

Abstract
Colorectal cancer (CRC) is a heterogeneous disease with genetic profiles and clinical outcomes dependent on the anatomic location of the primary tumor. How location impacts the molecular makeup of a tumor and how prognostic and predictive biomarkers differ between proximal versus distal colon cancers is not well established. We investigated the associations between tumor location, KRAS and BRAF mutation status, and the mRNA expression of proteins involved in major signaling pathways, including tumor growth (EGFR), angiogenesis (VEGFR2), DNA repair (ERCC1) and fluoropyrimidine metabolism (TS). FFPE tumor specimens from 431 advanced CRC patients were analyzed. The presence of 7 different KRAS base substitutions and the BRAF V600E mutation was determined. ERCC1, TS, EGFR and VEGFR2 mRNA expression levels were detected by RT-PCR. BRAF mutations were significantly more common in the proximal colon (p<0.001), whereas KRAS mutations occurred at similar frequencies throughout the colorectum. Rectal cancers had significantly higher ERCC1 and VEGFR2 mRNA levels compared to distal and proximal colon tumors (p=0.001), and increased TS levels compared to distal colon cancers (p=0.02). Mutant KRAS status was associated with lower ERCC1, TS, EGFR, and VEGFR2 gene expression in multivariate analysis. In a subgroup analysis, this association remained significant for all genes in the proximal colon and for VEGFR2 expression in rectal cancers. The mRNA expression patterns of predictive and prognostic biomarkers as well as associations with KRAS and BRAF mutation status depend on primary tumor location. Prospective studies are warranted to confirm these findings and determine the underlying mechanisms.
Keywords: BRAF, colorectal cancer, KRAS, predictive biomarkers, tumor location
INTRODUCTION
The translation of clinically relevant biomarkers into personalized medicine for colorectal cancer (CRC) patients has proven a challenging endeavor. For instance, while RAS mutant status predicts lack of response towards epidermal growth factor receptor (EGFR) directed antibodies[1–4], many patients with RAS wild-type tumors do not benefit from such therapy[5–7]. Similarly, the predictive utility of EGFR[3, 8–10] expression and the V600E BRAF mutation[11, 12] has been limited, and studies evaluating the predictive value of VEGFR2[13, 14] expression for vascular endothelial growth factor (VEGF) targeted drugs have yielded inconsistent results. Likewise, molecular determinants of response towards cytotoxic agents, including fluoropyrimidines (i.e. thymidylate synthase [TS]) and platinum agents (i.e. excision repair cross complement group 1 [ERCC1]), have been retrospectively associated with response rates and survival[15–18] but are not yet prospectively validated. Limitations impeding biomarker development include methodological differences across studies, redundancy in signaling as well as tumor heterogeneity.
Recent data suggests that the location of a colorectal tumor (i.e. proximal vs. distal colon vs. rectum) may impact its molecular landscape[19, 20] and clinical behavior[21, 22]. Microarray DNA analyses have revealed over 1,000 genes with different expression patterns between ascending and descending colon cancers[23], which partly reflect the distinct embryonic origin (i.e. midgut vs. hindgut) and vascular supply (i.e. superior vs. inferior mesenteric artery) of the proximal and distal colon[24]. Phenotypically, proximal tumors are prone to microsatellite instability[25, 26], BRAF mutations[27, 28] and poorly differentiated histology[29, 30], whereas distal tumors are characterized by loss of heterozygosity and TP53 mutations[29, 30]. Clinically, proximal tumors tend to present at later stages[31] and are associated with worse overall survival[32] relative to their distal counterparts.
Though the presence of anatomic based CRC gene signatures has been established, associations between predictive and prognostic biomarker expression and tumor location are not well understood. Such knowledge may shed insight on interactions linking tumor location and treatment response and outcomes which may guide personalized therapy in the future. On this premise, we used a commercially available database to determine the relationship between primary tumor site and the expression of biomarkers involved in major signaling pathways in advanced CRC patients. Specifically, we examined the associations between tumor location and gene expression levels of proteins involved in tumor growth (EGFR), angiogenesis (VEGFR2), DNA repair (ERCC1) and chemotherapy drug metabolism (TS) as well as KRAS and BRAF mutation status.
MATERIALS AND METHODS
Study Design and Patient Population
We conducted a retrospective analysis of data collected from a cohort of 578 patients with stage IV colorectal cancer, whose tumor tissue was submitted to Response Genetics Incorporated (Los Angeles, CA), a CLIA certified and CAP accredited laboratory, for comprehensive molecular testing (ColonDX™). Patient samples were submitted from both private and academic healthcare institutions across the United States between 2007 and 2010. Formalin-fixed paraffin embedded (FFPE) tumor specimens were tested for KRAS and BRAF mutation status, as well as mRNA expression levels of ERCC1, TS, EGFR and VEGFR2. Only patients whose specimens had sufficient tissue for analysis of at least one gene of interest (i.e. ERCC1, TS, EGFR, VEGFR2) and detection of either KRAS and/or BRAF mutations, as well as data regarding patient and tumor characteristics were included in this study. Tumor samples from metastatic sites, in which the primary tumor location was unknown, were excluded. A total of 431 patients were included in the final analysis.
Information regarding primary tumor location, patient age and gender, tumor grade and histology, were extracted from pathology reports submitted with the tissue specimens and recorded by two of the authors (M. K. M., D. L. H.). Specifically, the splenic flexure was used to distinguish proximal from distal tumors. Tumors within 15 cm of the anal verge were designated as originating in the rectum.
Tumor Tissue Preparation and Gene Expression Analysis
Tumor tissue from study patients was obtained at the time of diagnosis prior to surgery and at the time of surgical resection. Hematoxylin and eosin (H&E) stained sections of all FFPE specimens were evaluated by a board certified pathologist for tumor content.
Formalin-fixed paraffin-embedded tissues were dissected. Ten-micrometer-thick slides were obtained from the identified areas with the highest tumor concentration and were mounted on uncoated glass slides. For histologic diagnosis, three sections representative of the beginning, middle, and end of the tissue were stained with H&E, using the standard method. Before microdissection, sections were de-paraffinized in xylene for 10 minutes, hydrated with 100%, 95%, and 70% ethanol, and then washed in H2O for 30 seconds. Following microdissection of tumor cells, the sections were stained with nuclear fast red (American Master Tech Scientific, Inc.) for 20 seconds and rinsed in water for 30 seconds. Samples were then dehydrated with 70%, 95%, and 100% ethanol for 30 seconds each, followed by xylene for 10 min. The slides were then completely air-dried. Laser capture microdissection (PALM Microlaser Technologies AG) was carried out in all tumor samples to ensure that only tumor cells were dissected[33]. The dissected particles of tissue were transferred to a reaction tube containing 400 mL of RNA buffer for lysis of tumor cells.
After lysis of the tumor cells, RNA and DNA were isolated separately from the specimen. RNA isolation from paraffin-embedded samples was done according to a proprietary procedure defined by Response Genetics, Inc. (US Patent #6248535). The RNA was then reverse-transcribed to cDNA as described previously[34]. DNA was either directly extracted or back extracted from the organic phase, both with an RGI patented method (US Patent #6248535).
Quantitation of gene mRNA expression levels of ERCC1, TS, EGFR, VEGFR2, and an internal reference (β-actin) cDNA was done using a fluorescence-based real-time detection method [ABI PRISM 7900 Sequence detection System (TaqMan); Perkin-Elmer Applied Biosystem] as previously described(43)[35]. Isolated RNA was reverse-transcribed to cDNA, followed by RT-PCR using specific primers and probes. The PCR reaction mixture consisted of 1,200 nmol/L of each primer, a 200 nmol/L probe, 0.4 U of AmpliTaq Gold Polymerase, 200 nmol/L of dATP, dCTP, dGTP, dTTP; 3.5 mmol/L MgCl2, and 1X TaqMan Buffer A containing a reference dye added to a final volume of 20 mL (all reagents from PE Applied Biosystems). Cycling conditions were 50°C for 2 minutes, 95°C for 10 minutes, followed by 46 cycles at 95°C for 15 seconds and 60°C for 1 minute.
