

Abstract
Memories that last a lifetime are thought to be stored, at least in part, as persistent enhancement of the strength of particular synapses. The synaptic mechanism of these persistent changes, late long-term potentiation (L-LTP), depends on the state and number of specific synaptic proteins. Synaptic proteins, however, have limited dwell times due to molecular turnover and diffusion, leading to a fundamental question: how can this transient molecular machinery store memories lasting a lifetime? Because the persistent changes in efficacy are synapse-specific, the underlying molecular mechanisms must to a degree reside locally in synapses. Extensive experimental evidence points to atypical protein kinase C (aPKC) isoforms as key components involved in memory maintenance. Furthermore, it is evident that establishing long-term memory requires new protein synthesis. However, a comprehensive model has not been developed describing how these components work to preserve synaptic efficacies over time. We propose a molecular model that can account for key empirical properties of L-LTP, including its protein synthesis dependence, dependence on aPKCs, and synapse-specificity. Simulations and empirical data suggest that either of the two aPKC subtypes in hippocampal neurons, PKMζ and PKCι/λ, can maintain L-LTP, making the system more robust. Given genetic compensation at the level of synthesis of these PKC subtypes as in knockout mice, this system is able to maintain L-LTP and memory when one of the pathways is eliminated.
Amnesia and dementia, as well as addiction and post-traumatic stress disorder, are associated with deficient or pathophysiological memory induction and maintenance. To understand and treat these disorders, we must understand the mechanisms of memory. Experimental support for the hypothesis that the cellular process of long-term potentiation (LTP) is the basis of learning and memory is rapidly increasing (Bliss and Lømo 1973; Bliss and Collingridge 1993; Bear and Malenka 1994; Barco et al. 2006; Whitlock 2006; Matsuzaki 2007; Nabavi et al. 2014). The most commonly studied form of persistently enhanced synaptic plasticity, LTP, can be divided into at least two temporal phases, early-LTP (E-LTP) and late-LTP (L-LTP). E-LTP is defined as an increase in synaptic efficacies that lasts for up to a few hours; in contrast L-LTP is a long-lasting change that can be detected for months in vivo (Abraham et al. 2002). The induction of both L-LTP and long-lasting memory can be inhibited by the application of protein synthesis inhibitors (PSI). Once L-LTP is induced or long-term memory established, the same levels and durations of PSI that prevent L-LTP and long-term memory cannot reverse them (Flexner et al. 1965; McGaugh 1966; Fonseca et al. 2006; Abraham and Williams 2008).
The fundamental issue we address here is how memories can be maintained over extended periods of time despite protein turnover and diffusion (Crick 1984). In many biological systems similar problems of maintaining a biological signal exist, and the concept of a “molecular switch” has been proposed as a solution (Tyson et al. 2003; Verdugo et al. 2013). A molecular switch is a self-sustaining molecular network in which a brief signal can switch the system between long-lasting stable states. In many cases, it is implemented at the level of the whole cell, often at the level of transcription. Synaptic plasticity, however, must be synapse-specific in order to accomplish its role in learning, memory formation, and receptive field development. Therefore, despite strong evidence for transcriptional changes associated with L-LTP (Day and Sweatt 2011; Zovkic et al. 2013), we concentrate on local changes that can maintain synapse-specificity. A local molecular switch could be instantiated at the level of post-translational modifications (Lisman 1985; Routtenberg and Rekart 2005) or at the level of translation (Belelovsky et al. 2005; Abraham and Williams 2008; Aslam et al. 2009), as translation can occur and be regulated locally in dendritic compartments (Sutton and Schuman 2006; Santos et al. 2010; Batish et al. 2012; Leal et al. 2014).
There is substantial empirical evidence suggesting that a specific kinase located in dendrites and synapses, PKMζ, an atypical isoform of PKC, is essential for the maintenance of synaptic plasticity and memory, suggesting it is a substrate of such a molecular switch (Sacktor et al. 1993; Osten et al. 1996; Ling et al. 2002; Migues et al. 2010; Westmark et al. 2010; Ho et al. 2012; Shao et al. 2012). PKMζ is autonomously active, lacking the regulatory subunit that inhibits the activity of most other PKC isoforms. Electrophysiological and chemical protocols that induce L-LTP cause an increase in the concentration of two atypical PKCs (aPKCs): PKMζ and PKCι/λ (Kelly et al. 2007; Melemedjian et al. 2013). Furthermore, ζ inhibitory peptide (ZIP), a synthesized compound containing the auto-inhibitory sequence of the regulatory subunit of PKCζ and PKCι/λ, erases memory when injected in targeted areas such as hippocampus (spatial memory), insular cortex (taste memory), nucleus accumbens (addiction), and amygdala (fear memory) (Pastalkova 2006; Shema et al. 2007; Serrano et al. 2008; Li et al. 2011; Shabashov et al. 2011; Crespo et al. 2012; Kwapis et al. 2012). Thus, it is plausible that a molecular switch, based on persistent activation of one or both aPKCs, plays an important role in maintaining L-LTP and memory. We therefore developed a mathematical model of such a switch, based primarily on the properties of PKMζ. We then modified the model to fit properties of PKCι/λ in order to investigate compensation in a two-kinase model. To validate the single kinase model we selected several key experimental observations that our model must account for, in addition to forming a bistable molecular switch of an aPKC. These key observations are:
The amount of aPKC increases after inducing L-LTP. Biochemical measurements indicate that aPKCs increase by nearly 75% in the hippocampal CA1 tissue homogenate after L-LTP induction (Kelly et al. 2007) and in synaptoneurosomes after chemically induced L-LTP (Melemedjian et al. 2013).
L-LTP causes a minimal change in the fraction of phosphorylated aPKC. After induction, the proportion of total kinases that are fully (doubly) phosphorylated changes by <10% (Kelly et al. 2007).
Different outcomes result from kinase and synthesis inhibitors. Application of ZIP, a potent competitive inhibitor of PKMζ (Yao et al. 2013a), only during stimulation does not prevent formation of L-LTP (Ren et al. 2013), but when ZIP is applied after the induction phase, L-LTP is abolished (Ling et al. 2002; Serrano et al. 2005; Kelly et al. 2007). On the other hand, application of protein synthesis inhibitors prevents the induction of L-LTP, but the same inhibitor concentration and duration fails to reverse established L-LTP (Otani et al. 1989; Fonseca et al. 2006).
