mice exhibited no differences in open field exploratory behavior (Fig. 2G; two-way RM-ANOVA: F = 0.65, dfn = 1, dfd = 12, p = 0.4366 for genotype), suggesting that the change in thermotaxis behavior is not due to decreased overall activity. The shift in temperature preference could result from either increased sensitivity to heat or decreased sensitivity to cold. To test these possibilities, we measured responses to cold and heat stimuli in naive mice. NsERK2 KO mice exhibited increased withdrawal thresholds to cold stimuli compared with control littermates using the extended cold plantar assay (Brenner et al., 2012, 2014b) (Fig. 2B; two-way RM ANOVA with a Bonferroni post test: F = 7.55, dfn = 1, dfd = 14, *p = 0.0157 for genotype). However, NsERK2 KO mice showed no change in their withdrawal latency to radiant heat compared with control littermates (Fig. 2C; unpaired t test, p = 0.6796). In contrast, ERK1 KO mice did not exhibit a change in thermotaxis behavior (Fig. 2D; two-way RM ANOVA: no interaction between zone and genotype, F = 0.22, dfn = 19, dfd = 152, p = 0.9998), sensitivity to cold in the extended cold plantar assay (Fig. 2E; two-way RM ANOVA: F = 0.48, dfn = 1, dfd = 32, p = 0.5003 for genotype), or sensitivity to radiant heat (Alter et al., 2010). To determine whether mechanical sensation is altered by loss of either ERK1 or ERK2, we used calibrated von Frey filaments. Neither ERK1 KO (Alter et al., 2010) nor NsERK2 KO mice (Fig. 2F; unpaired t test, p = 0.2950) exhibited any changes in their withdrawal threshold compared with control littermates. Neither ERK isoform knock-out mouse exhibited a deficit in the accelerating rotarod test (Fig. 2H; two-way RM ANOVA: F = 0.03, dfn = 1, dfd = 48, p = 0.8739 for genotype) (Alter et al., 2010), indicating that gross motor function was intact. Overall, these data suggest that sensory neuron ERK2 regulates cold sensation.
Sensory neuron ERK2 plays a complex role in formalin-induced spontaneous pain behavior
ERK1/2 signaling is known to play a role in peripheral sensitization induced by chemical or inflammatory stimuli; however, ERK1 is dispensable for this sensitization (Obata et al., 2003; Alter et al., 2010). To test whether sensory neuron ERK2 drives chemical-induced peripheral sensitization, we injected formalin into the paw, which elicits a behavioral response known to rely, in part, on peripheral sensitization (Tjølsen et al., 1992; Puig and Sorkin, 1996). Control littermates exhibited the stereotypical biphasic response to 5% formalin intraplantar injection (Fig. 3A) characterized by an immediate behavioral response (Phase 1, 0–10 min) that is followed by a longer lasting delayed phase (Phase 2, 10–60 min). Unexpectedly, NsERK2 KO mice exhibited an increase in formalin-induced first phase spontaneous behavior compared with control littermates (Fig. 3B; unpaired t test, *p = 0.0280). Despite this increased first phase, the NsERK2 KO mice displayed decreased spontaneous behavior overall (0–60 min combined) (Fig. 3A; two-way RM ANOVA with a Bonferroni post test: significant interaction between time and genotype F = 2.80, dfn = 11, dfd = 121, **p = 0.0028) and in the second phase (Fig. 3B; unpaired t test, *p = 0.0119), suggesting that sensory neuron ERK2 plays a complex role in formalin-induced peripheral sensitization. Therefore, a more detailed behavioral study was required to understand the role of ERK2 in peripheral sensitization.
Sensory neuron ERK2 is necessary for CFA-induced mechanical, but not heat, hypersensitivity
To better understand the role of sensory neuron ERK2 in peripheral sensitization, we assessed the development and maintenance of CFA-induced hypersensitivity in NsERK2 KO mice. CFA-induced heat and mechanical hypersensitivity are both attenuated by pharmacological blockade of ERK1/2 signaling (Obata et al., 2003). Additionally, CFA-induced heat and mechanical hypersensitivity are unaffected by global deletion of ERK1 (Alter et al., 2010). Therefore, we hypothesized that sensory neuron ERK2 is necessary for CFA-induced hypersensitivity. Following intraplantar injection of CFA, NsERK2 KO mice exhibited attenuated mechanical hypersensitivity compared with control littermates (Fig. 3C; two-way RM ANOVA with a Bonferroni post test: F = 9.43, dfn = 1, dfd = 105, **p = 0.0078 for genotype). In contrast, NsERK2 KO mice displayed a similar magnitude of CFA-induced heat hypersensitivity compared with control littermates (Fig. 3D; two-way RM ANOVA: F = 0.12, dfn = 1, dfd = 119, p = 0.7380 for genotype). Importantly, NsERK2 KO and ERK1 KO mice exhibited CFA-induced paw edema comparable with that of littermate controls, suggesting that loss of either ERK isoform does not affect the magnitude of CFA-induced inflammation (Fig. 3E,F; two-way RM ANOVA for NsERK2 KO in 3E: F = 0.04, dfn = 1, dfd = 9, p = 0.8461 for genotype; two-way RM ANOVA for ERK1 KO in 3F: F = 0.26, dfn = 1, dfd = 8, p = 0.6209 for genotype). Overall, these results indicate that sensory neuron ERK2 plays an isoform-specific role in CFA-induced mechanical, but not heat, hypersensitivity.
Neither ERK1 nor sensory neuron ERK2 is necessary for NGF-induced mechanical hypersensitivity
To extend our understanding of the role of ERK isoforms in inflammation-induced pain, we assessed whether either ERK1 or ERK2 were necessary for NGF-induced hypersensitivity. Previously, a pharmacological study demonstrated a role for ERK1/2 in development of NGF-induced mechanical hypersensitivity (Malik-Hall et al., 2005). To determine whether isoform-specific functions exist for NGF-induced mechanical hypersensitivity, we assessed the development and maintenance of mechanical hypersensitivity after NGF in both ERK1 KO and NsERK2 KO mice compared with littermate controls. NsERK2 KO mice displayed similar NGF-induced mechanical hypersensitivity compared with their control littermates over the entire test (Fig. 4A; two-way RM ANOVA with a Bonferroni post test: for genotype, F = 0.25, dfn = 1, dfd = 23, p = 0.6208) and at 1–3 h following NGF injection (Fig. 4B; t test, p = 0.8007). Likewise, ERK1 KO mice exhibited comparable NGF-induced mechanical hypersensitivity to their control littermates over the entire test (Fig. 4A; two-way RM ANOVA with a Bonferroni post test: for genotype F = 0.50, dfn = 1, dfd = 24, p = 0.4877) and at 1–3 h following NGF injection (Fig. 4B; t test, p = 0.1789). Together, these data suggest that neither ERK1 nor ERK2 alone is necessary for the development and maintenance of NGF-induced mechanical hypersensitivity.