The ERCC1 primers and probe sequences used were as follows: forward primer, GGGAATTTGGCGACGTAATTC; reverse primer, GCGGAGGCTGAGGAACAG; probe, 6FAM-CACAGGTGCTCTGGCCCAGCACATA. The TS primers and probe sequences used were as follows: forward primer, GCCTCGGTGTGCCTTTCA; reverse primer, CCCGTGATGTGCGCAAT; probe, 6FAM-TCGCCAGCTACGCCCTGCTCA. The EGFR primers and probe sequences used were as follows: forward primer, TGCGTCTCTTGCCGGAAT; reverse primer GGCTCACCCTCCAGAAGGTT; probe, 6FAM-ACGCATTCCCTGCCTCGGCTG. The VEGFR2 primers and probe sequences used were as follows: forward primer, CCTGTGG CTCTGCGTGGA; reverse primer, CTGAGCCTGGGCAGAT CAAG; probe, 6FAM-CACTAGGCAAACCCACAGAGGCGGC. The β-actin primers and probe sequences used were as follows: forward primer, GAGCGCGGCTACAGCTT; reverse primer, TCCTTAATGTCACGCACGATTT; probe, 6FAM-ACCACCACGGCCGAGCGG.
For each sample, parallel TaqMan PCR reactions were carried out for each gene of interest and the β-actin reference gene to normalize for input cDNA. Results were obtained as a ratio of the PCR fluorescent signals of each gene of interest relative to the reference gene, β-actin.
KRAS mutation analysis was performed with a Response Genetics Inc. (RGI) designed mutation assay by RT-PCR using specifically designed primers and probes to detect each of the following mutations: Gly12Ala (GGT>GCT) 522; Gly12Asp (GGT>GAT) 521; Gly12Arg (GGT>CGT) 518; Gly12Cys (GGT>TGT) 516; Gly12Ser (GGT>AGT) 517; Gly12Val (GGT>GTT) 520; Gly13Asp (GGC>GAC) 532. After RT-PCR, all collected data was analyzed through an RGI Excel™ template and according to delta-CT values the mutation status was established. BRAF V600E mutations were detected by dye terminator sequencing.
Statistical Analysis
Messenger RNA expression levels of ERCC1, TS, EGFR, and VEGFR2 were summarized and analyzed by Wilcoxon two-sample tests to detect differences based on KRAS and BRAF mutation status within each tumor site. Pairwise differences between the expression of the four examined genes across tumor sites, independent of KRAS and BRAF mutation status, were then determined by Wilcoxon two-sample tests, with significance determined by Kruskal-Wallis testing. Bonferroni method was used to correct p value for multiple comparisons. All values were reported as medians and ranges, with a significance p-value cutoff ≤ 0.05. Analyses were performed using Statistical Analysis Software (SAS) version 9.3 (SAS Institute Inc. NC, USA).
RESULTS
Patient and Tumor Characteristics
Among 431 patients with advanced CRC, 58% were men and 42% were women, with a median age of 61 years (range 27–92 years old) (Table 1). The distribution of tumor site was as follows: 39% distal, 40% proximal, and 21% rectal. The frequency of KRAS and BRAF mutations amongst all patients was 42% and 8%, respectively. BRAF mutations were significantly more common in the proximal colon (14%), followed by distal colon (5%) and rectal tumors (2%) (p<0.001), whereas KRAS mutations occurred at similar frequencies throughout the colorectum (p=0.51) (Table 1).
Table 1.
Colorectal Cancer Patient and Tumor Characteristics (N = 431)
| Primary Tumor Location* | ||||
|---|---|---|---|---|
| Distal Colon | Proximal Colon | Rectum | ||
| Characteristic | (N=170) No. (%) |
(N=171) No. (%) |
(N=90) No. (%) |
P-value† |
| Age in Years median (range) | 59 (27–92) | 66 (29–89) | 59 (31–85) | <0.001 |
| Gender | 0.09 | |||
| Men | 97 (57%) | 92 (54%) | 61 (68%) | |
| Women | 73 (43%) | 79 (46%) | 29 (32%) | |
| Tumor Grade | 0.04 | |||
| Well Differentiated | 7 (4%) | 10 (6%) | 8 (9%) | |
| Moderately Differentiated | 106 (62%) | 88 (51%) | 52 (58%) | |
| Poorly Differentiated | 31 (18%) | 48 (28%) | 14 (16%) | |
| Unknown | 26 (15%) | 25 (15%) | 16 (18%) | |
| KRAS Mutation Status | 0.51 | |||
| Mutant | 69 (41%) | 78 (46%) | 33 (37%) | |
| Wildtype | 91 (54%) | 83 (49%) | 46 (51%) | |
| Unknown | 10 (6%) | 10 (6%) | 11 (12%) | |
| KRAS Mutation | 0.012 | |||
| Gly12Ala | 8 (12%) | 1 (1%) | 2 (6%) | |
| Gly12Asp | 20 (29%) | 31 (40%) | 9 (27%) | |
| Gly12Arg | 2 (3%) | 0 (0%) | 1 (3%) | |
| Gly12Cys | 9 (13%) | 5 (6%) | 3 (9%) | |
| Gly12Ser | 6 (9%) | 5 (6%) | 3 (9%) | |
| Gly12Val | 20 (29%) | 15 (19%) | 5 (15%) | |
| Gly13Asp | 4 (6%) | 21 (27%) | 10 (30%) | |
| BRAF Mutation Status | <0.001 | |||
| Mutant | 9 (5%) | 24 (14%) | 2 (2%) | |
| Wildtype | 147 (86%) | 133 (78%) | 81 (90%) | |
| Unknown | 14 (8%) | 14 (8%) | 7 (8%) | |
Proximal and distal colon tumors were demarcated by the splenic flexure. Rectal tumors were defined as those within 15 cm of the anal verge.
P value was based on Kruskal-Wallis Test for age and χ2 test for other characteristics. Patients with unknown characteristics were excluded.
ERCC1 Expression by Tumor Location and KRAS, BRAF Mutation Status
Rectal cancers demonstrated higher ERCC1 expression (median 1.11 [0.23–4.23]) than either distal (median 0.83 [0.17–6.66]) or proximal colon tumors (median 0.91 [0.26–3.06]) (p=0.001) (Figure 1A, Table 2).
Figure 1.
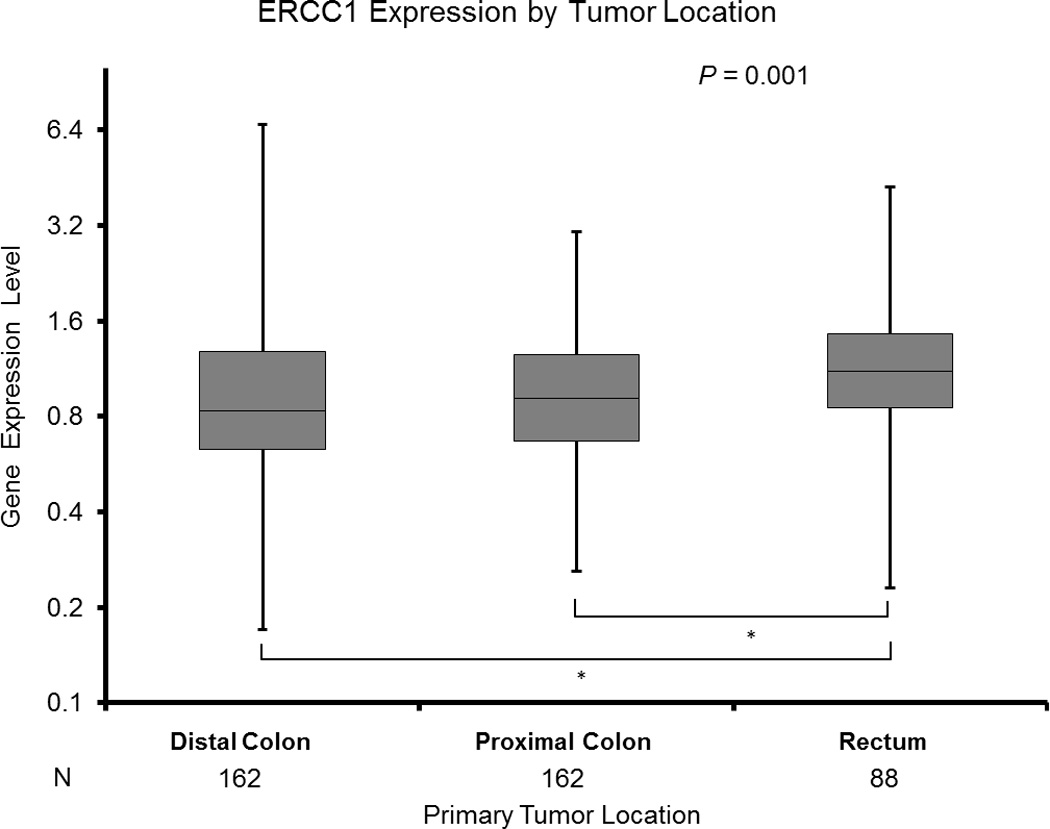
A: ERCC1 Expression by Tumor Location
* Based on Wilcoxon two - sample test for pairwise differences by two tumor sites adjusting for multiple comparisons.