In order to account for all of the above observations, we have developed a dynamical model composed of synthesis, degradation, and phosphorylation of an aPKC (Fig. 1A), the concentration of which is assumed to correlate with L-LTP as has been found empirically for PKMζ (Osten et al. 1996) and for PKCι/λ in ζ-knockout animals (P Tsokas, C Hsieh, Y Yao, EJC Wallace, P Serrano, L Pang, A Tcherepanov, D Jothianandan, D Tian, BR Hartley, et al. in prep.). We assume that the aPKC (denoted as K) can exist in one of two states, a singly phosphorylated (Kp) state or a doubly phosphorylated state (Kpp). The first site is phosphorylated by phosphoinositide-dependent protein kinase-1 (PDK1), and phosphorylation of the second site, denoted the “turn” site, is catalyzed either by another kinase, mTORC2, or the aPKC, itself (Standaert et al. 1997; Li and Gao 2014). Additional key assumptions are as follows: Positive feedback at the level of aPKC synthesis is assumed, such that higher levels of aPKC result in higher rates of synthesis (Hernandez et al. 2003; Tsokas et al. 2005, 2007; Kelly et al. 2007; Westmark et al. 2010). We also assume that the doubly phosphorylated kinase (Kpp) has a significantly larger dwell time in synapses than does the singly phosphorylated molecule (Kp) (Makuch et al. 2011; Li and Gao 2014; Vogt-Eisele et al. 2014). Kpp is also very stable, as shown in overexpression studies with mutated forms of PKMζ with kidney/brain protein (KIBRA), which is primarily located in the synapses (Johannsen et al. 2008). Finally, there is feedback at the level of aPKC autophosphorylation, because kinases in the Kp and Kpp states may facilitate autophosphorylation of Kp under some conditions (Standaert et al. 1997, 2001; Standaert 1999).
Figure 1.

Model of a molecular switch based on bistable aPKC dynamics. (A) Schematic diagram illustrates that the (hybrid) model has two forms of self-regulation: autophosphorylation (blue dashed arrows) and nonlinear modulation of protein synthesis (green arrow). Kp and Kpp are singly and doubly phosphorylated kinase with molecular turnover times, τp and τpp. C1, C2 and C3 are complexes resulting from binding of Kp with Kp, Kp with Kpp, and Kpp with free phosphatase P. α and γα are synthesis rates of Kp during pre- and post-potentiation; ɛ and βt are control parameters for feedback and feedforward (trans)autophosphorylation; βc controls rate of (cis)autophosphorylation or external kinase activity. H is a Hill function of order 4 used for nonlinear modulation of protein synthesis. Red arrows in panels B and C indicate the induction protocol: 15 min of elevated synthesis rates in the range 2–3.5 units sec−1. (B) Time evolution of total amount of the kinase, KT, with or without induction for γ = 22 (gray) and 30 (black). U and D indicate potentiated synaptic state or L-LTP and naïve or prepotentiated synaptic state, respectively. Solid lines result from a weaker induction protocol than dashed lines. Circles indicate equilibria achieved over time. (C) Bifurcation diagram showing dependence of equilibria on γ. Blue and black curves are composed of equilibria of type D and U. Circles indicate particular cases in panel B. Red dashed curve represents threshold or unstable equilibria. Green-shaded box indicates region of bistability (“molecular switch”). (D) Equilibria in terms of total kinase, KT, with respect to control parameter, ɛ, of feedback autophosphorylation when activity-dependent protein synthesis and other forms of phosphorylation are eliminated, γ = 0 and βt,c = 0. In all panels, shaded regions indicate bistability, blue lines are D states, black lines are U states, and red dashed lines are thresholds for switching between the two states. (E) For the same parameters as in panel D, equilibria in terms of fraction of phosphorylation, Fphos, is plotted. When present, vertical orange bars in all panels indicate change due to stimulation. (F) Same as panel E except βt = 1 or βc = 0.4. (G) Same type of plot as panel D, but ɛ is fixed at a value below the shaded region in panel D and γ is varied. (H) Same type of plot as panel E but parameters are as in panel G. (I) Same as panel E except βt = 1 or βc = 0.4.
We model two cases, with and without aPKC autophosphorylation because empirical evidence exists for both. In vitro charectarization of PKCζ using rat adipocytes clearly indicates autophosphorylation, although the site of autophoshorylation is not directly verified in these studies (Standaert et al. 1997, 2001). Because aPKCs have two phosphorylation sites, and one of them is clearly a PDK1 site, it is possible that autophosphorylation occurs in the turn site. However, using mouse embryonic fibroblasts, another group has reported that PKCζ does not autophosphorylate, and that the turn site is phosphorylated by mTORC2 (Li and Gao 2014). If no autophosphorylation is assumed, one must make an additional assumption to account for all of the experimental observations above. In this case, simulated ZIP destabilizes aPKC by preventing its binding to a stabilizing protein (Vogt-Eisele et al. 2014). In both our model cases, and for values of model parameters consistent with empirical data, bistability and a stimulus-induced switch to persistent high aPKC levels result.
In the paper we show that such a model can account for the above experimental observations, both with and without autophosphorylation, under slightly different and biologically feasible assumptions. Additionally, we will examine a two-kinase model representing the two types of aPKCs and show that this model can account for recent seemingly inconsistent experimental results (Lee et al. 2013; Volk et al. 2013) regarding the importance of aPKCs for the maintenance of synaptic plasticity and memory.
Results
Model description and simulated bistability in aPKC level
A schematic diagram of the molecular species and reactions in our model is presented in Figure 1A. This model is based on observations about the molecular interactions of aPKCs and is localized to a single synaptic compartment. For a dynamical system to describe a bistable molecular switch between low (basal) and high (stimulated) kinase activity, it must be bistable and bistability requires an explicit or implicit feedback loop. The primary form of feedback we assume is at the level of translation. We choose to model this feedback with a Hill function (H), which models the synthesis of Kp as a nonlinear function of the concentration of Kpp (Fig. 1A, inset). In a naïve synapse the basal aPKC synthesis rate is α, but this rate is up-regulated by a factor of γ in a persistently potentiated synapse, which maintains high protein levels following induction, the mechanism for which is indirect in our simulations. Another possible form of feedback is through autophosphorylation of aPKC. Autophosphorylation of the turn site can be of the intermolecular form (trans), which can be divided into two subclasses: a singly phosphorylated molecule can phosphorylate another singly phosphorylated molecule (feedforward), or a doubly phosphorylated one can phosphorylate a singly phosphorylated one (feedback). Autophosphorylation can also be of the intramolecular form (cis), which is regulated in the model by the parameter βc. The turn site could alternatively be phosphorylated by another kinase, which we assume to have constant activity, and as a result, based on mathematical homology, is equivalently regulated by βc. Although these are the only explicit forms of positive feedback that we assume, there is another indirect form of positive feedback, differential rates of degradation (see Results on differential dwell time).
