

Abstract
One-step synthesis of carbocycles substituted with amines from simple starting materials remains rare. We recently developed intermolecular [3+2] annulation of cyclopropylanilines with alkenes and alkynes that enable this one-step synthesis. Herein, we report our findings for a fullscale study of the annulation. Significant expansion of the substrate scope for both cyclopropylanilines and alkynes is achieved. A range of structurally diverse carbocycles substituted with amines is prepared.
Keywords: visible light, [3+2] annulation, photocatalysis, intermolecular, cyclopropylanilines, alkynes, enynes, diynes
Introduction
Cycloaddition or annulation reactions have been primarily used to construct carbo- or heterocycles with various sizes.[1] These types of reactions have gained increasing prominence in diversity-oriented synthesis (DOS) because it can efficiently increase small molecules’ structural diversity and complexity.[2] Both features are critical for the success of small molecule libraries as they enable small molecules to occupy more chemical space. Recently, the renaissance of visible light photocatalysis has brought new life to some existing reactions, including cycloaddition or annulation reactions.[3] Since the interaction of photoexcited catalysts and small molecules is often specific,[4] the photocatalyzed transformations are usually highly chemoselective and thus tolerate a variety of functional groups. Therefore, in principle, photocatalyzed cycloaddition or annulation reactions are ideal reactions for making small molecule libraries. We have recently developed a [3+2] annulation of cyclopropylanilines with alkenes and alkynes.[5] Although our preliminary results in these reactions gave a glimpse of their potential in DOS, constraints with respect to both reacting partners, cyclopropylanilines and alkenes or alkynes, existed. An aryl group was required on the cyclopropylamine moiety while only terminal alkenes and alkynes were reactive.[5] We intended to address both limitations with the ultimate goal of thoroughly investigating the scope of the [3+2] annulation. Herein we report our findings towards expansion of these reactions.
Results and Discussion
Reaction optimization
Although we previously optimized the conditions for the [3+2] annulation of cyclopropylanilines with alkynes using ruthenium polypyridyl complexes, the desired annulation products were obtained in only modest yields.[5b] We were motived to examine more conditions in order to improve the yields, as shown in Table 1. Using the [3+2] annulation of cyclopropylaniline 1a with phenylacetylene 2a as the model reaction, we first investigated the annulation in a continuous flow format[6] (entry 2). To our surprise, comparing against the batch format (entry 1), although the reaction time was shortened from 8 h to 3 h as expected, the yield did not improve. We also examined the use of cyclometalated iridium complexes to catalyze the annulation. These complexes are another important class of photocatalysts that finds rising numbers of synthetic applications in photochemistry.[7] Two iridium complexes were examined and neither provided a better yield than Ru(bpz)3(PF6)2 (entries 3 and 4). Lastly, we performed the reaction in the presence of TEMPO (entry 5). We previously proposed distonic ion 1a•+ as one of the key intermediates in the [3+2] annulation of cyclopropylanilines with alkenes and alkynes (Scheme 1). This ion presumably results from ring opening of cyclopropylanilines that is induced by one-electron oxidation of the parent amine by the excited state of Ru(bpz)3(PF6)2.[5a] The use of TEMPO was designed to intercept the radical moiety of the ion and thereby perturb the annulation. Indeed, the annulation product was isolated in a negligible yield, which lent credence to our argument for the involvement of distonic ion 1a•+. Since no improvement in the yield was achieved using the flow or the Ir catalysts, we settled the previously optimized conditions (entry 1, Table 1) as the standard conditions for the scope studies.
Scheme 1.
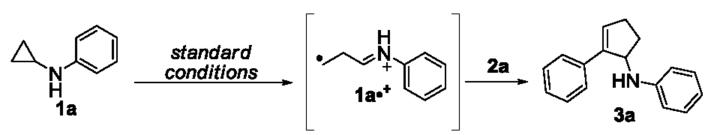
Proposed intermediate distonic ion in the [3+2] annulations.