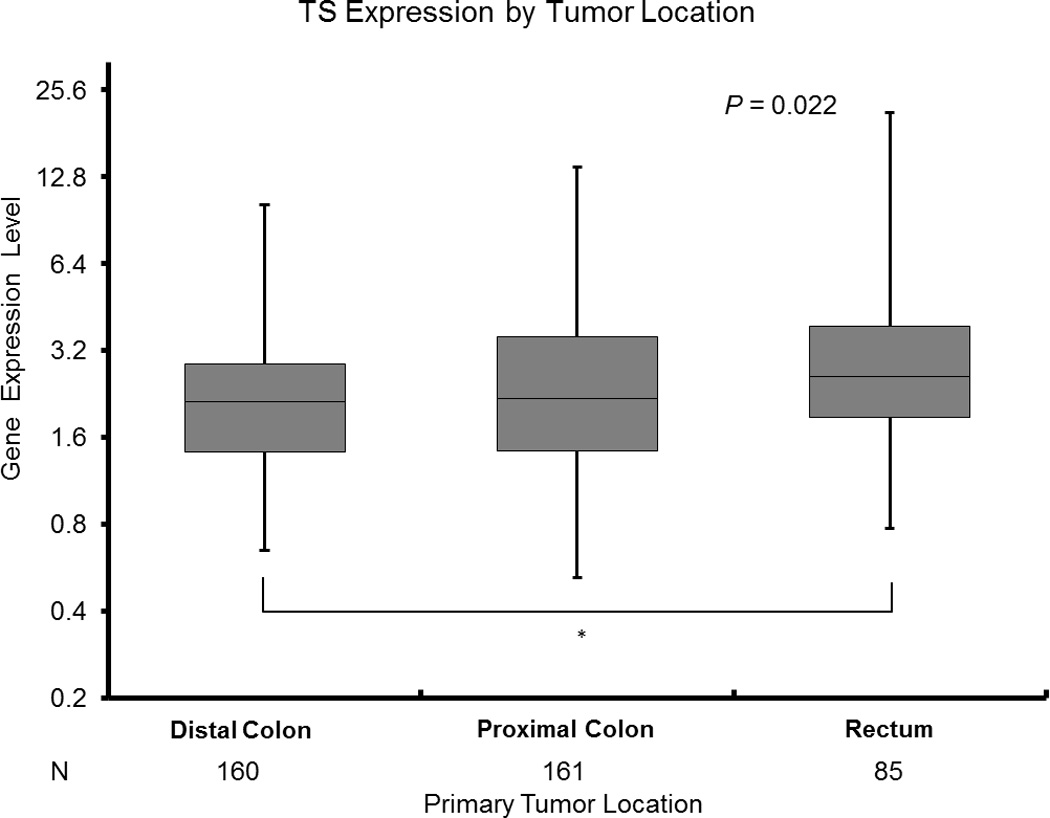
B: TS Expression by Tumor Location
* Based on Wilcoxon two - sample test for pairwise differences by two tumor sites adjusting for multiple comparisons.
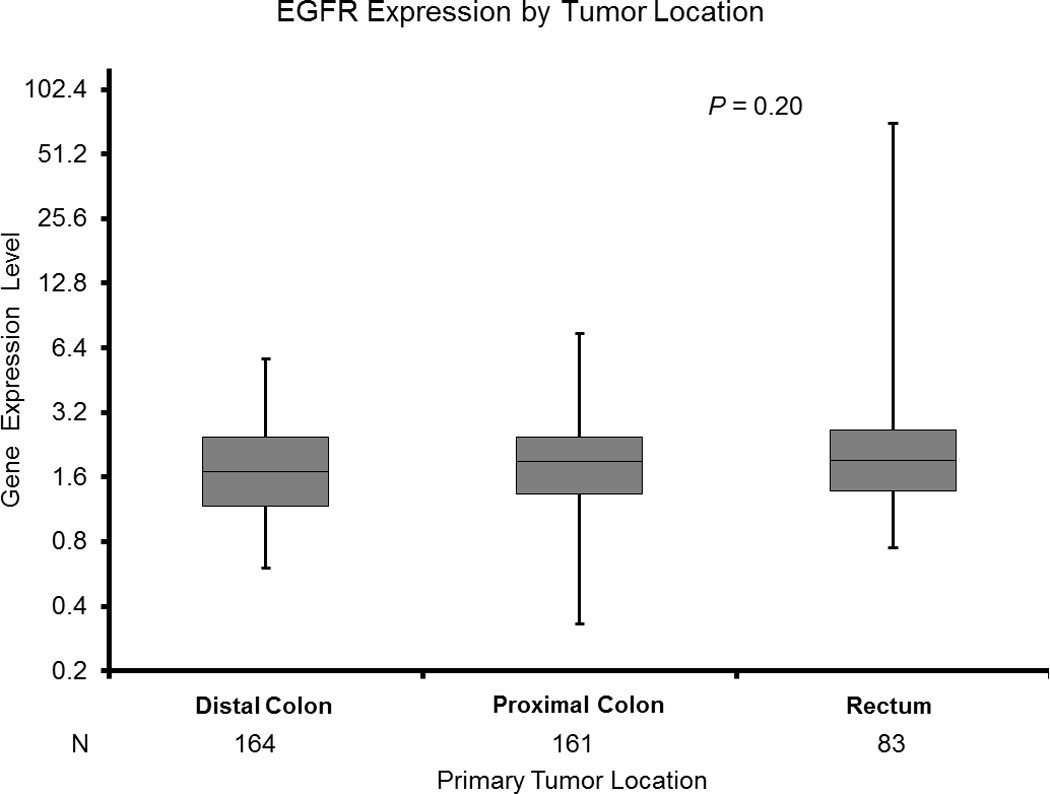
C: EGFR Expression by Tumor Location
* Based on Wilcoxon two - sample test for pairwise differences by two tumor sites adjusting for multiple comparisons.
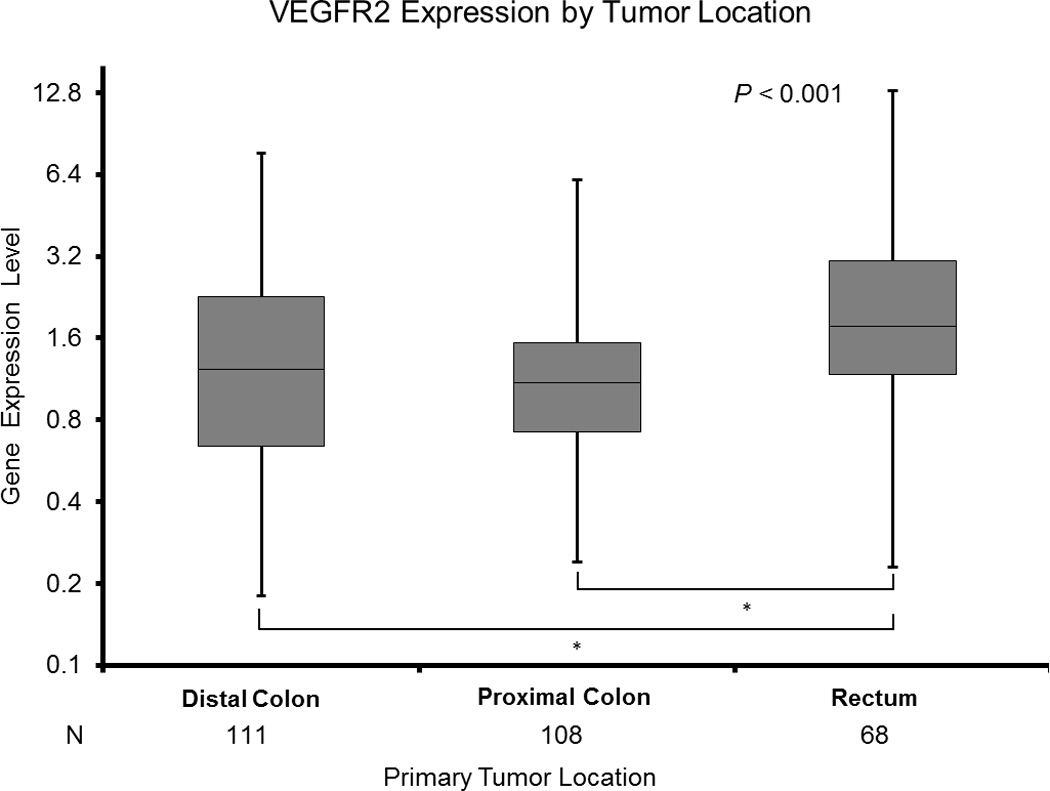
D: VEGFR2 Expression by Tumor Location
* Based on Wilcoxon two - sample test for pairwise differences by two tumor sites adjusting for multiple comparisons.