ERK1 deletion and sensory neuron ERK2 deletion differentially modulate NGF-induced heat hypersensitivity
A previous pharmacological study implicated ERK1/2 activity in the induction of heat hypersensitivity by NGF (Zhuang et al., 2004). Following intraplantar injection of NGF, control littermates developed heat hypersensitivity that persisted for 24 h. NsERK2 KO mice displayed blunted NGF-induced heat hypersensitivity compared with their control littermates over the entire test (Fig. 4C; two-way RM ANOVA with a Bonferroni post test: for genotype, F = 5.81, dfn = 1, dfd = 18, *p = 0.0268) and at 1–3 h following NGF injection (Fig. 4D; unpaired t test, **p = 0.0015). Unexpectedly, ERK1 KO mice developed increased heat hypersensitivity relative to their wild-type littermates during the entire test (Fig. 4C; two-way RM-ANOVA with a Bonferroni post test: for genotype, F = 4.51, dfn = 1, dfd = 22, *p = 0.0452) and at 1–3 h following NGF injection (Fig. 4D; unpaired t test, **p = 0.0098). Based on these findings, ERK1 and ERK2 play functionally distinct and opposing roles in the development of NGF-induced heat hypersensitivity.
Although ERK1 KO and NsERK2 KO mice had significantly altered NGF-induced heat hypersensitivity, both lines had normal CFA-induced heat hypersensitivity (Fig. 3D) (Alter et al., 2010). To further evaluate these findings, we tested heat hypersensitivity following other algogens. NsERK2 KO mice developed formalin- and mustard oil-induced heat hypersensitivity comparable with control littermates (Fig. 4E,F; unpaired t tests, p = 0.4871 and p = 0.7417, respectively). ERK1 KO mice exhibited attenuated formalin-induced heat hypersensitivity (Alter et al., 2010) but displayed similar mustard oil-induced heat hypersensitivity compared with control littermates (Fig. 4G; unpaired t test, p = 0.1379). Taken as a whole, these data suggest that ERK1 and ERK2 differentially contribute to heat hypersensitivity generated by different algogens.
Sensory neuron-specific ERK2 deletion causes basal hyperphosphorylation of ERK1
Because pharmacological studies support a role for ERK1/2 activity in both NGF-induced mechanical hypersensitivity and CFA-induced heat hypersensitivity (Obata et al., 2003; Malik-Hall et al., 2005), the absence of a phenotype in either ERK1 KO or NsERK2 KO mice may suggest functional redundancy (Figs. 3D and 4A) (Alter et al., 2010). Hyperphosphorylation of one ERK isoform has been observed in the absence of the other in various systems (Mazzucchelli et al., 2002; Lefloch et al., 2008; Xu et al., 2008; Alter et al., 2010; Otsubo et al., 2012) and may reflect a mechanism for functional compensation. Naive NsERK2 KO mice demonstrated increased levels of pERK1, but not pERK2, in DRG compared with control littermates (Fig. 5A; unpaired t tests, *p = 0.0232 for pERK1 and p = 0.7782 for pERK2). This change in pERK levels was not observed in SC (Fig. 5B; unpaired t tests, p = 0.8738 for pERK1 and p = 0.6854 for pERK2). Despite this compensatory hyperphosphorylation of ERK1 in DRG, NsERK2 KO mice fail to develop some inflammatory pain phenotypes, suggesting that sensory neuron ERK1 cannot completely compensate for loss of ERK2. However, in other inflammatory pain models, NsERK2 KO mice develop hypersensitivity normally, possibly due to compensation via hyperactivation of ERK1 in DRGs.
Figure 5.

Sensory neuron ERK2 deletion leads to basal hyperphosphorylation of ERK1. Representative Western blots of total DRG (A) and SC (B) lysates were probed with a pERK1/2 antibody and an ERK1/2 antibody in ERK2f/f (n = 7) and NsERK2 KO (n = 7). Integrated intensities of phosphorylated (activated) pERK1 and pERK2 were divided by integrated intensities of total ERK1 and ERK2, respectively, to determine the ratio of activated to total protein (pERK1/ERK1 and pERK2/ERK2). *p < 0.05 (unpaired t tests).
Sensory neuron-specific ERK1/2 double knock-out mice exhibit deficits in sensation and inflammatory pain
To assess functionally redundant roles of ERK1 and ERK2 in sensory neurons, we generated mice with global deletion of ERK1 and Nav1.8+ sensory neuron-specific deletion of ERK2 by crossing the NsERK2 KO and ERK1 KO mice. These mice are sensory neuron-specific ERK1/2 double knock-out mice in the context of ERK1 global deletion and, for simplicity of nomenclature, will be referred to as ERK dKO mice throughout the remainder of the paper. Naive ERK dKO mice did not exhibit any gross motor deficits (data not shown). Naive ERK dKO mice did exhibit decreased sensitivity to radiant heat on their hindpaw, but not their tails (Fig. 6A,C; one-way ANOVA, **p = 0.0025 and p = 0.6752, respectively). Naive ERK dKO mice also exhibited decreased sensitivity to mechanical stimuli applied to the tail, but not the hindpaw (Fig. 6D,B; one-way ANOVA, ***p < 0.0001 and p = 0.5025, respectively). Loss of mechanical sensitivity was only evident at the distal end of the tail, suggesting a length-dependent loss of sensation (Fig. 6E; two-way RM ANOVA with a Bonferroni post test: significant effect of genotype F = 20.00, dfn = 3, dfd = 44, ***p < 0.0001 compared with ERK2f/f). ERK dKO mice also displayed attenuated NGF-induced heat hypersensitivity compared with control littermates over the entire test (Fig. 7A; two-way RM ANOVA, F = 7.95, dfn = 1, dfd = 13, *p = 0.0145 for genotype) and at 1–3 h following NGF injection (Fig. 7B; unpaired t test, **p = 0.0093). Interestingly, all adult ERK dKO mice presented with diffuse injuries on their distal extremities, specifically on their tails and ears, first seen at ∼5 weeks of age (Fig. 8A,B). While these injuries could be due to either self-mutilation or fighting with littermates, the sites of injury were more consistent with self-mutilation behavior (autotomy). Such autotomy behavior may result from either dysesthesia or lack of sensation (Ossipov and Porreca, 2013). ERK dKO mice did not exhibit any overt signs of spontaneous scratching behavior, suggesting that the diffuse injuries were not the result of increased itch. Regardless of its cause, this self-injury phenotype confounds the interpretation of behavioral data in pain models; and, as such, no further behavioral testing was performed on ERK dKO mice. Importantly, neither ERK1 KO nor NsERK2 KO mice exhibited these injuries.