Diyne and enyne studies
We were intrigued by the possibility of using 1,3-conjugated diynes and enynes in the [3+2] annulation as they offer the potential to further enhance the value of the annulation in diversity-oriented synthesis (DOS). The expected annulation products would possess a 1,3-conjugated enyne moiety from 1,3-conjugated diynes or a 1,3-conjugated diene moiety from 1,3-conjugated enynes. Both moieties can be further engaged in cycloaddition reactions such as the Diels Alder reaction, and thereby enable cycloaddition cascades to rapidly assembly complex fused carbocycles or heterocycles. However, 1,3-conjugated diynes and enynes present more challenges than simple alkynes in the annulation. We previously reported that the annulation was sensitive to substitution on alkynes as well as their electronic characters. For example, alkyl substituted terminal alkynes and internal alkynes were found to be unreactive.[5b] It was not clear to us whether addition of one more pi bond would enhance the original pi bond’s reactivity enough to take part in the annulation. Moreover, prediction of regiochemistry in 1,3-conjugated diynes and enynes could be challenging. Finally, for unsymmetric diynes and enynes, since there are two different pi bonds, the issue of chemoselectivity may arise as addition to either or both pi bonds can occur.
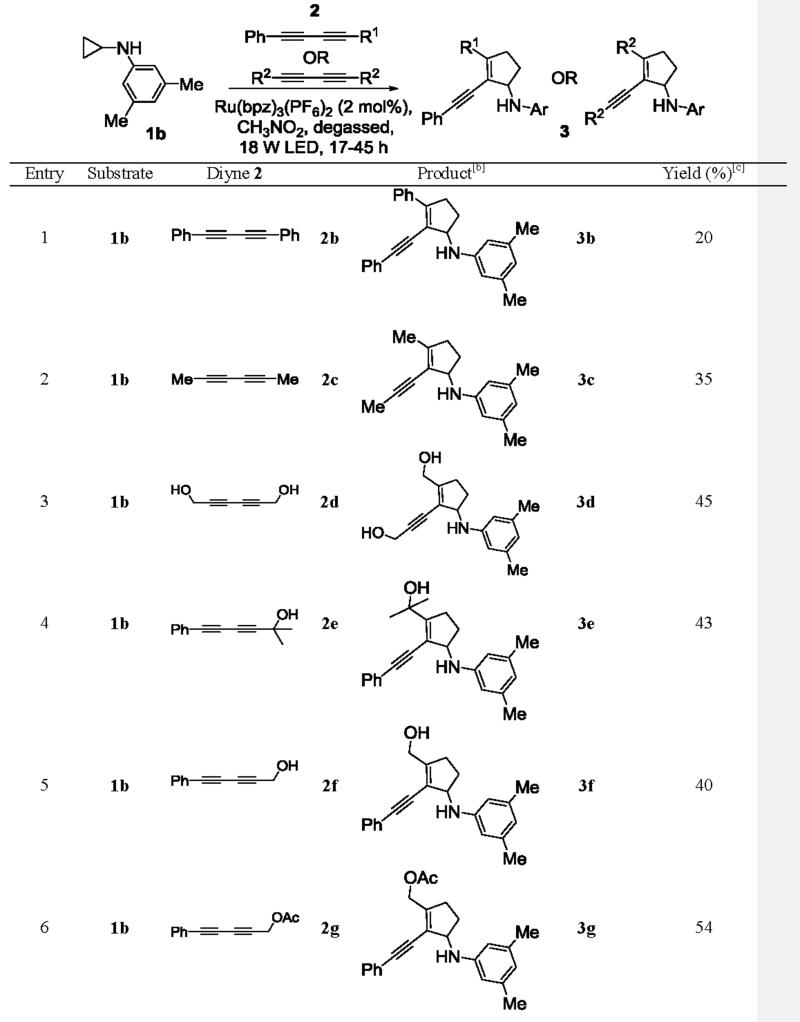
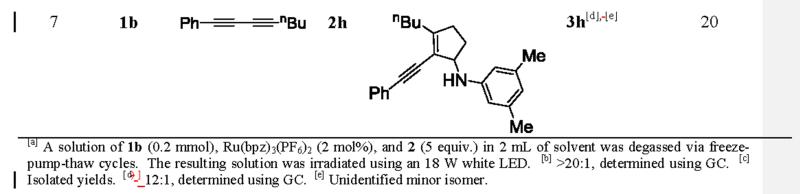
To our delight, under the standard conditions, symmetrical and asymmetrical 1,3-conjugated diynes successfully underwent the intermolecular [3+2] annulation with monocyclic cyclopropylaniline 1b to afford the conjugated cyclopentene adducts (Table 2). Symmetrical diynes bearing phenyl 2b, methyl 2c, and hydroxymethyl 2d groups