To mimic the induction of L-LTP and to validate that the proposed model could act as a molecular switch, aPKC synthesis was transiently elevated (Fig. 1B) for 15 min. Significant evidence shows that the induction of L-LTP requires translation of new protein locally at dendrites (Bradshaw et al. 2003; Tsokas et al. 2005; Sutton and Schuman 2006; Costa-Mattioli et al. 2009), and experimental protocols use time frames approximately consistent with that assumed in the model (Ling et al. 2002; Melemedjian et al. 2013). After induction of L-LTP, the total concentration of aPKCs increase, and the concentration of PKMζ remains at this elevated level (Osten et al. 1996; Kelly et al. 2007). Consistent with experiments, in our model (Fig. 1B) this induction also resulted in a permanent elevation of the total kinase, KT = (Kp + Kpp), from the basal (D) to the stimulated level (U). The concentration of KT at the U state depends on the protein synthesis feedback parameter, γ. The bifurcation diagram (Fig. 1C) shows that at low values of γ the system has a single equilibrium value. As γ is increased the system transitions abruptly to possessing three equilibrium values, the lower stable solution (D), the upper stable solution (U), and an unstable solution (dashed line). To induce L-LTP the concentration of the kinase needs to be elevated to above the unstable solution, the theoretical threshold for inducing L-LTP. The region with two stable solutions (the bistable region) is the region in which this network operates as a molecular switch.
Experiments have quantified increases, relative to the base levels, in aPKCs resulting from L-LTP (Kelly et al. 2007; Melemedjian et al. 2013); for our model to quantitatively match these results, we must take into account that only some synapses get potentiated after L-LTP. The empirical results are for hippocampal CA1 homogenates, which include both synapses that have and have not been potentiated, as well as nonsynaptic compartments. Translating the levels of D and U from the homogenate to the single synapse requires knowledge of what fraction of synapses were potentiated as a result of L-LTP, and what fraction of the homogenate arises from synaptic compartments. We currently lack the required parameters to quantitatively obtain D and U. However, given what we know about the small fraction of synapses that actually change their state due to L-LTP, it is reasonable to conclude that the single synapse U/D ratio (Supplemental Fig. S1) is much larger, possibly orders of magnitude larger than the ratio measured in the homogenate. Once the fraction of synapses potentiated as a result of L-LTP is determined, it would be possible to tune our model parameters to account for the actual U and D values as well. However, unlike previous models of a molecular switch that depend on post-translational modifications (Lisman 1985; Lisman and Zhabotinsky 2001; Miller et al. 2005), here the total kinase concentration (KT) is different in the U and D states, which is qualitatively consistent with experimental findings for aPKCs (Osten et al. 1996; Kelly et al. 2007; Melemedjian et al. 2013).
Two alternative pathways can maintain the “molecular switch” to regulate concentration and phosphorylation levels
The model in Figure 1 assumes two different feedback mechanisms, activation of translation and autophosphorylation, and we find that each of these pathways can generate bistability but with different degrees of robustness. A model with feedback only through (trans)autophosphorylation can be shown to be bistable (Fig. 1D–F) if we eliminate the activity-dependent protein synthesis completely (γ = 0). For a sufficiently high rate of (trans)autophosphorylation (ɛ ≈ 0.1 sec−1) bistability emerges, and if it is too large (ɛ ≈ 0.85 sec−1) the bistability is destroyed (Fig. 1D,E). Although for these parameters synthesis rate have not changed, experimental observation 1 can be met if τpp ≫ τp. For this case L-LTP results in a large increase in the fraction of doubly phosphorylated protein, Fphos = Kpp/(Kp + Kpp) (Fig. 1E). This large increase (≫10%), however, is inconsistent with experimental observation 2. The discrepancy with experimental observations can be addressed by increasing other forms of phosphorylation that are capable of raising Fphos in the basal level (Fig. 1F). When βt or βc is large enough L-LTP causes only a minimal increase in Fphos. However, the bistability region where this condition is upheld is small and the model therefore is not very robust even though valid (see Discussion). On the other hand, when bistability is generated by the alternate feedback mechanism, activity-dependent translation, the model is valid for a wide range of control parameter and is therefore very robust (Fig. 1G–I; Supplemental Figs. S1,S2). Similar to the previous model, reducing (trans)autophosphorylation feedback (ɛ < 0.1 sec−1) reveals bistability generated by activity-dependent synthesis (γ > 8). In this model, even when (τpp = τp), experimental observation 1 can be met, and if βt or βc are large enough one can account for observation 2 (Fig. 1I). In the rest of the paper, we use the hybrid model that includes both types of feedback pathways because it is more robust, and in addition exhibits tristability over some range of parameters, consistent with observations that after LTD induction the level of aPKC decreases below baseline (see below).
The differential effect of protein synthesis and kinase inhibitors depends on the different dwell times of singly and doubly phosphorylated protein
At first glance, it would seem that the effect of two types of inhibitors (experimental observation 3) should be similar, so why are the effects so different? A major cause of this difference is our empirically plausible assumption (Li and Gao 2014; Vogt-Eisele et al. 2014) that the synaptic dwell time of the doubly phosphorylated protein is much higher than that of the singly phosphorylated protein (τpp ≫ τp). Similar to empirical studies, eliminating synthesis in the model (α = γ → 0, PSI) prevents the induction of L-LTP (Fig. 2A) but does not reverse L-LTP when applied for the same duration (Fig. 2B). Because the L-LTP induction protocol we use is based on a transient increase in protein synthesis, clearly PSI should prevent the induction of L-LTP. However, when PSI is applied during maintenance it causes only an insignificant decay of total kinase levels (Fig. 2B); nearly all the protein is in the doubly phosphorylated state that decays very slowly at a rate determined by τpp, a time scale much longer than the duration of PSI application. In contrast, eliminating kinase-dependent pathways (ɛ = βt or c = γ → 0 or βc = γ → 0, ZIP) does not prevent the induction of L-LTP (Fig. 2C; Supplemental Fig. S4), but it can reverse previously established L-LTP (Fig. 2D). ZIP does not prevent the aforementioned transient increase in protein synthesis during induction, although it does shut down the feedback loops, which are essential for transitioning to U state. During induction in the presence of ZIP, the concentration of Kp increases as in the control, but once the short course of ZIP is finished the feedback loops are reactivated, making induction of L-LTP possible. Turning off autophosphorylation beyond induction causes the kinase to rapidly become dephosphorylated at the turn site, and the singly phosphorylated protein in this model degrades at a much faster rate. For the model variant in which the turn site was phosphorylated by an external kinase (Supplemental Fig. S4), ZIP (γ → 0 and τpp → τp) can also destabilize doubly phosphorylated kinase by preventing it from binding to a protective protein such as KIBRA (Vogt-Eisele et al. 2014). In either case, it is sufficient to apply ZIP on the order of τp (much shorter than τpp) in order to reverse L-LTP by the kinase inhibitor (Supplemental Fig. S5).
Figure 2.

Different time-dependent effects of kinase and protein synthesis inhibitors. (A) Gray line (control) indicates effect of induction without PSI; dark gray line (PSI outcome) indicates effect of induction with PSI. Arrows in all panels indicate stimulation as in Figure 1. Thick horizontal lines at the bottom indicate durations of inhibitor application in all panels. (B) Gray line (control) is previously induced U state, black dashed line (PSI outcome) indicates effect of PSI on previously potentiated state, and dark gray line (no induction) indicates D state. (C) Gray line as in panel B; black dashed line (ZIP outcome) is effect of induction in the presence of ZIP, which starts 1 h before the start of induction and lasts for 7.5 min from the start of induction. (D) Gray line (control) is previously potentiated synaptic state; black dashed line (ZIP outcome) is the effect of ZIP application on previously potentiated state.