Table 2.
ERCC1, TS, EGFR, VEGFR2 Gene Expression Levels by Tumor Site and KRAS Mutation Status
| Primary Tumor Location | |||||||
|---|---|---|---|---|---|---|---|
| Distal Colon | Proximal Colon | Rectum | |||||
| Gene | KRAS Mutation Status |
N | Median (range) | N | Median (range) | N | Median (range) |
| ERCC1 | Mutant | 67 | 0.79 (0.27 – 2.96) | 77 | 0.82 (0.26 – 2.95) | 32 | 0.96 (0.23 – 2.60) |
| Wildtype | 86 | 0.84 (0.17 – 6.66) | 79 | 0.97 (0.28 – 3.06) | 46 | 1.25 (0.37 – 4.23) | |
| P value* | 0.80 | 0.005 | 0.052 | ||||
| All | 162 | 0.83 (0.17 – 6.66) | 162 | 0.91 (0.26 – 3.06) | 88 | 1.11 (0.23 – 4.23) | |
| Pairwise‡ | × | √ | ×, √ | ||||
| P value† | 0.001 | ||||||
| TS | Mutant | 67 | 1.92 (0.81 – 9.26) | 76 | 1.90 (0.52 – 9.43) | 31 | 2.69 (0.77 – 21.43) |
| Wildtype | 84 | 2.25 (0.65 – 10.02) | 79 | 2.38 (0.65 – 13.81) | 46 | 2.66 (0.97 – 18.99) | |
| P value* | 0.31 | 0.03 | 0.80 | ||||
| All | 160 | 2.12 (0.65 – 10.26) | 161 | 2.18 (0.52 – 13.81) | 85 | 2.60 (0.77 – 21.43) | |
| Pairwise‡ | × | × | |||||
| P value† | 0.02 | ||||||
| EGFR | Mutant | 67 | 1.60 (0.79 – 5.24) | 76 | 1.60 (0.33 – 7.17) | 32 | 1.66 (0.78 – 4.08) |
| Wildtype | 88 | 1.78 (0.63 – 5.68) | 76 | 2.12 (0.63 – 7.45) | 44 | 2.20 (0.80 – 71.28) | |
| P value* | 0.11 | <0.001 | 0.11 | ||||
| All | 164 | 1.70 (0.60 – 5.68) | 161 | 1.88 (0.33 – 7.45) | 83 | 1.92 (0.75 – 71.28) | |
| Pairwise‡ | |||||||
| P value† | 0.20 | ||||||
| VEGFR2 | Mutant | 42 | 1.32 (0.25 – 7.66) | 50 | 0.91 (0.24 – 6.10) | 25 | 1.53 (0.23 – 3.62) |
| Wildtype | 66 | 1.21 (0.18 – 6.64) | 54 | 1.29 (0.33 – 5.94) | 38 | 2.21 (0.37 – 13.00) | |
| P value* | 0.58 | 0.01 | 0.02 | ||||
| All | 111 | 1.23 (0.18 – 7.66) | 108 | 1.10 (0.24 – 6.10) | 68 | 1.77 (0.23 – 13.00) | |
| Pairwise‡ | × | √ | ×, √ | ||||
| P value† | <0.001 | ||||||
Based on Wilcoxon two – sample test for differences by KRAS mutation status in each tumor site.
Based on Kruskal-Wallis test for differences across tumor sites.
Based on Wilcoxon two – sample test for pairwise differences by two tumor sites adjusting for multiple comparisons; includes patients with unknown KRAS mutation status. The same symbol represents a significant pairwise difference.
Among all patients, KRAS mutant status was associated with decreased ERCC1 expression (median 0.87 [0.23–2.96]) compared to KRAS wild-type status (median 0.97 [0.17–6.66]) (p=0.01) (Table 3). In subgroup analysis, this trend remained significant in proximal tumors (KRAS mutant median 0.82 [0.26–2.95]; KRAS wildtype median 0.98 [0.28–3.06]; p=0.005) (Table 2). A similar trend was seen in rectal tumors but did not reach statistical significance (KRAS mutant median 0.96 [0.23–2.60]; KRAS wildtype median 1.25 [0.37–4.23]; p=0.052) (Table 2).
Table 3.
ERCC1, TS, EGFR, VEGFR2 Gene Expression Levels by KRAS and BRAF Mutation Status
| KRAS Mutation Status | BRAF Mutation Status | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutant | Wildtype | Mutant | Wildtype | |||||||
| Gene | N | Median (range) | N | Median (range) | N | Median (range) | N | Median (range) | ||
| ERCC1 | 176 | 0.87 (0.23–2.96) | 211 | 0.98 (0.17–6.66) | 35 | 0.94 (0.28–2.47) | 356 | 0.91 (0.20–6.66) | ||
| P value* | 0.01 | 0.35 | ||||||||
| TS | 174 | 2.08 (0.52–21.43) | 209 | 2.38 (0.65–18.99) | 34 | 3.38 (1.01–13.81) | 352 | 2.17 (0.52–21.43) | ||
| P value* | 0.03 | <0.001 | ||||||||
| EGFR | 175 | 1.61 (0.33–7.17) | 208 | 2.06 (0.63–71.28) | 33 | 2.20(1.12–3.50) | 349 | 1.75 (0.33–71.28) | ||
| P value* | <0.001 | 0.002 | ||||||||
| VEGFR2 | 117 | 1.19 (0.23–7.66) | 158 | 1.35 (0.18–13.00) | 26 | 1.48 (0.29–3.41) | 252 | 1.25 (0.18–13.0) | ||
| P value* | 0.003 | 0.64 | ||||||||
Based on Kruskal-Wallis test.
There was no significant association between BRAF mutation status and ERCC1 expression (Table 3). In the subgroup of proximal tumors (n=156), there was a non-significant trend towards increased ERCC1 expression in BRAF mutant tumors (median 0.96 [0.28–2.47]) compared to BRAF wildtype tumors (median 0.88 [0.26–3.06]) (p=0.06).
TS Expression by Tumor Location and KRAS, BRAF Mutation Status
Rectal tumors had significantly higher TS expression levels (median 2.60 [0.77–21.43]) than distal colon cancers (median 2.12 [0.65–10.26]) (p=0.02) (Figure 1B, Table 2). There were no other differences detected in TS expression between tumor sites.
Patients with KRAS mutant tumors had significantly lower TS mRNA levels (median 2.08 [0.52–21.43]) than those with KRAS wild-type cancers (median 2.38 [0.65–18.99]) (p=0.03) (Table 3). When stratified by tumor location, this association remained significant only in proximal colon tumors (KRAS mutant median 1.90 [0.52–9.43]; KRAS wildtype median 2.38 [0.65–13.81]; p=0.03) (Table 2).
The BRAF V600E mutation was predictive of increased TS levels (BRAF mutant median 3.38 [1.01–13.81]; BRAF wildtype median 2.17 [0.52–21.43]; p<0.001) among all patients (Table 3). This association remained significant in proximal colon tumors (BRAF mutant median 3.68 [1.01–13.81]; BRAF wildtype median 2.07 [0.52–11.10]; p=0.003) (Figure 2A).
Figure 2.