proceeded in fair yields (entries 1–3). Asymmetrical diynes also produced the annulation products in similar yields (entries 4–7). These diynes 2e-h all bore a phenyl group on one end, while a variety of moieties, such as nBu and hydroxymethyl groups, were tolerated on the other end. A quaternary center adjacent to the reactive carbon center of diyne 2e was also well tolerated. Although the yields for the annulation products were modest, complete regiocontrol was observed universally in all but one example (entry 7). Even with the exception 3h, the regioselectivity was excellent (12:1). We rationalized the observed excellent regioselectivity based on the stability of radicals generated after the first C-C bond formation. DFT calculations on two regioisomeric radicals (5a and 5b) that model the proposed regioisomeric radicals (4a and 4b) in the annulation supported this argument (Figure 1). Radical 5a was found to be 44 kJ/mol more stable than radical 5b due to resonance. The stability trend of the two regioisomeric radicals was also observed in diyne 2b (see SI).
Figure 1.

Rationale for observed selectivity using DFT calculations.
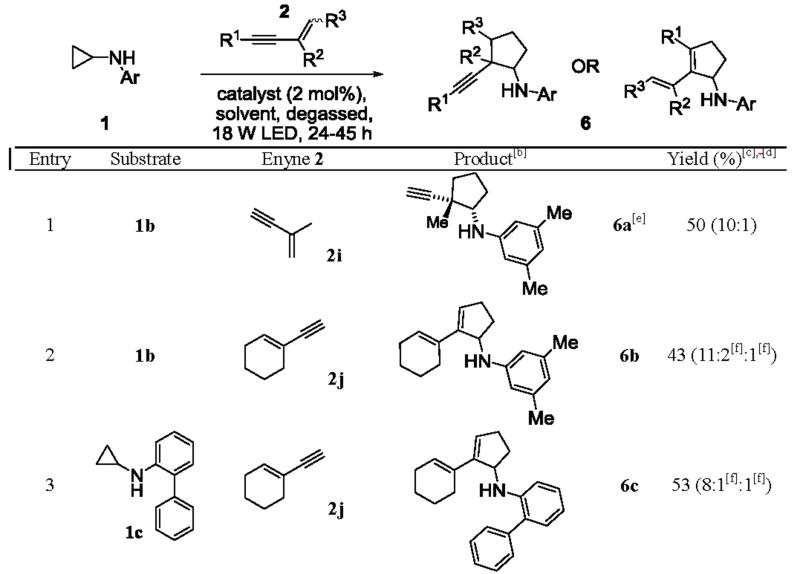
The annulation with enynes was expected to be more complicated than with diynes. In addition to regioselectivity, chemoselectivity has to be dealt with because of the competition between the alkene and alkyne moiety in the annulation. We found that in our previous work, alkenes are more reactive than alkynes in general.[5b] However, because of steric effect, this reactivity trend can be reversed if the alkene is more substituted than the alkyne. Furthermore, if the alkene moiety is more reactive than the alkyne moiety, the diastereoselectivity also needs to be addressed.