In short, observation 3 is consistent with our model, given that the model assumes feedback at the level of protein synthesis, phosphorylation at the turn site, and that the synaptic dwell times of the singly and doubly phosphorylated kinases are very different. Consequently, the model predicts that if an aPKC-selective PSI could be applied for much longer periods of time L-LTP would be reversed (Fig. 3A; Supplemental Fig. S5). In Figure 3B, we show that the time required to reverse L-LTP depends on the time constant of the doubly phosphorylated kinase. Furthermore, due to ongoing phosphatase activity in the presence of ZIP, our model predicts that phosphatase inhibitors would slow the reversal time of L-LTP due to ZIP (Supplemental Fig. S6).
Figure 3.
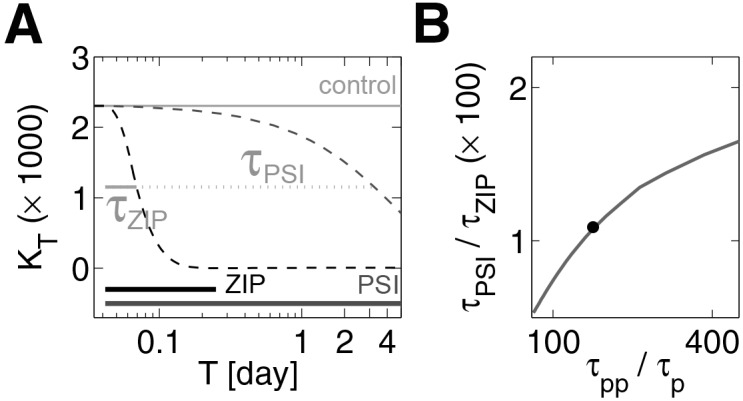
Both activity and protein synthesis inhibitors can reverse L-LTP but over different time scales. (A) Time evolution, on semi-log scale, of potentiated state with inhibitors. Gray line (control) indicates previously potentiated synaptic state. Blue dashed line (PSI outcome) indicates effect of PSI. Black dashed line (ZIP outcome) indicates effect of ZIP. Thick horizontal lines indicate durations of ZIP (black) and PSI (blue) application. Level of orange line indicates amount used to estimate exponential decay constants due to ZIP and PSI, τPSI (dotted plus solid line length) and τZIP (solid line length). (B) Ratio of decay times due to inhibitors plotted against synaptic lifetimes of each kinase form. Solid dot corresponds to the case in A.
Multistability can arise from the two parallel feedback mechanisms: implications for L-LTD
Many experimental results have shown that from baseline both L-LTP and L-LTD can be induced (Dudek and Bear 1992; Malenka and Bear 2004). This could be consistent with a binary synapse if at baseline some synapses are in the D state and others are in the U state. Although there are some indications that long-term plasticity might act by shifting synapses between two discrete states, there are also indications that synapses have a broad continuous distribution (Petersen et al. 1998; Feldmeyer et al. 2002; Montgomery and Madison 2004; O'Connor et al. 2005; Arellano et al. 2007; Enoki et al. 2009; Loewenstein et al. 2011). At the molecular level, experimental evidence indicates that the induction of L-LTD is followed by a decrease in the amount of PKMζ (Hrabetova and Sacktor 1996, 2001; Sajikumar and Frey 2003). In Figure 4, we show that it is possible to obtain a synapse with three stable fixed points (tristable), using the same model described above but in a different parameter range. For these parameters the model exhibits asymmetric transitions for potentiation and depression (Fig. 4A) consistent with experiments (Hrabetova and Sacktor 1996, 2001; Kelly et al. 2007). Such tristability requires that both feedback loops (based on autoactivation of aPKC synthesis, and on autophosphorylation) are operative. In Figure 4A, we show the dynamics resulting from our induction protocols and the application of ZIP. From the lowest state (L-LTD), a weak induction leads to an intermediate state (basal) because activation of autophosphorylation requires relatively low amount of new protein than the activation of the synthesis-based feedback in our model. Further stimulation by a strong pulse led to the highest state (L-LTP). Following induction, application of ZIP with sufficient duration led to reversal of each state because the stable form, Kpp, is lost due to ongoing phosphatase activity in the presence of ZIP. The existence of tristability was confirmed using bifurcation analysis (Fig. 4B), which identified steady states, both stable (solid lines) and unstable (dashed lines). L-LTD was revealed when ZIP lowered the total kinase below the levels (below red dashed lines in Fig. 4B) required for sustaining either of the feedback pathways. This model variant therefore accounts, at a single synapse level, for bidirectional plasticity from baseline.
Figure 4.
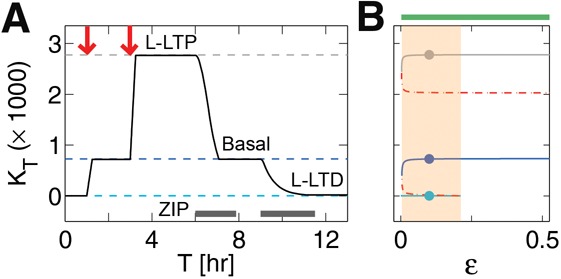
Two independent feedback loops, each of which can sustain bistability, can combine to give multistability. (A) Evolution in time with two 15-min long elevations of aPKC synthesis (red arrows) starting from low total kinase, KT, elicited three persistent states: L-LTP (gray dashed), basal (blue dashed), and L-LTD (cyan dashed). Simulated ZIP could both reverse L-LTP and induce L-LTD, depending on the initial concentration as well as on the concentration and duration. (B) Bifurcation diagram with respect to autophosphorylation rate, ɛ, shows this tristability is robust to ∼20% variation (shaded yellow) in the autophophorylation parameter. Solid lines are stable equilibriums resistant to changes in time; dashed lines are unstable equilibriums or thresholds.
A “molecular switch” resulting from the interaction between two kinases
Two types of aPKCs coexist in synapses, PKMζ and PKCι/λ. Both kinases might be involved in synaptic plasticity (Kelly et al. 2007; Sacktor 2010; Frankland and Josselyn 2013; Glanzman 2013; Matt and Hell 2013; Ren et al. 2013), and are likely to cross-phosphorylate. Recently, one of these kinases, PKMζ, was eliminated by genetic means, either constitutively or conditionally (Ling et al. 2002; Kelly et al. 2007; Lee et al. 2013; Volk et al. 2013). In these experiments memory and synaptic plasticity were claimed to be normal, contradicting an exclusive role in long-term plasticity and memory for PKMζ. Furthermore, in these knockout experiments plasticity and memory were still sensitive to ZIP. In these animals PKCι/λ, also a target of ZIP (Frankland and Josselyn 2013; Ren et al. 2013; Dong et al. 2014), was present. We postulate that the redundancy between these two kinases and possible compensation might account for the seeming inconsistency between these recent experiments and the plethora of previous experimental results.