Figure 8.
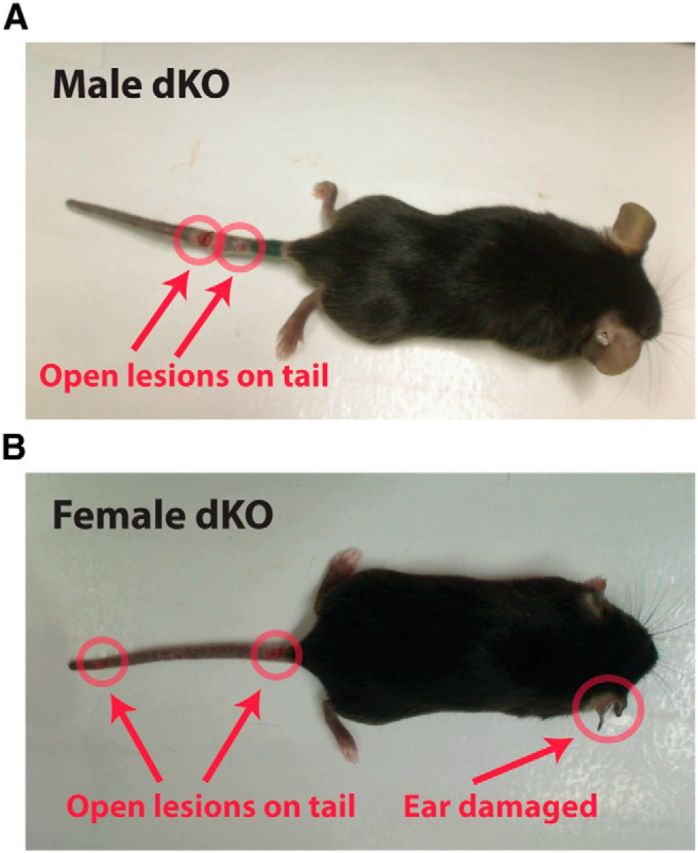
Sensory neuron-specific double knock-out mice exhibit injuries on their distal extremities. Representative images of adult male (A) and female (B) ERK dKO mice demonstrate injuries on distal extremities (e.g., tail and ear) highlighted in red.
Expression of at least one ERK isoform is necessary for target innervation
Because of the self-injury phenotype and baseline sensory deficits, we hypothesized that the anatomy of the peripheral nervous system was altered in ERK dKO mice. During embryogenesis, ERK1/2 signaling is known to play a predominant role in the development of the peripheral nervous system, particularly for DRG neuron target innervation and survival (Newbern et al., 2011). However, it is not known whether ERK1/2 signaling is necessary after birth for maintaining target innervation or for survival of sensory neurons. Because the Nav1.8-driven BAC transgenic Cre has been demonstrated to express Cre-recombinase starting at P0 (Agarwal et al., 2004), we can directly test whether sensory neuron ERK1/2 signaling is necessary for postnatal sensory neuron maintenance.
To assess target innervation in ERK dKO mice, we counted nociceptive free nerve endings in the epidermis. In the thick, glabrous skin of the hindpaw, both ERK1 KO and NsERK2 KO mice demonstrated that normal total IENFD was assessed by the pan-neuronal marker, βIII tubulin (Fig. 9A–C,I; unpaired t tests, p = 0.2970 for ERK1 KO and p = 0.7435 for NsERK2 KO). However, ERK dKO mice exhibited a decrease (∼40%) in total IENFD compared with control littermates, suggesting that expression of at least one ERK isoform is required for maintaining total IENFD (Fig. 9D,I; unpaired t test, ***p = 0.0009). To further investigate the molecular identity of the remaining fibers in ERK dKO mice, we assessed peptidergic IENFD that normally accounts for ∼40% of epidermal innervation by labeling fibers for CGRP (Zylka et al., 2005). Whereas ERK1 KO mice exhibited comparable CGRP+ IENFD to control littermates (Fig. 9F,J; unpaired t test, p = 0.5741), NsERK2 KO mice exhibited a small, but significant (∼25%), decrease (Fig. 9G,J; unpaired t test, *p < 0.05). Strikingly, ERK dKO mice exhibited almost total loss (∼85% decrease) of CGRP+ IENFD (Fig. 9H,J; unpaired t test, ***p < 0.001). Because peptidergic and nonpeptidergic fibers account for nearly all epidermal innervation (Zylka et al., 2005), the 40% decrease in total IENFD in ERK dKO mice is likely accounted for solely by the near-complete loss of peptidergic IENFD. In NsERK2 KO mice, the decrease in peptidergic IENFD is likely too small to be seen in total IENFD. Therefore, in both lines, nonpeptidergic IENFD likely remains normal.
Figure 9.
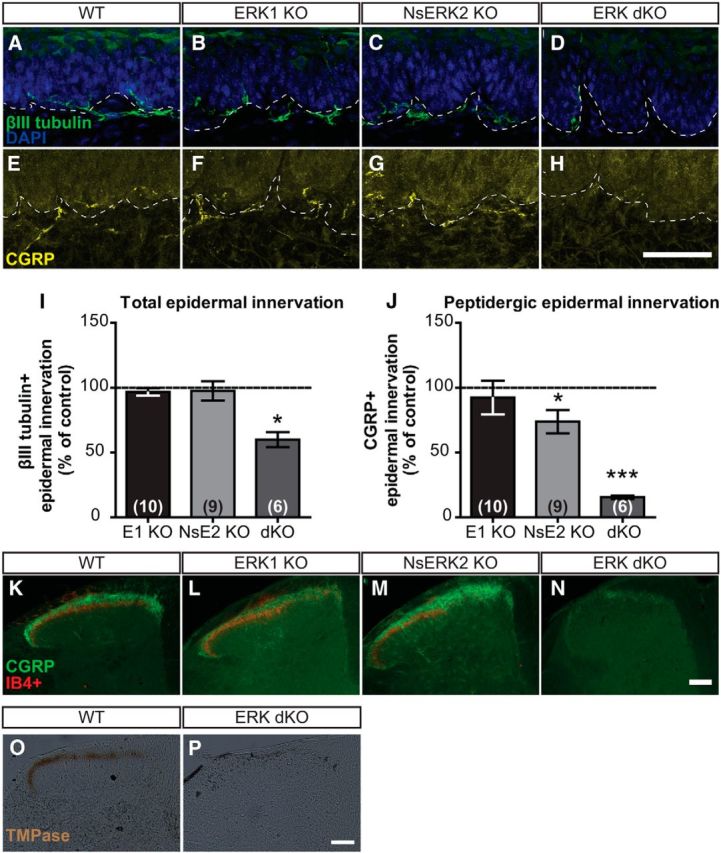
Expression of at least one ERK isoform is necessary for target innervation of epidermis and SCDH. A–D, Representative confocal images of total IENFD demonstrate βIII-tubulin+ fibers coursing through dermal–epidermal border in WT (A), ERK1 KO (B), NsERK2 KO (C), and ERK dKO (D) mice. E–H, Representative confocal images of peptidergic IENFD demonstrate CGRP+ fibers coursing through dermal–epidermal border in WT (E), ERK1 KO (F), NsERK2 KO (G), and ERK dKO (H) mice. Scale bar, 50 μm. I, J, In independent studies, average total (I) and peptidergic (J) IENFD (fibers/100 μm) were determined for NsERK2 KO (n = 9), ERK1 KO (n = 10), and ERK dKO (n = 6) mice, and then data were divided by the control (n = 6–10) littermates' IENFD for graphical illustration (mean ± SEM). K–N, Representative images of nociceptive central termination in the SCDH demonstrated CGRP+ (green) and IB4+ (red) nociceptive terminals in laminas I and II, respectively, for WT (K), ERK1 KO (L), NsERK2 KO (M), and ERK dKO (N) mice. Scale bar, 100 μm. TMPase assay assessed PAP activity present in nonpeptidergic fibers terminating in lamina II of SCDH in WT (O; n = 4). P, ERK dKO (n = 4) did not exhibit any TMPase activity indicative of nonpeptidergic terminals. Scale bar, 100 μm. *p < 0.05 and ***p < 0.001.