Indeed, 1,3-conjugated enynes 2i-l produced a mixture of isomeric products in the annulation (Table 3). Commercially available enyne 2i, possessing both a terminal alkyne and a terminal alkene, underwent the [3+2] annulation predominately on the alkene moiety to provide the annulation products in 50% combined yields (entry 1), with the minor isomer being identified as the annulation on the alkyne. The chemoselectivity was 10 to 1, favoring the alkene moiety. Another commercially available enyne, 2j, bearing a terminal alkyne and a trisubstituted alkene, was subjected to the annulation with cyclopropylanilines 1b and 1c, respectively, to afford the corresponding conjugated 1,3-dienes 6b and 6c in moderate yields (entries 2 and 3). In both cases, the annulation favored the terminal alkyne moiety of enyne 2j. It is worth noting that for these two enynes (2i and 2j), the catalyst system composed of Ir(ppy)2(dtb-bpy)(PF6) in a 1:1 mixture of CH3NO2 and CF3CH2OH proved to be more effective than the standard catalyst system (Ru(bpz)3(PF6)2 in CH3NO2). Likewise for synthetically prepared enyne 2k[8] bearing a terminal alkyne and a 1,2-disubstituted alkene, the furnished annulated product 6d was isolated in 59% combined yields, with the 1,3-diene as the major isomer (entry 4). Lastly, enyne 2l, possessing both an internal alkyne and a 1,2-disubstituted alkene, was prepared according to a literature protocol[9] and then treated with cyclopropylaniline 1b under the standard conditions. The major products 6e, composed of four diastereomers derived from the annulation on the alkene moiety, was obtained in 66% combined yields (entry 5).
Substituted cyclopropanes
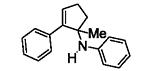
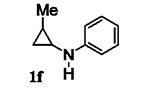
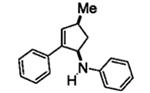
Next, we shifted our attention to focus on the scope of cyclopropylanilines. To maximize the potential of the [3+2] annulation, it was imperative for us to examine substituents on the nitrogen atom and the cyclopropyl ring. Particularly for the latter, if succeeded, it would allow us to decorate the cyclopentene ring of the annulation products. Using phenylacetylene 2a as the model alkyne, we first investigated the annulation of N-cyclopropyl-N-methylaniline 1d to form 7a and no reaction was observed (entry 1, Table 4). This is in accordance with the result reported by Tanko and coworkers in which the ring opening of the amine radical cation derived from 1d was found to be very sluggish.[10] Despite this observation, substitution on the cyclopropyl ring was generally tolerated. N-(1-methylcyclopropyl)aniline 1e provided the annulation product 7b bearing a quaternary carbon center in 33% yield when subjected to the standard conditions (entry 2). The [3+2] annulation was also effective using 2-substituted cyclopropyl rings, as demonstrated with methyl 1f and phenyl 1g substituents (entries 3 and 4). In both examples, the ring opening was completely regioselective, presumably forming the more substituted carbon radical. Modest diastereoselectivity was observed, providing the cis isomer as the major product in both cases.
Cleavage of N-aryl groups
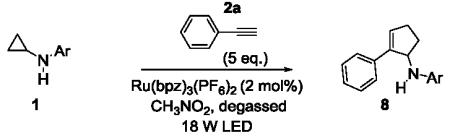
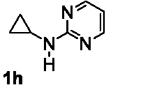
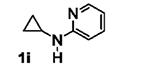
The requirement of an aniline moiety has been a limitation to the [3+2] annulation, as it lowers the generality of the reaction. To circumvent this limitation, we were interested in installing a removable aryl or heteroaryl group that is also capable of mediating the annulation. Some of these potential candidates include pyrimidine 1h,[11] pyridine 1i,[12] and para-methoxyphenyl (PMP, 1k)[13] groups. With this in mind, we synthesized N-aryl cyclopropylamines 1h-k bearing these groups and subjected them to the standard conditions with phenylacetylene 2a.