In order to demonstrate how two similar kinases can compensate, we examined a model composed of two kinases and find significant change in their individual behavior is possible due to interactions. We chose a hybrid model to describe each kinase independently but the autophosphorylation pathway in each can be replaced with that of external kinase with corresponding parameter adjustments. The latter kinase denoted L was in general assumed to be less efficient than kinase K (Fig. 5A). Turnover rate for kinase L was also assumed to be faster than that of kinase K, similar to what was found for PKCs compared with PKMs (Le Good and Brindley 2004). In the two-kinase model, Kp and Kpp could bind Lp and Lpp, leading to phosphorylation of both the singly phosphorylated species. Both kinase species dephosphorylate by binding to a single species of phosphatase. The competition for a single phosphatase is effectively a negative feedback. In the presence of K, we find L stabilized post-stimulus (Fig. 5B), which could explain why Kelly et al. (2007) found persistent increase in both of the aPKCs in the wild-type animals. The time evolution of kinase L after stimulation, in Figure 5C, shows decline when kinase K is eliminated unless hand tuned translational compensation is allowed (Fig. 5D). The bifurcation diagram, in Figure 5E, shows that an increase in the protein synthesis rate of PKCι/λ converts the kinase L-only network from a monostable to a bistable system (Supplemental Figs. S7,S8). This implies normal L-LTP could arise in PKMζ knockout animals from up-regulation of PKCι/λ synthesis (P Tsokas, C Hsieh, Y Yao, EJC Wallace, P Serrano, L Pang, A Tcherepanov, D Jothianandan, D Tian, BR Hartley, et al. in prep.).
Figure 5.
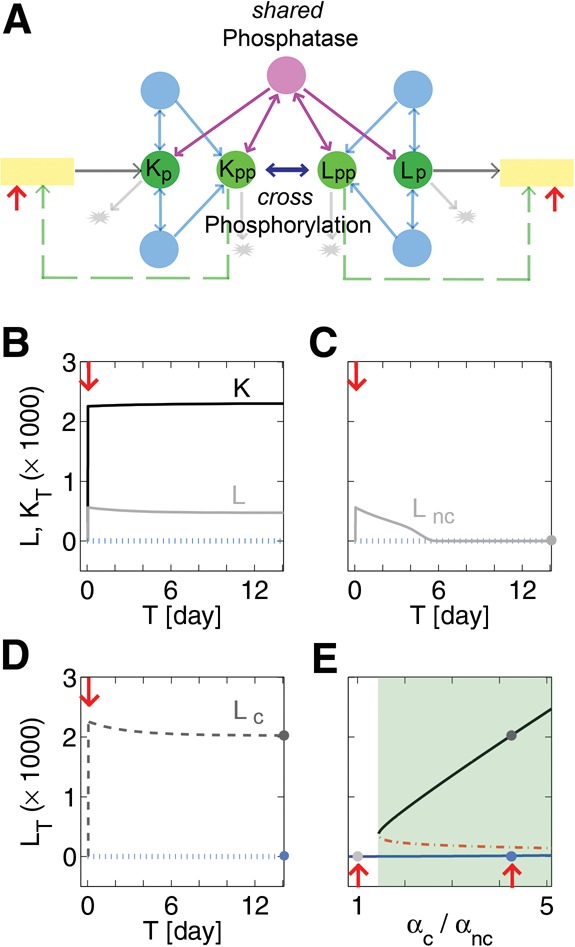
Two-kinase model with or without compensation. (A) Schematic diagram of the model. For simplicity each molecular species is color-coded as Figure 1A and additional complexes are not visible; the two forms of interactions between the kinases are symbolically shown. Details are in Materials and Methods. (B) Black curve labeled K is the potentiated level for the stronger kinase, which has higher levels of activity-dependent synthesis and autophosphorylation, than does the weaker kinase L (for which the lower gray curve indicates its potentiated level). Blue dotted line indicates virtually identical amounts of K and L in the naïve state. Red arrows in all panels indicate induction as mentioned previously. Circles indicate equilibrium achieved over time in all panels. (C) Gray line indicates kinase L that receives the same induction as in panel A but in the absence of kinase K. Blue dotted line indicates level of L in the naïve state. (D) Dark gray dashed line indicates kinase L after potentiation when compensation in the form of increased basal synthesis of L is taken into account. Blue dotted line indicates naïve state. (E) Compensatory rate of basal synthesis of L, αc, is varied compared with the noncompensatory rate αnc. Blue curve is composed of naïve states, dark gray curve is composed of potentiated states, and red dashed curve is the threshold. Circles indicate cases in panels C and D.
Discussion
A central challenge in understanding the synaptic basis of learning and memory is the problem of maintenance—how can memory be preserved for periods of time that are orders of magnitude larger than the lifetime of the proteins encoding these memories (Crick 1984)? Although this problem seems to be similar to the problem of cell differentiation, it differs in that synaptic plasticity subserving learning and memory must be synapse-specific. Synapse-specificity has two consequences: first, what limits information storage is not the lifetime of proteins in the cell, but the dwell time of proteins in the synapse; second, whole-cell mechanisms such as regulation of transcription are not appropriate solutions, however, see Chen et al. (2014). Over the last several decades, the contribution of many molecular pathways to maintenance has been studied (Sanes and Lichtman 1999; Bosch et al. 2014). By far, the most comprehensive evidence points to the dependence of maintenance on aPKCs (Osten et al. 1996; Ling et al. 2002; Serrano et al. 2005; Kelly et al. 2007; Sacktor 2010; Hsieh et al. 2013). These findings have been challenged recently (Lee et al. 2013; Volk et al. 2013), but we show that these new results can still be consistent with a major role for aPKCs. However, knowing that aPKCs are a major component in the mechanisms of maintenance does not explain how aPKCs can overcome the problem of limited protein dwell times.
Here we propose a model that explains how aPKCs may not only account for maintenance on a general conceptual level, but can account specifically for a large set of experimental results. We explore a hybrid model, in which feedback pathways at both the translational and post-translational levels are assumed, and additionally, we assume that the doubly phosphorylated form of aPKC has a significantly larger synaptic dwell time than the single phosphorylated form. With this set of assumptions, our model can account for protein synthesis dependence, associated phosphorylation levels and effects of different inhibitors on L-LTP—in short, a comprehensive set of experimental results.
Evidence for different assumptions
Apart from demonstrating that the consequences of our molecular model are consistent with the physiological experimental observations, we must show that our assumptions are indeed consistent with experimental results at the molecular level. Obviously, we do not have sufficient evidence for proof, but there is extensive experimental evidence that supports our assumptions.