To determine whether central terminals of DRG neurons were affected, we assessed their termination in the spinal cord dorsal horn (SCDH) from ERK mutants. Distinct patterns of nociceptive fiber inputs occur in the SCDH such that peptidergic (CGRP+) fibers project to laminas I, IIi, and deeper lamina, and nonpeptidergic (isolectin B4 binding, IB4+) fibers project to lamina IIo as seen in control littermates (Fig. 9K). In both ERK1 KO and NsERK2 KO mice, this pattern remained intact (Fig. 9L,M). However, ERK dKO mice displayed decreased CGRP+ terminals and loss of IB4+ binding, suggesting that there was a loss of peptidergic and nonpetidergic terminals (Fig. 9N). To assess another marker of nonpetidergic terminals, we examined whether the SCDH of ERK dKO mice exhibited TMPase activity seen specifically in nonpeptidergic fibers (Zylka et al., 2008). No TMPase activity remained in the ERK dKO SCDH, whereas control littermates showed TMPase activity in lamina IIo (Fig. 9O,P). Together, this suggests that expression of at least one ERK isoform is required for central termination of nociceptive fibers.
Expression of at least one ERK isoform is necessary for DRG sensory neuron survival
In prior studies, prenatal loss of ERK1/2 in sensory neurons led to loss of epidermal innervation and subsequent neuronal cell death presumably due to a lack of trophic support (Newbern et al., 2011). Given these observations, we hypothesized that the decreased IENFD in ERK dKO mice was associated with DRG neuron loss. To test this hypothesis, L5 DRGs from both single and double knock-out mice were analyzed for total neuron number using the pan-neuronal marker, βIII tubulin. Consistent with unchanged IENFD, neither ERK1 KO nor NsERK2 KO mice exhibited changes in total L5 DRG neuron number compared with control littermates (Tables 1, 2; unpaired t tests). However, ERK dKO mice showed an ∼67% decrease in L5 DRG neuron number (Fig. 10A,B; unpaired t test, ***p = 0.0004). Notably, ERK dKO mice exhibited a decrease in neurons of various sizes (Fig. 10C; two-way RM ANOVA, two-way RM ANOVA with a Bonferroni post test; interaction between genotype and size, **p = 0.0011) but still had a similar cumulative distribution of DRG neuron sizes compared with control littermates (Fig. 10D; Kologorov–Smirnov test). These data suggest that expression of at least one ERK isoform is required for postnatal survival of both small- and large-diameter sensory neurons. Importantly, loss of ∼67% of DRG neurons is consistent with loss of most Nav1.8+ sensory neurons (Shields et al., 2012). Because Nav1.8+ sensory neurons are critical for noxious mechanical and inflammatory pain (Abrahamsen et al., 2008), our behavioral experiments in the ERK dKO mice likely demonstrate the necessity of Nav1.8+ sensory neurons rather than the role of sensory neuron ERK1/2 signaling in pain.
Table 1.
NsERK2 KO versus ERK2f/f (control)a
| ERK2f/f | NsERK2 KO | p | |
|---|---|---|---|
| Total neurons | 9879 ± 724 (4) | 9190 ± 316 (4) | 0.4167 |
| CGRP (%) | 33.11 ± 4.45% (6) | 27.71 ± 3.12% (6) | 0.3442 |
| TrkA (%) | 26.17 ± 2.65% (5) | 23.90 ± 2.71% (6) | 0.5680 |
| IB4+ (%) | 14.89 ± 1.76% (4) | 14.71 ± 1.24% (4) | 0.9384 |
| NF200+ (%) | 31.36 ± 5.58%b (3) | 31.97 ± 3.78% (4) | 0.8131 |
a Neither ERK2 nor ERK1 alone is necessary for survival and normal distribution of DRG neuron subpopulations. Data are DRG neuron distributions in NsERK2 KO compared with the distributions in control littermates. Total neuron data represent total neuron counts in L5 DRGs. For specific markers, the percentage of marker-specific positive neurons is reported as measured in one ganglion from L3-L5 DRGs. Data represent mean ± SEM; values in parentheses are the number of animals for each experiment.
b The same control (ERK2f/f) used for NsERK2 KO and ERK1 KO littermates from ERK dKO colony.
Table 2.
ERK1 KO versus WTa
| WT | ERK1 KO | p | |
|---|---|---|---|
| Total neurons | 7789 ± 979 (6) | 5641 ± 340 (6) | 0.0650 |
| CGRP (%) | 17.08 ± 4.32% (3) | 19.29 ± 3.38% (3) | 0.5130 |
| TrkA (%) | 12.00 ± 1.28% (4) | 14.73 ± 1.30% (4) | 0.1867 |
| IB4+ (%) | 20.73 ± 2.28% (6) | 24.73 ± 2.75% (6) | 0.2884 |
| NF200+ (%) | 31.36 ± 5.58%b (3) | 25.06 ± 2.44% (4) | 0.3034 |
a Neither ERK2 nor ERK1 alone is necessary for survival and normal distribution of DRG neuron subpopulations. Data are DRG neuron distributions in ERK1 KO compared with the distributions in control littermates. Total neuron data represent total neuron counts in L5 DRGs. For specific markers, the percentage of marker-specific positive neurons is reported as measured in one ganglion from L3-L5 DRGs. Data represent mean ± SEM; values in parentheses are the number of animals for each experiment.
b The same control (ERK2f/f) used for NsERK2 KO and ERK1 KO littermates from ERK dKO colony.