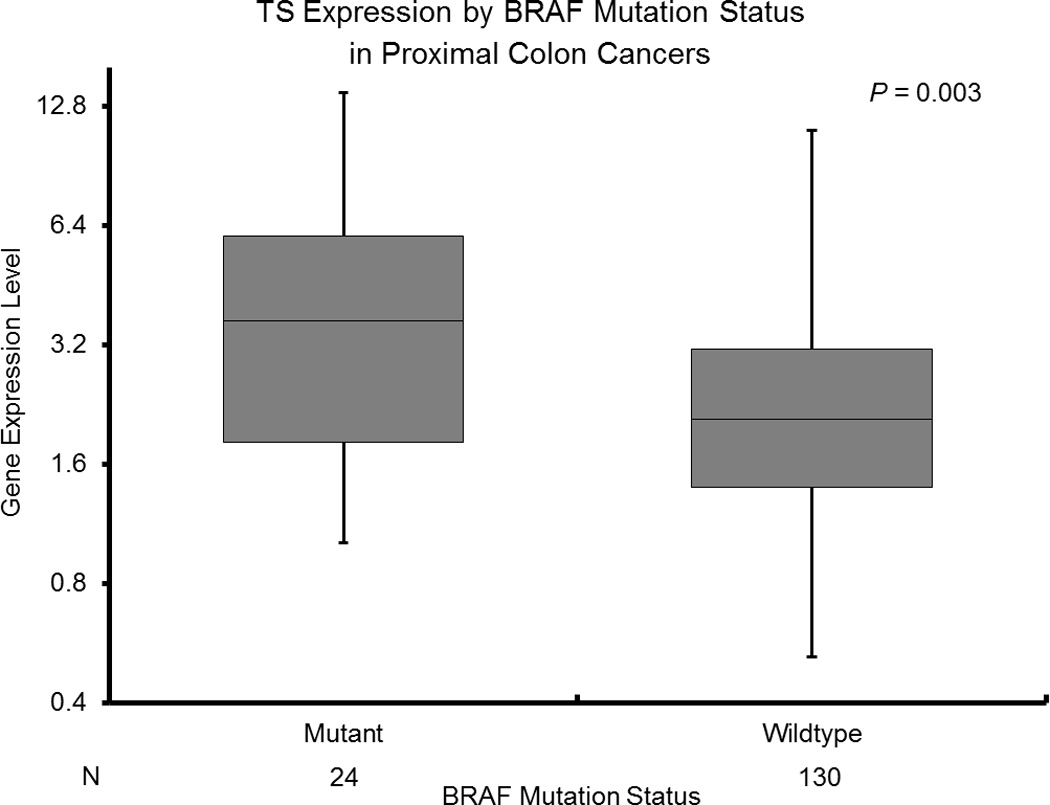
A: TS Expression by BRAF Mutation Status in Proximal Colon Cancers
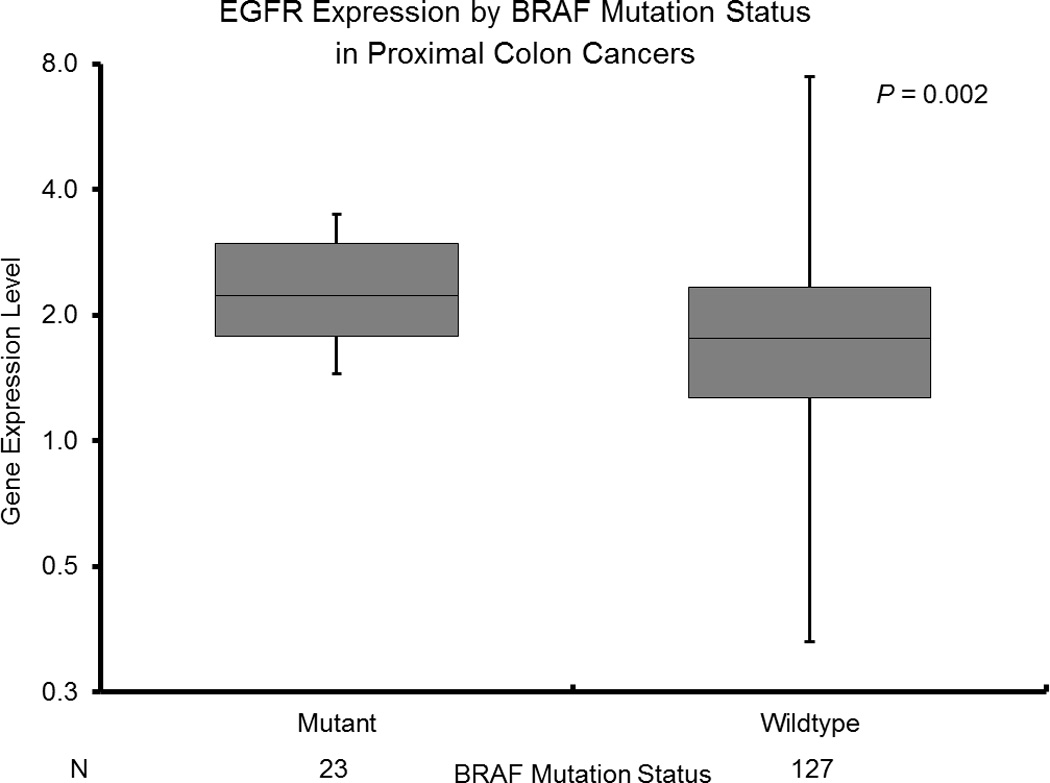
B: EGFR Expression by BRAF Mutation Status in Proximal Colon Cancers
EGFR Expression by Tumor Location and KRAS, BRAF Mutation Status
EGFR expression was similar across all tumor sites (p=0.20; Figure 1C, Table 2). However, EGFR expression was decreased in KRAS mutated cancers (median 1.61 [0.33–7.17]) compared to KRAS wildtype cancers (median 2.06 [0.63–71.28]) (p<0.001) among all patients (Table 3). In subgroup analysis, this difference remained significant in proximal colon cancers (KRAS mutant median 1.60 [0.33–7.17]; KRAS wildtype median 2.12 [0.63–7.45]; p<0.001) (Table 2).
BRAF mutant status was associated with increased EGFR levels across all patients (BRAF mutant median 2.20 [1.12–3.50]; BRAF wildtype median 1.75 [0.33–71.28]; p=0.002) (Table 3). This association remained significant in the proximal colon cohort (BRAF mutant median 2.23 [1.45–3.50]; BRAF wildtype median 1.76 [0.33–7.45]; p=0.002) (Figure 2B).
VEGFR2 Expression by Tumor Location and KRAS, BRAF Mutation Status
Rectal tumors demonstrated significantly higher VEGFR2 expression (1.77 [0.23–13.00]) than distal (1.24 [0.18–7.66]) and proximal (1.10 [0.24–6.10]) tumors (p<0.001 for both comparisons; Figure 1D, Table 2).
Furthermore, VEGFR2 mRNA levels were significantly lower in mutated KRAS tumors (median 1.19 [0.23–7.66]) than in wild-type tumors (median 1.35 [0.18–13.00]) (p=0.003) (Table 3). In subgroup analysis, this relationship persisted in proximal (KRAS mutant median 0.91 [0.24–6.10]; KRAS wildtype median 1.29 [0.33–5.94]; p=0.01) and rectal (KRAS mutant median 1.53 [0.23–3.62]; KRAS wildtype median 2.21 [0.37–13.00]; p=0.02) cancers (Table 2).
There was no significant association between BRAF mutation status and VEGFR2 expression (Table 3). In subgroup analysis, proximal tumors (n=106) with the BRAF V600E mutation trended towards increased VEGFR2 expression (median 1.48 [0.56–2.63]) compared to BRAF wildtype tumors (median 1.06 [0.24–6.10]) (p=0.07).
DISCUSSION
Colorectal cancer subsites are characterized by distinct genetic and histopathological features, but the association between tumor location and prognostic and predictive biomarker expression is not well delineated. We evaluated whether primary tumor site influences KRAS and BRAF mutation status and the mRNA expression of biomarkers reflecting DNA repair, fluoropyrimidine metabolism, tumor cell growth, and angiogenesis in advanced CRC. Our analysis revealed that each tumor site has a unique molecular phenotype which may predict chemotherapeutic and antibody drug sensitivity as well as clinical outcomes (Figure 3). Furthermore, we found that KRAS and BRAF mutation status is associated with biomarker gene expression, and these relationships depend upon tumor location.
Figure 3.