Control of translation: Induction of L-LTP causes an increase in the level of PKMζ, but when PKMζ is inhibited this increase is substantially smaller, consistent with the notion that PKMζ activity affects its own translation (Kelly et al. 2007). Further, experimental evidence indicates that such feedback at the level of translation is at least partially mediated by Pin1 (Westmark et al. 2010).
Autophosphorylation/Phosphorylation: Atypical PKCs are kinases with at least two phosphorylation sites. In biochemical assays, purified PKMζ undergoes phosphorylation implicating autophosphorylation (Sacktor et al. 1993). PKCζ (Standaert et al. 1997) and PKCι/λ (Akimoto et al. 1994) have also been observed to autophosphorylate despite their regulatory domains. PKCι/λ may be found in similar quantities to PKMζ (Melemedjian et al. 2013), and the phosphorylation of its autophosphorylation site increases by a lower percentage than that of PKMζ (Kelly et al. 2007), suggesting a slower autophosphorylation process. In addition, we considered alternate pathways for phosphorylation such as mTORC2 phosphorylation of the turn site, as has been proposed (Ikenoue et al. 2008; Li and Gao 2014).
Different synaptic dwell times: In general, molecular turnover can depend on phosphorylation states as demonstrated for typical forms of PKC (Le Good and Brindley 2004). For PKMζ, complex formation with KIBRA (Vogt-Eisele et al. 2014) or PKCι/λ with p62 (Ren et al. 2013) can stabilize aPKCs in the synaptic compartment and protects against their degradation by the proteasome.
Difference from previous implementations of a molecular switch
We find that different synaptic dwell times together with only autophosphorylation feedback can account for a concentration increase (Supplemental Fig. S9), unlike most previous molecular models of a synapse that are based on post-translational modifications (Crick 1984; Lisman 1985; Miller and Kennedy 1986; Lisman and Goldring 1988; Lisman and Fallon 1999; Lisman and Zhabotinsky 2001; Miller et al. 2005). Alternative to these models are trafficking based models (Hayer and Bhalla 2005; Shouval 2005), which can explain an increase in local concentrations of specific proteins in a synaptic compartment, but not the experimentally observed increase in total hippocampal tissue. To explain the latter increase, the concept of feedback at the level of translation has been proposed (Lisman and Fallon 1999; Richter and Klann 2009) and implemented in models (Hayer and Bhalla 2005; Aslam et al. 2009). But in most of these studies, the modeled molecule is assumed to be CaMKII, the structural role of which remains to be tested (Sanhueza et al. 2011). Previous models of PKMζ also cannot account for the set of experimental results we consider (Clopath et al. 2008; Ogasawara and Kawato 2010; Zhang et al. 2010; Smolen et al. 2012).
In comparison with other models, which are either very molecularly complex or exceedingly simple, ours falls in the intermediate level such that it can account for specific experimental results and can be experimentally tested. Because late-LTP and memory storage involve many complex processes and a large variety of biomolecules (Sanes and Lichtman 1999; Citri and Malenka 2008), one could model as many of the complete signal transduction pathway related to LTP and LTD as possible (Bhalla and Iyengar 1999; Hayer and Bhalla 2005). Analyzing such complex models mathematically is, however, very difficult and even if the model can account for some experimental data it is often difficult to understand how the model accomplishes this. In contrast, modeling specific pathways implicitly make models mathematically very simple and insightful but they cannot be experimentally tested. Here we constructed an intermediate level model with only a few key proteins and used a hybrid of translational and post-translational feedbacks. Under some conditions this model can be multistable due to two feedback loops, also commonly found in other molecular switch models in biology (Macía et al. 2009; Thomson and Gunawardena 2009; Tyson and Novák 2010; Rodrigo et al. 2011; Feng and Wang 2012; Lu et al. 2013), and, more specifically, of L-LTP (Hayer and Bhalla 2005). Finally, unlike translation-only models, which consider only a single state of a kinase (Supplemental Figs. S10,S11), the hybrid model has two states due to post-translational modification. We use these states to implement a differential degradation rate hypothesis, which helps us to account for a number of key experimental observations and predict several testable experiments.
Redundancy and compensation can account for PKMζ knockout experiments
Recent experimental results have cast doubt on the basic idea that PKMζ is critical for maintenance of L-LTP and long-term memory. In two recent experiments, conventional and conditional knockout animals have been found to have seemingly normal L-LTP (Volk et al. 2013), and conventional knockout animals displayed normal memory (Lee et al. 2013; Volk et al. 2013). These results stand in apparent contradiction to a plethora of previous experimental results using ZIP and other inhibitors, dominant negative mutations, and overexpression strategies (Drier et al. 2002; Ling et al. 2002; Serrano et al. 2005; Shema et al. 2007, 2009; Migues et al. 2010; Cai et al. 2011; Barry et al. 2012; Ho et al. 2012; Kwapis et al. 2012). Strikingly, ZIP was still able to reverse LTP and erase long-term memory in the constitutive knockout.
However, PKMζ is not the only aPKC that can be inhibited by ZIP. We postulated that the redundancy in aPKC proteins might be able to address the apparent discrepancy between the different experimental results. Therefore, we extended our single kinase model to a two-kinase model with two similar but nonidentical aPKCs, which could interact with each other (Fig. 5). We show that if compensation occurs (Tsokas 2013; Yao et al. 2013b), such that concentration of the remaining kinase type increases via synthesis up-regulation (equivalent to downregulation of degradation) when the other is knocked out, then the two-kinase model can account for the new experimental results and the apparent discrepancy between different experiments.
Testing model predictions
The model was designed to account for a set of key experimental findings and the assumptions used were restricted to those consistent with experimental findings when available. Apart from accounting for existing experimental results, there are many experiments that could be carried out to test both the predictions and assumptions of the model. Readily testable experiments include:
Long-term effect of PSI: Specific inhibition of PKMζ synthesis should be able to reverse L-LTP in wild-type animals, if applied over a sufficiently long period of time (Fig. 3), provided there is no compensatory up-regulation of PKCι/λ synthesis in this time frame.
Impact of phosphatase inhibitors: Our model predicts that in order to account for the rate of L-LTP reversal with kinase inhibitors, the dephosphorylation of aPKC at the turn site or on PKMζ-stabilizing substrates such as KIBRA should occur in the order of ∼1 h. Consequently, the inhibition of dephosphorylation, via an appropriate phosphatase inhibitor, should slow down the reversal of L-LTP and memory due to application of protein kinase inhibitors such as ZIP (Supplemental Fig. S6).
Compensation: We predict that in PKMζ knockout animals in which L-LTP and long-term memory are preserved there is compensation, at the level of hippocampal neurons, due to increased expression of PKCι/λ.
These experiments are quite feasible and if they yield inconsistent results, the model in its current form will be invalidated. To summarize, we have proposed a comprehensive model that accounts for the major experimental observations in the field of LTP maintenance, and how redundancy and compensation can account for the recent results that seem to cast doubt on the role of aPKCs.