Figure 10.

Expression of at least one ERK isoform is necessary for DRG neuron survival. A, Representative images of L5 DRG demonstrate total DRG neurons identified using the pan-neuronal marker, βIII tubulin, in WT (A) and ERK dKO (B) mice. Scale bar, 100 μm. B, Total neuron counts for WT (n = 6) and ERK dKO (n = 6) mice were performed assessing neuronal number in the L5 DRGs using the pan-neuronal marker, βIII tubulin, to identify neurons. C, D, Size distribution data of βIII tubulin+ neurons were acquired for WT (n = 3) and ERK dKO (n = 3) mice using MetaMorph software to manually measure cross-sectional area of neurons in 18-μm-thick sections. Data are graphed both as a total number of neurons per bins (C) and cumulative frequency of neuron sizes (D). Data are mean ± SEM. *p < 0.05 and ***p < 0.001.
Expression of at least one ERK isoform is necessary for survival of various Nav1.8+ neuronal subpopulations
Given the widespread loss of sensory neurons, we further characterized the subpopulations of sensory neurons lost by ERK1/2 deletion. Consistent with central fiber termination, neither ERK1 KO nor NsERK2 KO L4-L5 DRGs displayed changes in the percentages of IB4+, CGRP+, or TrkA+ neurons compared with control littermates (Tables 1 and 2; unpaired t tests). Strikingly, ERK dKO L5 DRGs presented with few, if any, IB4+ nonpeptidergic neurons (Fig. 11A; unpaired t test, ***p < 0.0001). To determine whether nonpeptidergic nociceptors remained, we assessed Ret expression in small- and large-diameter neurons that correspond to nociceptors and low-threshold mechanoreceptors, respectively. ERK dKO L5 DRGs exhibited a profound decrease in Ret+ neurons compared with control littermates (Fig. 11B; unpaired t test, ***p = 0.0003) that was limited to the small-diameter subset (data not shown). Consistent with epidermal innervation, ERK dKO L4 DRGs also exhibited a decrease in CGRP+ pepidergic neurons compared with control littermates (Fig. 11C; unpaired t test, *p = 0.0117). Likewise, ERK dKO L4 DRGs demonstrated a similar reduction in TrkA+ peptidergic neurons compared with control littermates (WT: 675 ± 95.43 TrkA+ neurons and ERK dKO: 406 ± 21.57 TrkA+ neurons; unpaired t test, *p = 0.0333).
Figure 11.
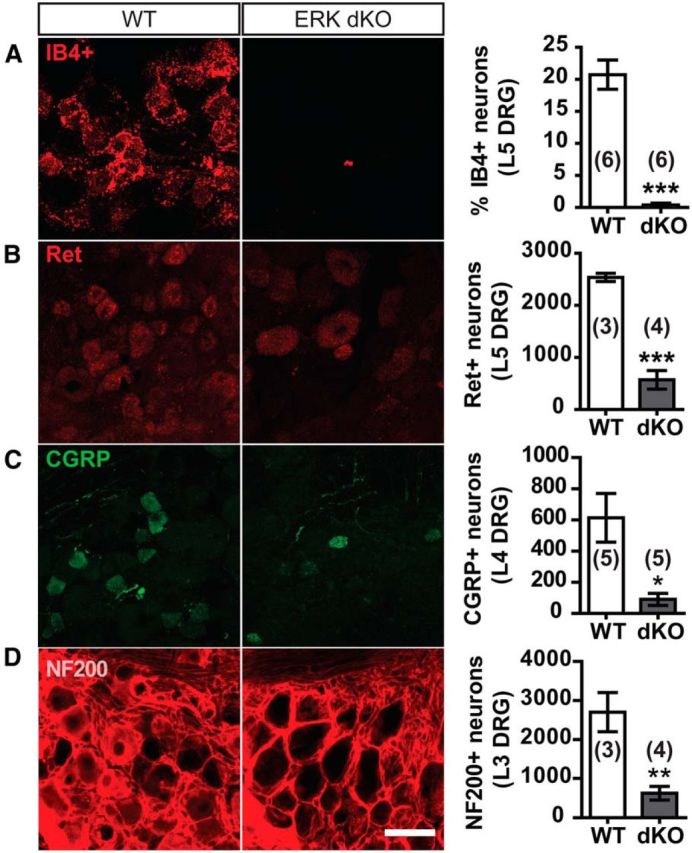
Expression of at least one ERK isoform is necessary for survival of various Nav1.8+ neuronal subpopulations. A, Representative confocal images illustrate IB4+ binding in L5 DRG neurons of WT and ERK dKO mice. Percentage of IB4+ neurons as a function of total βIII tubulin+ neurons were quantified for WT and ERK dKO littermates. ***p < 0.001. B, Representative confocal images illustrate Ret+ immunostaining in L5 DRG neurons of WT and ERK dKO mice. Total Ret+ neurons were quantified for WT and ERK dKO littermates. ***p < 0.001. C, Representative confocal images illustrate CGRP+ immunostaining in L4 DRG neurons of WT and ERK dKO mice. Total CGRP+ neurons were quantified for WT and ERK dKO littermates. *p < 0.05. D, Representative confocal images illustrate NF200+ immunostaining in L3 DRG neurons of WT and ERK dKO mice. Total NF200+ neurons were quantified for WT and ERK dKO littermates. **p < 0.01. Scale bar, 50 μm. Sample size varied from 3 to 6 per group as indicated on the graphs. Data are mean ± SEM.
Our neurochemical analysis shows that expression of at least one ERK isoform is required for maintenance of peptidergic and nonpeptidergic nociceptors. However, size-distribution analysis also suggests loss of neurofilament 200+ (NF200+), large-diameter myelinated Aβ-sensory neurons comprised of low-threshold mechanoreceptors and proprioceptors (Lawson and Waddell, 1991). Both ERK1 KO and NsERK2 KO L3 DRG show normal percentages of NF200+ neurons compared with control littermates (Tables 1 and 2; unpaired t tests). In contrast, ERK dKO mice exhibited an ∼77% decrease in NF200+ neurons compared with control littermates (Fig. 11D; unpaired t test, **p = 0.0068). Overall, these data demonstrate a redundant role for ERK1 and ERK2 in sensory neurons requiring expression of at least one ERK isoform for the survival of both small- and large-diameter sensory neurons.