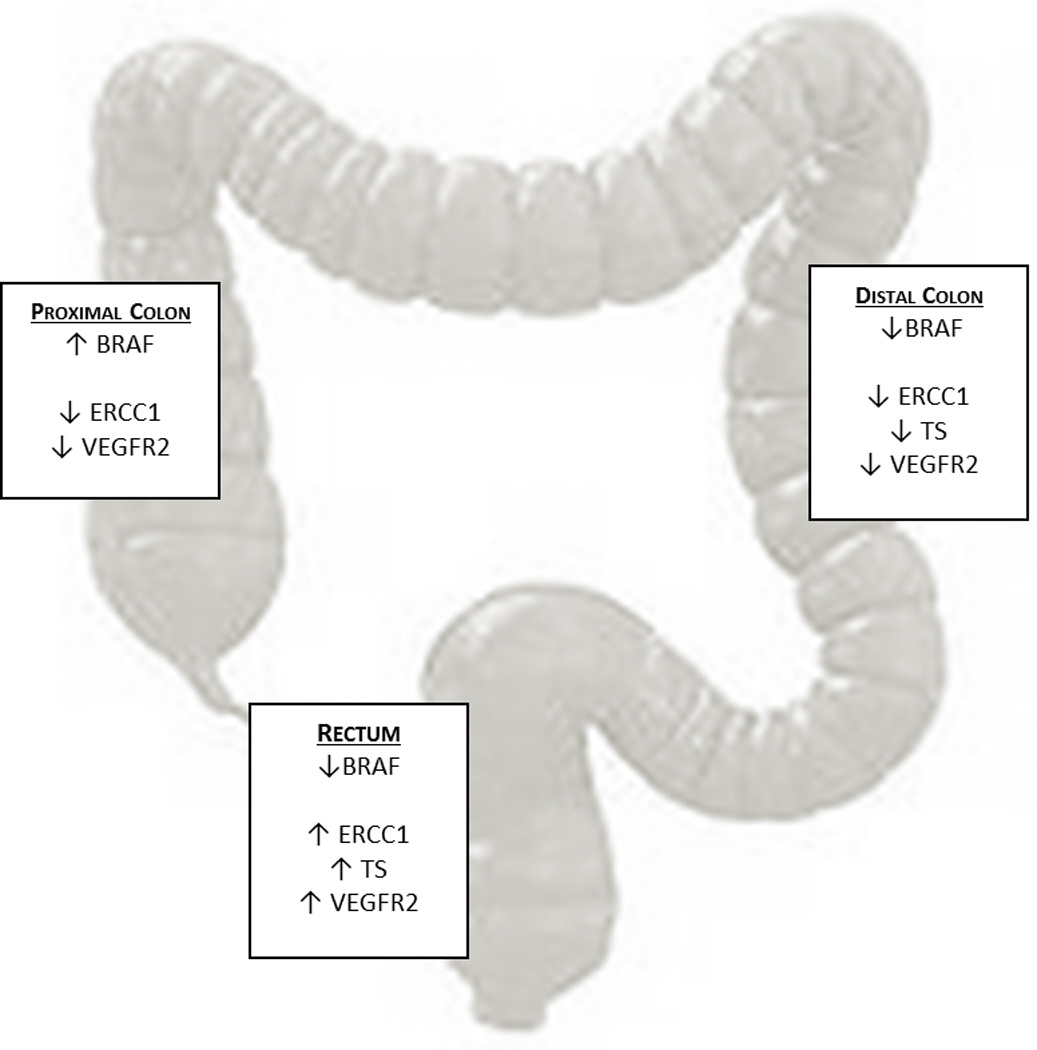
Gene Expression Profiles by Primary Tumor Location
KRAS and BRAF Mutation Status by Tumor Location
The distribution of KRAS mutations was similar across tumor sites. While others[36–38] have demonstrated more frequent KRAS mutations in the proximal colon, the data are not consistent[39], and this may reflect methodologic variation as well as different KRAS mutations being examined between studies. Conversely, BRAF V600E mutations were more common in proximal cancers in our cohort, which is consistent with prior data[12, 28, 37]. The predilection of BRAF mutations for the proximal colon partly reflects increased microsatellite instability (MSI) in this region[26, 40, 41], as microsatellite unstable tumors are more enriched with BRAF mutations[42, 43].
ERCC1, TS, EGFR, and VEGFR2 Expression by Tumor Location
Though prior investigations have demonstrated divergent patterns of gene expression, metastatic spread, and response to therapy between rectal and colonic tumors[28, 37], ours is the first study to reveal anatomic-based differences in ERCC1 and VEGFR2 expression. Irrespective of KRAS or BRAF mutation status, rectal cancers had significantly higher ERCC1 and VEGFR2 mRNA levels compared to distal and proximal colon tumors, in addition to increased TS levels compared to distal colon cancers. Our group and others have previously shown contrasting TS expression patterns by CRC subsite[44–46], though the different assays used (i.e. enzymatic vs. nuclear or cytoplasmic protein expression) confounds interpretation. In sum, our findings suggest that the efficacy of oxaliplatin, anti-angiogenic, and fluoropyrimidine agents may differ between proximal and distal tumors, and this hypothesis warrants prospective validation in the clinical trial setting. Furthermore, stratification by specific anatomic site (rather than broad categories of proximal vs. distal tumors) may offer more useful predictive and prognostic information[37].
Influence of KRAS and BRAF Mutation Status on ERCC1, TS, EGFR, and VEGFR2 Expression Depends on Tumor Location
Mutant KRAS status was associated with lower expression of ERCC1, TS, EGFR, and VEGFR2 among all patients. In multivariate analysis, this association remained significant for all biomarkers in the proximal colon. In addition, among rectal cancers, KRAS mutant tumors had decreased VEGFR2 expression than KRAS wildtype ones. That the association between KRAS status and biomarker gene expression varied by tumor location supports distinct carcinogenic mechanisms across tumor sites. It also suggests the presence of a heterogeneous intestinal microenvironment, and that tumor-stromal interactions and epigenetic modifications are critical in mediating the effects of cytotoxic and targeted agents.
One such epigenetic association may exist between the KRAS and nucleotide excision repair (NER) pathways, particularly in proximal colonic tumors. In vitro studies in COLO320DM colon cancer cell lines[47] have demonstrated that KRAS suppression by small interfering RNAs (siRNAs) leads to ERCC1 overexpression and oxaliplatin resistance, whereas KRAS activation may decrease ERCC1 gene expression through upregulation of DNA methyltransferase 3 beta (DNMT3B) and subsequent ERCC1 hypermethylation, promoting oxaliplatin sensitivity[47]. It follows that one anatomic-based link between KRAS and NER may lie in methylation differences between tumor sites. The CpG island methylator phenotype (CIMP) is more common in proximal[48, 49] and KRAS-mutant[50] tumors. Our findings may also help explain clinical outcome data from the PRIME[51] and OPUS[52] trials. Patients from these studies with KRAS mutant tumors who were treated with FOLFOX alone showed a trend towards improved progression-free survival, relative to those with KRAS wildtype tumors. Such a relationship was not observed in the CRYSTAL trial[53] which used irinotecan-based chemotherapy. Identifying the regulatory mechanisms between KRAS activation, NER, and tumor location may offer novel and more personalized drug targets in future investigations.
TS expression was also influenced by KRAS and BRAF mutation status in a location-dependent manner. The lower TS mRNA levels in KRAS mutated proximal colon tumors suggest increased fluoropyrimidine sensitivity in this cohort. Previous studies have not shown a significant association between KRAS mutation status and response to fluoropyrimidines[54] or TS expression[54–56] regardless of tumor location, though these studies employed enzymatic rather than mRNA assays, which limits direct comparison with our results. In contrast, the BRAF V600E mutation was associated with increased TS expression among proximal cancers. This may provide a potential explanation for the inferior outcomes seen in patients with BRAF-mutated tumors treated with 5-FU based regimens[57]. Both CIMP[58, 59] and MSI[60, 61] status have also been independently linked with lack of clinical benefit from 5-FU, though examinations of the predictive utility of CIMP[62] and MSI[63, 64], and the correlation between MSI and TS expression[65–70] have yielded conflicting data. As such, further studies are needed to better define the relationship between methylation patterns, mismatch repair, and TS expression in proximal tumors.
Among proximal colon cancers, KRAS mutated tumors had significantly decreased EGFR and VEGFR2 levels, compared to wildtype tumors. These findings suggest that in addition to constitutive KRAS activation, downregulation of EGFR and angiogenic pathways may provide another reason for diminished response towards targeted antibodies in these patients. Indeed, in a recent subgroup analysis of the FIRE-3 trial[71], patients with proximal tumors benefited less from anti-EGFR directed therapies than those with distal cancers. It also suggests that the EGFR and VEGF signaling pathways share regulatory pathways[72], including the MAPK/PI3K, STAT3 and hypoxia-inducible factor (HIF) signaling cascades, as demonstrated in cell line and xenograft models[73]. Within the rectal cancer cohort, KRAS mutant tumors had decreased VEGFR2 expression compared to the KRAS wildtype group. This is consistent with clinical data showing significantly improved pathologic complete response rates among KRAS wildtype rectal cancer patients receiving cetuximab-based neoadjuvant chemoradiation, and whose tumors had increased intratumoral VEGFR2 expression[74].
In contrast to KRAS mutant tumors, BRAF mutated cancers had increased EGFR expression in the proximal colon subgroup. As our samples came from un-treated patients, this supports the hypothesis that EGFR overexpression may be an inherent rather than acquired resistance mechanism towards BRAF inhibitors[75, 76] in CRC patients.
Our study has its limitations, the first of which is its retrospective design. We could not account for potential confounding variables, including history of prior cancers, patient ethnicity, and MSI status, any of which could have independently influenced biomarker expression. An extended RAS analysis examining mutations outside of exon 2 may have yielded additional associations between tumor location and gene expression. Furthermore, information regarding prior treatment, particularly in patients who may have had liver-limited metastases and received chemotherapy prior to surgical resection is not known. In addition, we used the splenic flexure as the dividing line between proximal and distal sided tumors since the precise location of tumors within the transverse colon (i.e. proximal two-thirds vs. distal one-third) was not always documented, and this does not reflect boundaries based on blood supply and embryonic origin. Importantly, the lack of outcome data precludes definitive conclusions about the prognostic significance of the demonstrated anatomic-based associations in gene expression.