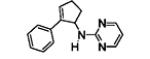
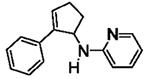
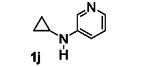
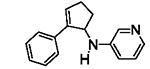
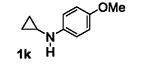
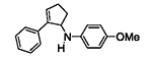
The results are summarized in Table 5. No reaction was observed with 2-pyrimidyl-substituted cyclopropylamine 1h (entry 1). Surprisingly, 2-pyridyl-substituted cyclopropylamine 1i afforded the annulation product in a low yield (13%), which was not synthetically useful (entry 2). In comparison, a much higher yield (48%) was obtained with 3-pyridyl-substituted cyclopropylamine 1j (entry Unfortunately, 3-pyridyl group was not cleavable. A similar yield (47%) was achieved with PMP-substituted cyclopropylamine 1k (entry 4). Therefore, we opted to focus on studying the cleavage of the PMP group in 8d.
The PMP group is generally cleaved under oxidative conditions, and a number of oxidants are suitable for the cleavage. [13] We subjected 8d to these oxidants, including ceric ammonium nitrate (CAN), periodic acid (H5IO6), trichloroisocyanuric acid (TCCA), and CAN/Fe(bpy)3(PF6)2[14]. Among the conditions examined, CAN in sulfuric acid gave the highest yield. The cleavage product was initially isolated as the HCl salt 9 and then immediately acylated in the presence of Et3N and acetyl chloride to give the corresponding acetamide 10 in 68% isolated yield over 2 steps (Scheme 2). The successful removal of the PMP group circumvents the previous limitation of our chemistry and allows for more structural diversification of the [3+2] annulation products.
Scheme 2.

Removal of para-methoxyphenyl (PMP) group.
Conclusion
In summary, we have significantly expanded our studies on the [3+2] annulation of cyclopropylanilines with alkynes. These studies were conducted on multiple fronts including catalyst optimization, mechanistic studies, expansion of the substrate scope for both cyclopropylanilines and alkynes, and identification of a removable N-aryl group for cyclopropylamines. Using simple building blocks, the [3+2] annulation enables rapid assembly of diverse cyclic allylic amine derivatives. Most of these amines possess embedded functional groups that allow for further structural diversification. We anticipate that this method will find usage particularly in diversity-oriented synthesis (DOS).
Experimental Section
General procedure for the [3+2] annulation of cyclopropylanilines with alkyne, enyne, and diyne
an oven-dried test tube (16 × 125 mm) equipped with a stir bar was charged with catalyst (2 mol %), cyclopropylaniline (0.2 mmol), alkyne, enyne, or diyne (1.0 mmol), and dry solvent (2 mL). The test tube was sealed with a Teflon screw cap. The reaction mixture was degassed by Freeze–Pump–Thaw cycles and then irradiated at room temperature with one white LED (18 watts) positioned 8 cm from the test tube. After the reaction was complete as monitored by TLC, the mixture was diluted with diethyl ether and filtered through a short pad of silica gel. The filtrate was concentrated in vacuum and purified by silica gel flash chromatography to afford the desired product.
Procedure for the deprotection of PMP
To a pre-cooled solution of para-methoxyphenyl amine (51 mg, 0.2 mmol) in 3:1 CH3CN : H2O (2 mL) was slowly added concentrated H2SO4 (22 L, 0.385 mmol) at 0 °C. Ceric ammonium nitrate (220 mg, 0.4 mmol) was then added in one portion, and the mixture was stirred for one hour at 0 °C. The resulting mixture was then diluted with water (2 mL) and separated. The aqueous phase was then washed with Et2O (3 × 5 mL). The combined organic phase was then extracted with 0.1 N HCl (1 × 15 mL), and the obtained aqueous phase was added to the previous aqueous mixture, which was immediately basified to pH 14 using 5 N KOH. The basic aqueous layer was then extracted with Et2O (2 × 30 mL). The combined organic layer was then acidified to pH 1 using hydrogen chloride (2 M in Et2O). The resulting solution was then dried over MgSO4 and concentrated to give the HCl salt as a dark brown oil. The crude HCl salt was used in the next step without further purification.