Discussion
This study is the first to identify functional differences between ERK1 and ERK2 explicitly in sensory neurons. Conditional deletion of ERK2 in sensory neurons attenuated cold sensation and behavioral sensitization in several inflammatory pain models. Characterization of ERK dKO mice revealed a role for ERK1/2 signaling in postnatal survival of DRG neurons. Yet, expression of either isoform alone is sufficient to maintain DRG neuron survival. Thus, ERK1 and ERK2 exhibit both functionally distinct and redundant roles in sensory neurons.
ERK2 regulates peptidergic epidermal innervation, cold adaptation, and inflammatory pain
ERK1/2 signaling plays a critical role in the development of epidermal innervation that begins at ∼E15 and is completed by birth (Coggeshall et al., 1994; Jackman and Fitzgerald, 2000; Newbern et al., 2011). Genetic knock-out studies demonstrated that a conserved Raf/MEK/ERK signaling cascade downstream of NGF/TrkA is critical for establishing epidermal target innervation during embryogenesis (Patel et al., 2000; Zhong et al., 2007; Newbern et al., 2011). Our NsERK2 KO mice exhibit a small decrease in epidermal innervation. Because ERK2 deletion in NsERK2 KO mice is initiated at P0 after innervation is completed (Coggeshall et al., 1994; Jackman and Fitzgerald, 2000; Agarwal et al., 2004), our data show that ERK2 is required for maintenance of epidermal innervation. Because NGF is required throughout adulthood for maintaining epidermal innervation (Bennett et al., 1998), we speculate that ERK2 signals downstream of NGF in a subset of peptidergic neurons to maintain epidermal target innervation throughout adulthood.
Our data suggest that ERK2 functions differently in distinct sensory neuron populations. Peptidergic fibers are required for formalin, NGF, and CFA-induced heat hypersensitivity (Peterson et al., 1997; Chen et al., 2007; McCoy et al., 2013). Thus, the complicated phenotype of NsERK2 KO mice in these three models may reflect the decreased peptidergic innervation instead of a deficit in their sensitization. Indeed, ERK2 may be dispensable for sensitization of peptidergic fibers because CFA-induced heat hypersensitivity is preserved in NsERK2 KO mice. However, our data suggest that ERK2 is required for sensitization in nonpeptidergic neurons. Sensory neuron ERK2 deletion does not affect nonpeptidergic innervation but does decrease CFA-induced mechanical hypersensitivity, which has been shown to require nonpeptidergic neurons (Cavanaugh et al., 2009). Based on our previous study of cold adaptation, the observed impairment in cold sensation in NsERK2 KO mice suggests a deficit in TRPM8-dependent cold adaptation (Brenner et al., 2014a). Interestingly, an upstream kinase, protein kinase C, is required for cold adaptation downstream of TRPM8-dependent Ca2+ influx in vitro (Premkumar et al., 2005; Abe et al., 2006). Because ERK1/2 is activated in TRPM8+ sensory neurons by noxious cold application to the hindpaw (Mizushima et al., 2006), it is possible that ERK2 may act downstream of protein kinase C in an isoform-specific manner to mediate TRPM8-dependent cold adaptation in vivo. Thus, sensory neuron ERK2 may be the predominant mediator of ERK-dependent mechanisms of nociception in both nonpeptidergic and cold-sensitive neurons.
A few possibilities could explain why sensory neuron ERK2 may not drive all inflammation-induced peripheral sensitization, although pharmacological inhibition of ERK1/2 signaling abrogates both NGF-induced mechanical and CFA-induced heat hypersensitivity (Obata et al., 2003; Malik-Hall et al., 2005). Activation of ERK1/2 signaling in non-neuronal cells or in the SCDH neurons may be required to drive some types of inflammatory pain (Obata et al., 2003; Xu et al., 2008). Another possibility is that ERK1 hyperphosphorylation in the DRG compensates for the loss of ERK2. Alternatively, other signaling cascades, including p38, JNK, or PI3 kinase, which are known to be involved in inflammation-induced peripheral sensitization, may compensate for loss of sensory neuron ERK2 (Ji et al., 2002; Gao and Ji, 2008; Melemedjian et al., 2010). Compensation from these signaling cascades could underlie the paradoxical observation of increased spontaneous pain behavior in the first phase of the formalin test.
ERK1 may play an antinociceptive role in NGF-induced heat hypersensitivity. ERK1 KO mice exhibited increased NGF-induced heat hypersensitivity that, unlike NsERK2 KO mice, could not be explained by a change in epidermal innervation. Similar gain-of-function effects due to ERK1 deletion have been observed in vitro (Vantaggiato et al., 2006) and in vivo (Mazzucchelli et al., 2002). In these cases, gain-of-function is associated with ERK2 hyperphosphorylation, raising the possibility that deleting ERK1 may reveal gain-of-function phenotypes via elevated ERK2 activity. Consistent with this hypothesis, we found that ERK1 deletion increases ERK2 phosphorylation (Alter et al., 2010). Alternatively, ERK1 could have antinociceptive effects in primary afferents, or global deletion of ERK1 could affect either CNS neurons or non-neuronal cells to enhance NGF-induced heat hypersensitivity.
Functionally redundant roles for sensory neuron ERK1 and ERK2 in postnatal DRG sensory neuron survival
Sensory neuron-specific ERK1/2 deletion revealed functionally redundant roles for ERK1 and ERK2 in postnatal sensory neuron survival. We hypothesize that neuron loss in ERK dKO mice results from decreased epidermal trophic support. This hypothesis is supported by several observations. ERK1/2 signaling is activated downstream of TrkA and Ret, the receptors for the epidermal-derived trophic factors NGF and the GDNF family ligands, respectively (Airaksinen and Saarma, 2002; Sah et al., 2003). We have previously shown that deletion of Ret in Nav1.8+ neurons results in an ∼30% decrease in the number of DRG neurons by adulthood (Golden et al., 2010). Additionally, target-derived NGF/TrkA signaling is required for DRG neuron survival during early postnatal life (Lewin and Mendell, 1994). Prenatal deletion of ERK1/2 starting at E12.5 results in loss of epidermal innervation at P3 followed by cell loss at P18 (Newbern et al., 2011), consistent with the requirement for target-derived trophic support for sensory neuron survival. Our findings support and extend these results by demonstrating a requirement for expression of at least one ERK isoform for neuron survival after the establishment of epidermal innervation.