CONCLUSIONS
Colorectal cancer comprises a spectrum of tumors with unique carcinogenic mechanisms, stromal interactions, and clinical outcomes based on primary site. Our study is the first to demonstrate that the mRNA expression of predictive and prognostic biomarkers and their relationship with KRAS and BRAF mutation status are contingent on anatomic location. Our analysis further emphasizes the distinct biology among colorectal cancers and that tumor location should be included in clinical decision-making. Prospective studies ought to confirm our findings and incorporate the role of tumor site in understanding CRC molecular heterogeneity and evolving phenotypes with treatment. A more refined use of biomarkers should advance clinical trial design, drug development, and patient outcomes.
Acknowledgments
Funded by P30CA014089-27S1. Supported by Dhont Foundation, Wunderglo Foundation
Footnotes
Preliminary results presented at the American Society of Clinical Oncology Annual Meeting, May 30 – June 3, 2013, Chicago, IL
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 2.Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 3.Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 4.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 5.Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–1762. doi: 10.1200/JCO.2011.38.0915. [DOI] [PubMed] [Google Scholar]
- 6.Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Primrose J, Falk S, Finch-Jones M, et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis: the New EPOC randomised controlled trial. Lancet Oncol. 2014 doi: 10.1016/S1470-2045(14)70105-6. [DOI] [PubMed] [Google Scholar]
- 8.Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–1810. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 9.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, Mirtsching B, Cohn AL, Pippas AW, Azarnia N, Tsuchihashi Z, Mauro DJ, Rowinsky EK. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–4921. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 10.Vallbohmer D, Zhang W, Gordon M, Yang DY, Yun J, Press OA, Rhodes KE, Sherrod AE, Iqbal S, Danenberg KD, Groshen S, Lenz HJ. Molecular determinants of cetuximab efficacy. J Clin Oncol. 2005;23:3536–3544. doi: 10.1200/JCO.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 11.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Kohne CH. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–1475. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 12.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Azuma M, Lurje G, Gordon MA, Yang D, Pohl A, Ning Y, Bohanes P, Gerger A, Winder T, Hollywood E, Danenberg KD, Saltz L, Lenz HJ. Molecular predictors of combination targeted therapies (cetuximab, bevacizumab) in irinotecan-refractory colorectal cancer (BOND-2 study) Anticancer Res. 2010;30:4209–4217. [PubMed] [Google Scholar]
- 14.Schimanski CC, Zimmermann T, Schmidtmann I, Gockel I, Lang H, Galle PR, Moehler M, Berger MR. K-ras mutation status correlates with the expression of VEGFR1, VEGFR2, and PDGFRalpha in colorectal cancer. Int J Colorectal Dis. 2010;25:181–186. doi: 10.1007/s00384-009-0843-7. [DOI] [PubMed] [Google Scholar]
- 15.Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, Lenz HJ, Leichman CG, Leichman L, Diasio RB, Danenberg PV. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–1327. [PubMed] [Google Scholar]
- 16.Uchida K, Danenberg PV, Danenberg KD, Grem JL. Thymidylate synthase, dihydropyrimidine dehydrogenase, ERCC1, and thymidine phosphorylase gene expression in primary and metastatic gastrointestinal adenocarcinoma tissue in patients treated on a phase I trial of oxaliplatin and capecitabine. BMC Cancer. 2008;8:386. doi: 10.1186/1471-2407-8-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SH, Kwon HC, Oh SY, Lee DM, Lee S, Lee JH, Roh MS, Kim DC, Park KJ, Choi HJ, Kim HJ. Prognostic value of ERCC1, thymidylate synthase, and glutathione S-transferase pi for 5-FU/oxaliplatin chemotherapy in advanced colorectal cancer. Am J Clin Oncol. 2009;32:38–43. doi: 10.1097/COC.0b013e31817be58e. [DOI] [PubMed] [Google Scholar]
- 18.Shirota Y, Stoehlmacher J, Brabender J, Xiong YP, Uetake H, Danenberg KD, Groshen S, Tsao-Wei DD, Danenberg PV, Lenz HJ. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol. 2001;19:4298–4304. doi: 10.1200/JCO.2001.19.23.4298. [DOI] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Triff K, Konganti K, Gaddis S, Zhou B, Ivanov I, Chapkin RS. Genome-wide analysis of the rat colon reveals proximal-distal differences in histone modifications and proto-oncogene expression. Physiol Genomics. 2013;45:1229–1243. doi: 10.1152/physiolgenomics.00136.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benedix F, Kube R, Meyer F, Schmidt U, Gastinger I, Lippert H. Comparison of 17,641 patients with right- and left-sided colon cancer: differences in epidemiology, perioperative course, histology, and survival. Dis Colon Rectum. 2010;53:57–64. doi: 10.1007/DCR.0b013e3181c703a4. [DOI] [PubMed] [Google Scholar]
- 22.Sinicrope FA, Mahoney MR, Smyrk TC, Thibodeau SN, Warren RS, Bertagnolli MM, Nelson GD, Goldberg RM, Sargent DJ, Alberts SR. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31:3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glebov OK, Rodriguez LM, Nakahara K, Jenkins J, Cliatt J, Humbyrd CJ, DeNobile J, Soballe P, Simon R, Wright G, Lynch P, Patterson S, Lynch H, Gallinger S, Buchbinder A, Gordon G, Hawk E, Kirsch IR. Distinguishing right from left colon by the pattern of gene expression. Cancer Epidemiol Biomarkers Prev. 2003;12:755–762. [PubMed] [Google Scholar]
- 24.Sadler TW, (Thomas W.) Langman, Jan. Langman's Medical Embryology 11th ed. 2010 [Google Scholar]
- 25.Forster S, Sattler HP, Hack M, Romanakis K, Rohde V, Seitz G, Wullich B. Microsatellite instability in sporadic carcinomas of the proximal colon: association with diploid DNA content, negative protein expression of p53, and distinct histomorphologic features. Surgery. 1998;123:13–18. [PubMed] [Google Scholar]
- 26.Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–156. [PMC free article] [PubMed] [Google Scholar]
- 27.De Roock W, Biesmans B, De Schutter J, Tejpar S. Clinical biomarkers in oncology: focus on colorectal cancer. Mol Diagn Ther. 2009;13:103–114. doi: 10.1007/BF03256319. [DOI] [PubMed] [Google Scholar]
- 28.Minoo P, Zlobec I, Peterson M, Terracciano L, Lugli A. Characterization of rectal, proximal and distal colon cancers based on clinicopathological, molecular and protein profiles. Int J Oncol. 2010;37:707–718. doi: 10.3892/ijo_00000720. [DOI] [PubMed] [Google Scholar]
- 29.Bufill JA. Colorectal cancer: evidence for distinct genetic categories based on proximal or distal tumor location. Ann Intern Med. 1990;113:779–788. doi: 10.7326/0003-4819-113-10-779. [DOI] [PubMed] [Google Scholar]
- 30.Distler P, Holt PR. Are right- and left-sided colon neoplasms distinct tumors? Dig Dis. 1997;15:302–311. doi: 10.1159/000171605. [DOI] [PubMed] [Google Scholar]
- 31.Nawa T, Kato J, Kawamoto H, Okada H, Yamamoto H, Kohno H, Endo H, Shiratori Y. Differences between right- and left-sided colon cancer in patient characteristics, cancer morphology and histology. J Gastroenterol Hepatol. 2008;23:418–423. doi: 10.1111/j.1440-1746.2007.04923.x. [DOI] [PubMed] [Google Scholar]
- 32.Weiss JM, Pfau PR, O'Connor ES, King J, LoConte N, Kennedy G, Smith MA. Mortality by stage for right- versus left-sided colon cancer: analysis of surveillance, epidemiology, and end results--Medicare data. J Clin Oncol. 2011;29:4401–4409. doi: 10.1200/JCO.2011.36.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S, Liotta LA. Laser capture microdissection: molecular analysis of tissue. Science. 1997;278:1481–1483. doi: 10.1126/science.278.5342.1481. [DOI] [PubMed] [Google Scholar]
- 34.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 35.Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 36.Rosty C, Young JP, Walsh MD, Clendenning M, Walters RJ, Pearson S, Pavluk E, Nagler B, Pakenas D, Jass JR, Jenkins MA, Win AK, Southey MC, Parry S, Hopper JL, Giles GG, Williamson E, English DR, Buchanan DD. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol. 2013;26:825–834. doi: 10.1038/modpathol.2012.240. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower C, Chan AT, Giovannucci E, Fuchs C, Ogino S. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61:847–854. doi: 10.1136/gutjnl-2011-300865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samowitz WS, Curtin K, Schaffer D, Robertson M, Leppert M, Slattery ML. Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000;9:1193–1197. [PubMed] [Google Scholar]