Materials and Methods
Biochemical reactions
Reaction kinetics for kinase K that interacts with kinase L within the two-kinase model, are presented below. Kinase L has analogous kinetics with different parameters. Kp and Lp are singly phosphorylated, and Kpp and Lpp are doubly phosphorylated. Complexes are denoted by C with superscripts K or L indicating the particular kinase forming them; subscripts 1 and 2 indicate feedforward and feedback autophosphorylation, ×1 and ×2 indicate cross-phosphorylations, 3 indicates bound phosphatase, P. For the single kinase model, terms involving L were omitted. Parameter values for the simulations in Figures 1–5 are given in Tables 1 and 2. Matlab code for all figures can be found in the model database at http://www.ebi.ac.uk/biomodels-main/MODEL1505130000 and http://www.ebi.ac.uk/biomodels-main/MODEL1505130001 (see Chelliah et al. 2015).
Table 1.

Table 2.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
Simulation of induction protocol
Induction protocol was simulated by 15 min of an elevated synthesis rate of aPKC, where strong inductions were 2 (Fig. 1B, gray), 2.5 (Fig. 4A), or 3.5 (Fig. 1B, black) units sec−1 and weak induction was 0.1 units sec−1 (Fig. 4A).
Simulation of dynamics and steady states
Reaction kinetics were converted to a system of ordinary differential equations (ODEs), which were solved using Matlab routine ode15s. Equilibriums or steady states of single kinase models were computed based on mathematical reduction of the full system to a one-dimensional equation (nullcline), which was numerically solved using the Matlab command fzero. Aside from long-term simulation indicating stability of the persistent states, mathematical evaluation using eigenvalues of the Jacobian of the full system was used to verify stability of states that appear persistent in the time evolution of solutions. Finding of steady states analytically for the two-kinase model is shown in the Supplemental Material.
Supplementary Material
Acknowledgments
This work is supported by NIH funding RO1 DA034979 (H.Z.S., T.C.S.), RO1 MH53576 (T.C.S.), 2R37 MH057068 (T.C.S.), and the LightFighter Trust (T.C.S.).
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.038844.115.
References
- Abraham WC, Williams JM. 2008. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem 89: 260–268. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Logan B, Greenwood JM, Dragunow M. 2002. Induction and experience-dependent consolidation of stable long-term potentiation lasting months in the hippocampus. J Neurosci 22: 9626–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto K, Mizuno K, Osada S, Hirai S, Tanuma S, Suzuki K, Ohno S. 1994. A new member of the third class in the protein kinase C family, PKC λ, expressed dominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. J Biol Chem 269: 12677–12683. [PubMed] [Google Scholar]
- Arellano JI, Benavides-Piccione R, DeFelipe J, Yuste R. 2007. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci 1: 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam N, Kubota Y, Wells D, Shouval HZ. 2009. Translational switch for long-term maintenance of synaptic plasticity. Mol Syst Biol 5: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Bailey CH, Kandel ER. 2006. Common molecular mechanisms in explicit and implicit memory. J Neurochem 97: 1520–1533. [DOI] [PubMed] [Google Scholar]
- Barry JM, Rivard B, Fox SE, Fenton AA, Sacktor TC, Muller RU. 2012. Inhibition of protein kinase Mζ disrupts the stable spatial discharge of hippocampal place cells in a familiar environment. J Neurosci 32: 13753–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batish M, van den Bogaard P, Kramer FR, Tyagi S. 2012. Neuronal mRNAs travel singly into dendrites. Proc Natl Acad Sci 109: 4645–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. 1994. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4: 389–399. [DOI] [PubMed] [Google Scholar]
- Belelovsky K, Elkobi A, Kaphzan H, Nairn AC, Rosenblum K. 2005. A molecular switch for translational control in taste memory consolidation. Eur J Neurosci 22: 2560–2568. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. 1999. Emergent properties of networks of biological signaling pathways. Science 283: 381–387. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lømo T. 1973. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232: 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y. 2014. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82: 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw KD, Emptage NJ, Bliss TVP. 2003. A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci 18: 3150–3152. [DOI] [PubMed] [Google Scholar]
- Cai D, Pearce K, Chen S, Glanzman DL. 2011. Protein kinase M maintains long-term sensitization and long-term facilitation in aplysia. J Neurosci 31: 6421–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelliah V, Juty N, Ajmera I, Ali R, Dumousseau M, Glont M, Hucka M, Jalowicki G, Keating S, Knight-Schrijver V, et al. 2014. BioModels: ten-year anniversary. Nucl Acids Res 43: D542–D548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Cai D, Pearce K, Sun PY, Roberts AC, Glanzman DL. 2014. Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. Elife 3: e03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC. 2008. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33: 18–41. [DOI] [PubMed] [Google Scholar]
- Clopath C, Ziegler L, Vasilaki E, Büsing L, Gerstner W. 2008. Tag-trigger-consolidation: a model of early and late long-term-potentiation and depression. PLoS Comput Biol 4: e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. 2009. Translational control of long-lasting synaptic plasticity and memory. Neuron 61: 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JA, Stöckl P, Ueberall F, Jenny M, Saria A, Zernig G. 2012. Activation of PKCζ and PKMζ in the nucleus accumbens core is necessary for the retrieval, consolidation and reconsolidation of drug memory. PLoS One 7: e30502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. 1984. Memory and molecular turnover. Nature 312: 101. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. 2011. Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiol Learn Mem 96: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, Peng Y, Zhou L, He W, Wu X, et al. 2014. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest 125: 234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drier EA, Tello MK, Cowan M, Wu P, Blace N, Sacktor TC, Yin JCP. 2002. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat Neurosci 5: 316–324. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. 1992. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci 89: 4363–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoki R, Hu Y, Hamilton D, Fine A. 2009. Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: optical quantal analysis. Neuron 62: 242–253. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Lübke J, Silver RA, Sakmann B. 2002. Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signalling within a cortical column. J Physiol 538: 803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Wang J. 2012. A new mechanism of stem cell differentiation through slow binding/unbinding of regulators to genes. Sci Rep 2: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner LB, Flexner JB, Stellar E. 1965. Memory and cerebral protein synthesis in mice as affected by graded amounts of puromycin. Exp Neurol 13: 264–272. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Bonhoeffer T. 2006. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci 9: 478–480. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Josselyn SA. 2013. Neuroscience: memory and the single molecule. Nature 493: 312–313. [DOI] [PubMed] [Google Scholar]