Widespread loss of large-diameter, NF200+ neurons in ERK dKO is unexpected because Nav1.8-Cre driven ERK2 deletion should only occur in approximately one-third of these neurons (Agarwal et al., 2004; Shields et al., 2012). In neurons expressing both NF200 and Nav1.8, ERK1 and ERK2 play functionally redundant roles to promote survival; however, the remaining NF200+ neurons may be lost as a result of non–cell-autonomous effects. BDNF is expressed in small-diameter TrkA+ peptidergic neurons and may promote postnatal survival of both small- and large-diameter neurons through paracrine signaling, which would be lost in ERK dKO mice due to loss of peptidergic neurons (Valdés-Sánchez et al., 2010). Another possibility is that, due to global loss of ERK1, Nav1.8-driven Cre expression is altered, leading to changes in its timing and/or its cell type specificity. However, we do not favor this interpretation because ERK1 has not been shown to affect Nav1.8 expression.
With widespread neuron loss, it is interesting that some epidermal innervation remains. The decrease in total IENFD in ERK dKO mice initially appeared to be accounted for solely by loss of peptidergic IENFD. However, the majority of nonpeptidergic neurons are lost in ERK dKO mice. Thus, the cellular origin of the remaining fibers is unclear because nonpeptidergic fibers normally account for the remaining epidermal innervation (Zylka et al., 2005). Previous studies suggest that the amount of available trophic factors in target fields regulates innervation density (Kessler et al., 1983; Zwick et al., 2002; Elitt et al., 2006; Golden et al., 2010). Therefore, other afferents dependent on GDNF family ligands or NGF may provide collaterals or de novo innervation to this hypoinnervated epidermal region in ERK dKO mice.
Functional redundancy model of ERK1 and ERK2
The functional redundancy hypothesis presumes that ERK1 and ERK2 share roles because of their high sequence homology (Boulton and Cobb, 1991; Boulton et al., 1991); however, ERK1 and ERK2 have functionally distinct roles in various cell types (Pagès et al., 1999; Nekrasova et al., 2005; Satoh et al., 2007, 2011a; Newbern et al., 2008; Samuels et al., 2008; Alter et al., 2010; Fyffe-Maricich et al., 2011; Otsubo et al., 2012). This study is the first to demonstrate a distinct functional role for ERK2 in sensory neurons. The molecular basis of this isoform-specific function remains unclear. ERK isoforms could be differentially expressed in sensory neuron subtypes; however, loss of Nav1.8+ neurons only in the ERK dKO and not in single isoform KO mice suggests that both isoforms are expressed in these cells. Alternatively, it is possible that ERK1 and ERK2 are differentially localized in nerve terminals such that ERK2 specifically regulates ERK-dependent changes in membrane excitability (Stamboulian et al., 2010) or local nascent protein translation (Price and Géranton, 2009; Melemedjian et al., 2010). Faster nuclear shuttling of ERK2 could also contribute to the predominant role of ERK2 in peripheral sensitization (Marchi et al., 2008). Last, as previously postulated, isoform-specific roles for ERK1 and ERK2 may not exist because total ERK activity may be paramount (Lefloch et al., 2008). However, loss of both isoforms leads to novel phenotypes in different systems, including in sensory neurons as we have shown, indicating that there are both functionally distinct and fully redundant roles for ERK1 and ERK2 (Newbern et al., 2011; Satoh et al., 2011b; Yasuda et al., 2011; Ishii et al., 2012, 2013).
Footnotes
D.E.O. was supported by National Science Foundation Grant DGE-1143954 and National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant F31NS083207. B.J.A. was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant F30NS061398. This work was supported by National Institutes of Health Grants NS48602, NS042595, and DE022000 to R.W.G. We thank Dr. Rohini Kuner for generously providing SNS-cre mice; members of the R.W.G. laboratory for valuable comments on the manuscript, especially Daniel Brenner and Dr. Vijay Samineni; Dr. Bryan Copits for helpful advice with confocal microscopy; and Dr. Gina Story for assistance with the thermal gradient experiments.
The authors declare no competing financial interests.
References
- Abe J, Hosokawa H, Sawada Y, Matsumura K, Kobayashi S. Ca2+-dependent PKC activation mediates menthol-induced desensitization of transient receptor potential M8. Neurosci Lett. 2006;397:140–144. doi: 10.1016/j.neulet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Alter BJ, Zhao C, Karim F, Landreth GE, Gereau RW., 4th Genetic targeting of ERK1 suggests a predominant role for ERK2 in murine pain models. J Neurosci. 2010;30:11537–11547. doi: 10.1523/JNEUROSCI.6103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Koltzenburg M, Priestley JV, Shelton DL, McMahon SB. Endogenous nerve growth factor regulates the sensitivity of nociceptors in the adult rat. Eur J Neurosci. 1998;10:1282–1291. doi: 10.1046/j.1460-9568.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2:357–371. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-J. [DOI] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, Gereau RW., 4th A novel behavioral assay for measuring cold sensation in mice. PLoS One. 2012;7:e39765. doi: 10.1371/journal.pone.0039765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, Vogt SK, Dhaka A, Story GM, Gereau RW., IV A dynamic set point for thermal adaptation requires phospholipase C-mediated regulation of TRPM8 in vivo. Pain. 2014a;155:2124–2133. doi: 10.1016/j.pain.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DS, Vogt SK, Gereau RW. A technique to measure cold adaptation in freely behaving mice. J Neurosci Methods. 2014b;236C:86–91. doi: 10.1016/j.jneumeth.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain oerception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A. 2009;106:9075–9080. doi: 10.1073/pnas.0901507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, He X, Wang Y, Wen WW, You HJ, Arendt-Nielsen L. Roles of capsaicin-sensitive primary afferents in differential rat models of inflammatory pain: a systematic comparative study in conscious rats. Exp Neurol. 2007;204:244–251. doi: 10.1016/j.expneurol.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Pover CM, Fitzgerald M. Dorsal root ganglion cell death and surviving cell numbers in relation to the development of sensory innervation in the rat hindlimb. Dev Brain Res. 1994;82:193–212. doi: 10.1016/0165-3806(94)90163-5. [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K. Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci. 2002;22:7737–7745. doi: 10.1523/JNEUROSCI.22-17-07737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A. TRPM8 is required for cold sensation in mice. Neuron. 2007;54:371–378. doi: 10.1016/j.neuron.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Elitt CM, McIlwrath SL, Lawson JJ, Malin SA, Molliver DC, Cornuet PK, Koerber HR, Davis BM, Albers KM. Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. J Neurosci. 2006;26:8578–8587. doi: 10.1523/JNEUROSCI.2185-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci. 2011;31:843–850. doi: 10.1523/JNEUROSCI.3239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett. 2008;437:180–183. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden JP, Hoshi M, Nassar MA, Enomoto H, Wood JN, Milbrandt J, Gereau RW, 4th, Johnson EM, Jr, Jain S. RET signaling is required for survival and normal function of nonpeptidergic nociceptors. J Neurosci. 