To a solution of the HCl salt (35 mg, 0.18 mmol) in dry CH2Cl2 (5 mL) was added Et3N (56 μL, 0.4 mmol) dropwise at room temperature. Acetyl chloride (16 μL, 0.22 mmol) was slowly added and the reaction was stirred for 6 hours at room temperature. The reaction was quenched with water (10 mL) and the layers were separated.
The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated to give the crude product. Purification using silica gel chromatography (1:1 hexanes : EtOAc) provided the desired acylated product (26.5 mg, 68% over 2 steps).
Supplementary Material
Table 1.
Reaction optimization.[a]
| Entry | Catalyst | Solvent | Light | GC Yield (%)[b] |
|---|---|---|---|---|
| 1 | Ru(bpz)3(PF6)2 | CH3NO2 | 18 W LED | 69 (68)[c] |
| 2 | Ru(bpz)3(PF6)2 | CH3NO2 | Flow[d] | 60 |
| 3 | Ir(ppy)2(dtb-bpy)(PF6) | CF3CH2OH | 18 W LED | 60 |
| 4 | Ir(dF(CF3)ppy)2(dtb-bpy)(PF6) | CF3CH2OH | 18 W LED | 36 |
| 5[e][f] | Ru(bpz)3(PF6)2 | CH3NO2 | 18 W LED | 7 |
A solution of 1a (0.2 mmol) , catalyst (2 mol%), and 2a (5 equiv.) in 2 mL of solvent was degassed via freeze-pump-thaw cycles. The resulting solution was irradiated using an 18 W white LED for 8 hours.
Using dodecane as an internal standard.
Isolated yield.
Flow reaction time of 3 hours.
19 hour reaction.
Using 1 equiv. TEMPO.
Table 2.
Scope studies using symmetrical and unsymmetrical diynes.[a]
|
|
Table 3.
Scope studies using enynes.[a]
|
|
Table 4.
[3+2] Annulation of substituted cyclopropylanilines.[a]
| Entry | Substrate | Time (h) | Product | Yield (%)[b] | |
|---|---|---|---|---|---|
| 1 |
|
16 |
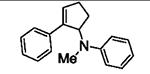
|
7a | 0[c] |
| 2 |
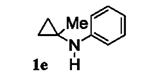
|
15 |
|
7b | 33 |
| 3 |
|
18 |
|
7c [d] | 46[e] |
| 4 |
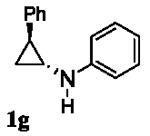
|
36 |
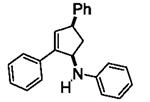
|
7d [d] | 30[f] |
A solution of 1 (0.2 mmol), Ru(bpz)3(PF6)2 (2 mol%), and 2a (5 equiv.) in 2 mL of CH3NO2 was degassed via freeze-pump-thaw cycles. The resulting solution was irradiated using an 18 W white LED.
Isolated yields.
Determined using GC-MS.
Major isomer shown.
2:1 cis:trans, determined by GC-MS.
4:1 cis:trans, determined by GC-MS.
Table 5.
[3+2] Annulation of various aryl amines.[a]
A solution of 1 (0.2 mmol), Ru(bpz)3(PF6)2 (2 mol%), and 2a (5 equiv.) in 2 mL CH3NO2 was degassed via freeze-pump-thaw cycles. The resulting solution was irradiated using an 18 W white LED.
Monitored by TLC.
Isolated yields.
Determined using GC-MS.
Acknowledgments
This publication was supported by the University of Arkansas, Arkansas Bioscience Institute, Grant Number P30 GM103450 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH), NSF Career Award under Award Number CHE-1255539. We thank Jiang Wang, Soumitra Maity, and Mingzhao Zhu for early experimental assistance of this work as well as Derrel Walters and Professor Peter Pulay for the DFT calculations.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201.