- 39.Benedix F, Meyer F, Kube R, Kropf S, Kuester D, Lippert H, Roessner A, Kruger S. Influence of anatomical subsite on the incidence of microsatellite instability, and KRAS and BRAF mutation rates in patients with colon carcinoma. Pathol Res Pract. 2012;208:592–597. doi: 10.1016/j.prp.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Lleonart ME, Garcia-Foncillas J, Sanchez-Prieto R, Martin P, Moreno A, Salas C, Ramon y Cajal S. Microsatellite instability and p53 mutations in sporadic right and left colon carcinoma: different clinical and molecular implications. Cancer. 1998;83:889–895. [PubMed] [Google Scholar]
- 41.Samowitz WS, Slattery ML, Kerber RA. Microsatellite instability in human colonic cancer is not a useful clinical indicator of familial colorectal cancer. Gastroenterology. 1995;109:1765–1771. doi: 10.1016/0016-5085(95)90742-4. [DOI] [PubMed] [Google Scholar]
- 42.Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, Qian ZR, Morikawa T, Shen J, Meyerhardt JA, Fuchs CS, Ogino S. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105:1151–1156. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O, Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lenz HJ, Danenberg KD, Leichman CG, Florentine B, Johnston PG, Groshen S, Zhou L, Xiong YP, Danenberg PV, Leichman LP. p53 and thymidylate synthase expression in untreated stage II colon cancer: associations with recurrence, survival, and site. Clin Cancer Res. 1998;4:1227–1234. [PubMed] [Google Scholar]
- 45.Sulzyc-Bielicka V, Domagala P, Majdanik E, Chosia M, Bielicki D, Kladny J, Kaczmarczyk M, Safranow K, Domagala W. Nuclear thymidylate synthase expression in sporadic colorectal cancer depends on the site of the tumor. Virchows Arch. 2009;454:695–702. doi: 10.1007/s00428-009-0787-x. [DOI] [PubMed] [Google Scholar]
- 46.Wong NA, Brett L, Stewart M, Leitch A, Longley DB, Dunlop MG, Johnston PG, Lessells AM, Jodrell DI. Nuclear thymidylate synthase expression, p53 expression and 5FU response in colorectal carcinoma. Br J Cancer. 2001;85:1937–1943. doi: 10.1054/bjoc.2001.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin YL, Liau JY, Yu SC, Ou DL, Lin LI, Tseng LH, Chang YL, Yeh KH, Cheng AL. KRAS mutation is a predictor of oxaliplatin sensitivity in colon cancer cells. PLoS One. 2012;7:e50701. doi: 10.1371/journal.pone.0050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O'Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–1387. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 49.van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut. 2002;51:797–802. doi: 10.1136/gut.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U S A. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 52.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 53.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 54.Rosty C, Chazal M, Etienne MC, Letoublon C, Bourgeon A, Delpero JR, Pezet D, Beaune P, Laurent-Puig P, Milano G. Determination of microsatellite instability, p53 and K-RAS mutations in hepatic metastases from patients with colorectal cancer: relationship with response to 5-fluorouracil and survival. Int J Cancer. 2001;95:162–167. doi: 10.1002/1097-0215(20010520)95:3<162::aid-ijc1028>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 55.Etienne MC, Chazal M, Laurent-Puig P, Magne N, Rosty C, Formento JL, Francoual M, Formento P, Renee N, Chamorey E, Bourgeon A, Seitz JF, Delpero JR, Letoublon C, Pezet D, Milano G. Prognostic value of tumoral thymidylate synthase and p53 in metastatic colorectal cancer patients receiving fluorouracil-based chemotherapy: phenotypic and genotypic analyses. J Clin Oncol. 2002;20:2832–2843. doi: 10.1200/JCO.2002.09.091. [DOI] [PubMed] [Google Scholar]
- 56.Etienne-Grimaldi MC, Formento JL, Francoual M, Francois E, Formento P, Renee N, Laurent-Puig P, Chazal M, Benchimol D, Delpero JR, Letoublon C, Pezet D, Seitz JF, Milano G. K-Ras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clin Cancer Res. 2008;14:4830–4835. doi: 10.1158/1078-0432.CCR-07-4906. [DOI] [PubMed] [Google Scholar]
- 57.Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–5937. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- 58.Jover R, Nguyen TP, Perez-Carbonell L, et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology. 2011;140:1174–1181. doi: 10.1053/j.gastro.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 60.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 61.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen L, Catalano PJ, Benson AB, 3rd, O'Dwyer P, Hamilton SR, Issa JP. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin Cancer Res. 2007;13:6093–6098. doi: 10.1158/1078-0432.CCR-07-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Des Guetz G, Uzzan B, Nicolas P, Schischmanoff O, Perret GY, Morere JF. Microsatellite instability does not predict the efficacy of chemotherapy in metastatic colorectal cancer. A systematic review and meta-analysis. Anticancer Res. 2009;29:1615–1620. [PubMed] [Google Scholar]
- 64.Elsaleh H, Joseph D, Grieu F, Zeps N, Spry N, Iacopetta B. Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet. 2000;355:1745–1750. doi: 10.1016/S0140-6736(00)02261-3. [DOI] [PubMed] [Google Scholar]
- 65.Ricciardiello L, Ceccarelli C, Angiolini G, Pariali M, Chieco P, Paterini P, Biasco G, Martinelli GN, Roda E, Bazzoli F. High thymidylate synthase expression in colorectal cancer with microsatellite instability: implications for chemotherapeutic strategies. Clin Cancer Res. 2005;11:4234–4240. doi: 10.1158/1078-0432.CCR-05-0141. [DOI] [PubMed] [Google Scholar]
- 66.Jensen SA, Vainer B, Kruhoffer M, Sorensen JB. Microsatellite instability in colorectal cancer and association with thymidylate synthase and dihydropyrimidine dehydrogenase expression. BMC Cancer. 2009;9:25. doi: 10.1186/1471-2407-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Odin E, Wettergren Y, Nilsson S, Carlsson G, Gustavsson B. Colorectal carcinomas with microsatellite instability display increased thymidylate synthase gene expression levels. Clin Colorectal Cancer. 2007;6:720–727. doi: 10.3816/CCC.2007.n.042. [DOI] [PubMed] [Google Scholar]
- 68.Popat S, Wort R, Houlston RS. Inter-relationship between microsatellite instability, thymidylate synthase expression, and p53 status in colorectal cancer: implications for chemoresistance. BMC Cancer. 2006;6:150. doi: 10.1186/1471-2407-6-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sinicrope FA, Rego RL, Halling KC, Foster NR, Sargent DJ, La Plant B, French AJ, Allegra CJ, Laurie JA, Goldberg RM, Witzig TE, Thibodeau SN. Thymidylate synthase expression in colon carcinomas with microsatellite instability. Clin Cancer Res. 2006;12:2738–2744. doi: 10.1158/1078-0432.CCR-06-0178. [DOI] [PubMed] [Google Scholar]
- 70.Merkelbach-Bruse S, Hans V, Mathiak M, Sanguedolce R, Alessandro R, Ruschoff J, Buttner R, Houshdaran F, Gullotti L. Associations between polymorphisms in the thymidylate synthase gene, the expression of thymidylate synthase mRNA and the microsatellite instability phenotype of colorectal cancer. Oncol Rep. 2004;11:839–843. [PubMed] [Google Scholar]
- 71.V H, Modest DP, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmuller CA, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, Held S, Giessen CA, Jung A, Kirchner T, Stintzing S. Gender and tumor location as predictors for efficacy: Influence on endpoints in first-line treatment with FOLFIRI in combination with cetuximab or bevacizumab in the AIO KRK 0306 (FIRE3) trial. Journal of Clinical Oncology. 2014;32 Abstract 3600. [Google Scholar]
- 72.Ruzzo A, Graziano F, Canestrari E, Magnani M. Molecular predictors of efficacy to anti-EGFR agents in colorectal cancer patients. Curr Cancer Drug Targets. 2010;10:68–79. doi: 10.2174/156800910790980205. [DOI] [PubMed] [Google Scholar]
- 73.Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther. 2011;131:80–90. doi: 10.1016/j.pharmthera.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 74.Grimminger PP, Danenberg P, Dellas K, Arnold D, Rodel C, Machiels JP, Haustermans K, Debucquoy A, Velenik V, Sempoux C, Bracko M, Holscher AH, Semrau R, Yang D, Danenberg K, Lenz HJ, Vallbohmer D. Biomarkers for cetuximab-based neoadjuvant radiochemotherapy in locally advanced rectal cancer. Clin Cancer Res. 2011;17:3469–3477. doi: 10.1158/1078-0432.CCR-10-2273. [DOI] [PubMed] [Google Scholar]
- 75.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M, Engelman JA. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]