2010;30:3983–3994. doi: 10.1523/JNEUROSCI.5930-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci. 2012;32:8855–8864. doi: 10.1523/JNEUROSCI.0137-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Bansal R. Sustained activation of ERK1/2 MAPK in oligodendrocytes and Schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J Neurosci. 2013;33:175–186. doi: 10.1523/JNEUROSCI.4403-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman A, Fitzgerald M. Development of peripheral hindlimb and central spinal cord innervation by subpopulations of dorsal root ganglion cells in the embryonic rat. J Comp Neurol. 2000;418:281–298. doi: 10.1002/(SICI)1096-9861(20000313)418:3<281::AID-CNE4>3.0.CO%3B2-9. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW., 4th Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein kinase kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JA, Bell WO, Black IB. Interactions between the sympathetic and sensory innervation of the iris. J Neurosci. 1983;3:1301–1307. doi: 10.1523/JNEUROSCI.03-06-01301.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, Waddell PJ. Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J Physiol. 1991;435:41–63. doi: 10.1113/jphysiol.1991.sp018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28:511–527. doi: 10.1128/MCB.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Regulation of cutaneous C-fiber heat nociceptors by nerve growth factor in the developing rat. J Neurophysiol. 1994;71:941–949. doi: 10.1152/jn.1994.71.3.941. [DOI] [PubMed] [Google Scholar]
- Malik-Hall M, Dina OA, Levine JD. Primary afferent nociceptor mechanisms mediating NGF-induced mechanical hyperalgesia. Eur J Neurosci. 2005;21:3387–3394. doi: 10.1111/j.1460-9568.2005.04173.x. [DOI] [PubMed] [Google Scholar]
- Marchi M, D'Antoni A, Formentini I, Parra R, Brambilla R, Ratto GM, Costa M. The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One. 2008;3:e3873. doi: 10.1371/journal.pone.0003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knock-out of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/S0896-6273(02)00716-X. [DOI] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ. Peptidergic CGRPα primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron. 2013;78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30:15113–15123. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima T, Obata K, Katsura H, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Mashimo T, Noguchi K. Noxious cold stimulation induces mitogen-activated protein kinase activation in transient receptor potential (TRP) channels TRPA1- and TRPM8-containing small sensory neurons. Neuroscience. 2006;140:1337–1348. doi: 10.1016/j.neuroscience.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Nekrasova T, Shive C, Gao Y, Kawamura K, Guardia R, Landreth G, Forsthuber TG. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J Immunol. 2005;175:2374–2380. doi: 10.4049/jimmunol.175.4.2374. [DOI] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD. Specific functions for ERK/MAPK signaling during PNS development. Neuron. 2011;69:91–105. doi: 10.1016/j.neuron.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O'Loughlin B, Wikenheiser J, Gargesha M, Doughman YQ, Charron J, Ginty DD, Watanabe M, Saitta SC, Snider WD, Landreth GE. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci U S A. 2008;105:17115–17120. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Activation of extracellular signal-regulated protein kinase in the dorsal root ganglion following inflammation near the nerve cell body. Neuroscience. 2004;126:1011–1021. doi: 10.1016/j.neuroscience.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Porreca F. Animal models of experimental neuropathic pain. In: McMahon SB, Koltzenburg M, Tracey I, Turk DC, editors. Wall and Melzack's textbook of pain. Ed 6. Philadelphia: Elsevier; 2013. pp. 889–900. [Google Scholar]
- Otsubo Y, Satoh Y, Kodama M, Araki Y, Satomoto M, Sakamoto E, Pagès G, Pouysségur J, Endo S, Kazama T. Mechanical allodynia but not thermal hyperalgesia is impaired in mice deficient for ERK2 in the central nervous system. Pain. 2012;153:2241–2252. doi: 10.1016/j.pain.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. doi: 10.1016/S0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Peterson MA, Basbaum AI, Abbadie C, Rohde DS, McKay WR, Taylor BK. The differential contribution of capsaicin-sensitive afferents to behavioral and cardiovascular measures of brief and persistent nociception and to Fos expression in the formalin test. Brain Res. 1997;755:9–16. doi: 10.1016/S0006-8993(97)00068-1. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Raisinghani M, Pingle SC, Long C, Pimentel F. Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J Neurosci. 2005;25:11322–11329. doi: 10.1523/JNEUROSCI.3006-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Géranton SM. Translating nociceptor sensitivity: the role of axonal protein synthesis in nociceptor physiology. Eur J Neurosci. 2009;29:2253–2263. doi: 10.1111/j.1460-9568.2009.06786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1996;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4:964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah DW, Ossipo MH, Porreca F. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat Rev Drug Discov. 2003;2:460–472. doi: 10.1038/nrd1107. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory: ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Nakata T, Kobayashi Y, Yamada K, Ikeda T, Takeuchi A, Hiramoto T, Watanabe Y, Kazama T. ERK2 contributes to the control of social behaviors in mice. J Neurosci. 2011a;31:11953–11967. doi: 10.1523/JNEUROSCI.2349-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Kobayashi Y, Takeuchi A, Pagès G, Pouysségur J, Kazama T. Deletion of ERK1 and ERK2 in the CNS causes cortical abnormalities and neonatal lethality: Erk1 deficiency enhances the impairment of neurogenesis in Erk2-deficient mice. J Neurosci. 2011b;31:1149–1155. doi: 10.1523/JNEUROSCI.2243-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153:2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjølsen A, Berge OG, Hundskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Valdés-Sánchez T, Kirstein M, Pérez-Villalba A, Vega JA, Fariñas I. BDNF is essentially required for the early postnatal survival of nociceptors. Dev Biol. 2010;339:465–476. doi: 10.1016/j.ydbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:1–15. doi: 10.1186/jbiol33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund's adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–14096. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Kometani K, Takahashi N, Imai Y, Aiba Y, Kurosaki T. ERKs induce expression of the transcriptional repressor Blimp-1 and subsequent plasma cell differentiation. Sci Signal. 2011;4:ra25. doi: 10.1126/scisignal.2001592. [DOI] [PubMed] [Google Scholar]
- Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat Neurosci. 2007;10:598–607. doi: 10.1038/nn1898. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick M, Davis BM, Woodbury CJ, Burkett JN, Koerber HR, Simpson JF, Albers KM. Glial cell line-derived neurotrophic factor is a survival factor for isolectin B4-positive, but not vanilloid receptor 1-positive, neurons in the mouse. J Neurosci. 2002;22:4057–4065. doi: 10.1523/JNEUROSCI.22-10-04057.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25. doi: 10.1016/j.neuron.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, Vihko P. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60:111–122. doi: 10.1016/j.neuron.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]