References
- [1].Knochel P, Molander GA, editors. Comprehensive Organic Synthesis. 2nd ed. Vol. 5. Elsevier; 2014. [Google Scholar]
- [2].For selected recent reviews: G. Muncipinto G. In: Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology. Trabocchi A, editor. John Wiley & Sons, Hoboken; 2013. pp. 59–95. O’Connor CJ, Beckmann HSG, Spring DR. Chem. Soc. Rev. 2012;41:4444–4456. doi: 10.1039/c2cs35023h.
- [3].For selected recent reviews: Douglas JJ, Nguyen JD, Cole KP, Stephenson CRJ. Aldrichimica Acta. 2014;47:15–25. Yoon TP. ACS Catal. 2013;3:895–902. doi: 10.1021/cs400088e. Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. Xuan J, Lu L-Q, Chen J-R, Xiao W-J. Eur. J. Org. Chem. 2013:6755–6770.
- [4].a) Kalyanasundaram K. Coord. Chem. Rev. 1982;46:1159–244. [Google Scholar]; b) Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top. Curr. Chem. 2007;281:143–203. [Google Scholar]
- [5].a) Maity S, Zhu M, Shinabery RS, Zheng N. Angew. Chem. 2012;124:226–230. doi: 10.1002/anie.201106162. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:222–226. [Google Scholar]; b) Nguyen TH, Maity S, Zheng N. Beilstein. J. Org. Chem. 2014;10:975–980. doi: 10.3762/bjoc.10.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nguyen JD, Reiss B, Dai C, Stephenson CRJ. Chem. Commun. 2013;49:4352–4354. doi: 10.1039/c2cc37206a. [DOI] [PubMed] [Google Scholar]
- [7].Lowry MS, Bernhard S. Chem. Eur. J. 2006;12:7970–7977. doi: 10.1002/chem.200600618. [DOI] [PubMed] [Google Scholar]
- [8].Charpenay M, Boudhar A, Hulot C, Blond G, Suffert J. Tetrahedron. 2013;69:7568–7591. [Google Scholar]
- [9].Zhou H, Moberg C. J. Am. Chem. Soc. 2012;134:15992–15999. doi: 10.1021/ja3070717. [DOI] [PubMed] [Google Scholar]
- [10].Li X, Grimm ML, Igarashi K, Castagnoli N, Jr., Tanko JM. Chem. Commun. 2007:2648–2650. doi: 10.1039/b702157g. [DOI] [PubMed] [Google Scholar]
- [11].a) Ackermann L, Lygin AV. Org. Lett. 2011;13:3332–3335. doi: 10.1021/ol2010648. [DOI] [PubMed] [Google Scholar]; b) Ackermann L, Lygin AV. Org. Lett. 2012;14:764–767. doi: 10.1021/ol203309y. [DOI] [PubMed] [Google Scholar]
- [12].a) Jana CK, Grimme S, Studer A. Chem. Eur. J. 2009;15:9078–9084. doi: 10.1002/chem.200901331. [DOI] [PubMed] [Google Scholar]; b) Dastbaravardeh N, Schnürch M, Mihovilovic MD. Org. Lett. 2012;14:1930–1933. doi: 10.1021/ol300627p. [DOI] [PubMed] [Google Scholar]; c) Wang Z, Reinus BJ, Dong G. Chem. Commun. 2014;50:5230–5232. doi: 10.1039/c3cc47556e. [DOI] [PubMed] [Google Scholar]
- [13].a) Verkade JMM, van Hemert LJC, Quaedflieg PJLM, Alsters PL, van Delft FL, Rutjes FPJT. Tetrahedron Lett. 2006;47:8109–8113. [Google Scholar]; b) De Lamo Marin S, Martens T, Mioskowski C, Royer J. J. Org. Chem. 2005;70:10592–10595. doi: 10.1021/jo051867o. [DOI] [PubMed] [Google Scholar]
- [14].Bergonzini G, Schindler CS, Wallentin C-J, Jacobsen EN, Stephenson CRJ. Chem. Sci. 2014;5:112–116. doi: 10.1039/C3SC52265B. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.