- Glanzman DL. 2013. PKM and the maintenance of memory. F1000 Biol Rep 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer A, Bhalla US. 2005. Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Comput Biol 1: e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AI, Blace N, Crary JF, Serrano PA, Leitges M, Libien JM, Weinstein G, Tcherapanov A, Sacktor TC. 2003. Protein kinase Mζ synthesis from a brain mRNA encoding an independent protein kinase Cζ catalytic domain. Implications for the molecular mechanism of memory. J Biol Chem 278: 40305–40316. [DOI] [PubMed] [Google Scholar]
- Ho S-Y, Chen C-H, Liu T-H, Chang H-F, Liou J-C. 2012. Protein kinase Mζ is necessary for cocaine-induced synaptic potentiation in the ventral tegmental area. Biol Psychiatry 71: 706–713. [DOI] [PubMed] [Google Scholar]
- Hrabetova S, Sacktor TC. 1996. Bidirectional regulation of protein kinase Mζ in the maintenance of long-term potentiation and long-term depression. J Neurosci 16: 5324–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetova S, Sacktor TC. 2001. Transient translocation of conventional protein kinase C isoforms and persistent downregulation of atypical protein kinase Mζ in long-term depression. Mol Brain Res 95: 146–152. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Tsokas P, Hartley BR, Serrano PA, Fenton AA, Sacktor TC. 2013. Persistent increases of PKMz correlate with the retention and duration of spatial long term memory. Soc Neurosci Abstr 233: H1. [Google Scholar]
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan K-L. 2008. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 27: 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsen S, Duning K, Pavenstädt H, Kremerskothen J, Boeckers TM. 2008. Temporal-spatial expression and novel biochemical properties of the memory-related protein KIBRA. Neuroscience 155: 1165–1173. [DOI] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC. 2007. Regulation of protein kinase M synthesis by multiple kinases in long-term potentiation. J Neurosci 27: 3439–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Jarome TJ, Gilmartin MR, Helmstetter FJ. 2012. Intra-amygdala infusion of the protein kinase Mζ inhibitor ZIP disrupts foreground context fear memory. Neurobiol Learn Mem 98: 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal G, Comprido D, Duarte CB. 2014. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 76: 639–656. [DOI] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. 2013. Prkcz null mice show normal learning and memory. Nature 493: 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good J, Brindley D. 2004. Molecular mechanisms regulating protein kinase Cζ turnover and cellular transformation. Biochem J 378: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gao T. 2014. mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep 15: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-Y, Ko H-G, Chen T, Collingridge GL, Kaang B-K, Zhuo M. 2011. Erasing injury-related cortical synaptic potentiation as a new treatment for chronic pain. J Mol Med 89: 847–855. [DOI] [PubMed] [Google Scholar]
- Ling DSF, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. 2002. Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat Neurosci 5: 295–296. [DOI] [PubMed] [Google Scholar]
- Lisman JE. 1985. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci 82: 3055–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Fallon JR. 1999. What maintains memories? Science 285: 339–340. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Goldring MA. 1988. Feasibility of long-term storage of graded information by the Ca2+/calmodulin-dependent protein kinase molecules of the postsynaptic density. Proc Natl Acad Sci 85: 5320–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. 2001. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31: 191–201. [DOI] [PubMed] [Google Scholar]
- Loewenstein Y, Kuras A, Rumpel S. 2011. Multiplicative dynamics underlie the emergence of the log-normal distribution of spine sizes in the neocortex in vivo. J Neurosci 31: 9481–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Jolly MK, Gomoto R, Huang B, Onuchic J, Ben-Jacob E. 2013. Tristability in cancer-associated microRNA-TF chimera toggle switch. J Phys Chem B 117: 13164–13174. [DOI] [PubMed] [Google Scholar]
- Macía J, Widder S, Solé R. 2009. Why are cellular switches Boolean? General conditions for multistable genetic circuits. J Theor Biol 261: 126–135. [DOI] [PubMed] [Google Scholar]
- Makuch L, Volk L, Anggono V, Johnson RC, Yu Y, Duning K, Kremerskothen J, Xia J, Takamiya K, Huganir RL. 2011. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron 71: 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. 2004. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M. 2007. Factors critical for the plasticity of dendritic spines and memory storage. Neurosci Res 57: 1–9. [DOI] [PubMed] [Google Scholar]
- Matt L, Hell JW. 2013. PKCλ: a new player in LTP coming to the rescue of PKCζ’s faltering role in LTP? EMBO J 32: 1348–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh J. 1966. Time-dependent procesesses in memory storage. Science 153: 1351–1358. [DOI] [PubMed] [Google Scholar]
- Melemedjian OK, Tillu DV, Asiedu MN, Mandell EK, Moy JK, Blute VM, Taylor CJ, Ghosh S, Price TJ. 2013. BDNF regulates atypical PKC at spinal synapses to initiate and maintain a centralized chronic pain state. Mol Pain 9: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. 2010. PKMζ maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci 13: 630–634. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. 1986. Regulation of brain type II Ca2+ /calmodulin- dependent protein kinase by autophosphorylation: a Ca2+ -triggered molecular switch. Cell 44: 861–870. [DOI] [PubMed] [Google Scholar]
- Miller P, Zhabotinsky AM, Lisman JE, Wang X-J. 2005. The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS Biol 3: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV. 2004. Discrete synaptic states define a major mechanism of synapse plasticity. Trends Neurosci 27: 744–750. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. 2014. Engineering a memory with LTD and LTP. Nature 511: 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DH, Wittenberg GM, Wang SS-H. 2005. Graded bidirectional synaptic plasticity is composed of switch-like unitary events. Proc Natl Acad Sci 102: 9679–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara H, Kawato M. 2010. The protein kinase Mζ network as a bistable switch to store neuronal memory. BMC Syst Biol 4: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Valsamis L, Harris A, Sacktor TC. 1996. Protein synthesis-dependent formation of protein kinase Mζ in long-term potentiation. J Neurosci 16: 2444–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Marshall CJ, Tate WP, Goddard GV, Abraham WC. 1989. Maintenance of long-term potentiation in rat dentate gyrus requires protein synthesis but not messenger RNA synthesis immediately post-tetanization. Neuroscience 28: 519–526. [DOI] [PubMed] [Google Scholar]
- Pastalkova E. 2006. Storage of spatial information by the maintenance mechanism of LTP. Science 313: 1141–1144. [DOI] [PubMed] [Google Scholar]
- Petersen CC, Malenka RC, Nicoll RA, Hopfield JJ. 1998. All-or-none potentiation at CA3-CA1 synapses. Proc Natl Acad Sci 95: 4732–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S-Q, Yan J-Z, Zhang X-Y, Bu Y-F, Pan W-W, Yao W, Tian T, Lu W. 2013. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J 32: 1365–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Klann E. 2009. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev 23: 1–11. [DOI] [PubMed] [Google Scholar]
- Rodrigo G, Carrera J, Jaramillo A. 2011. Computational design of synthetic regulatory networks from a genetic library to characterize the designability of dynamical behaviors. Nucleic Acids Res 39: e138–e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtenberg A, Rekart JL. 2005. Post-translational protein modification as the substrate for long-lasting memory. Trends Neurosci 28: 12–19. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. 2010. How does PKMζ maintain long-term memory? Nat Rev Neurosci 12: 9–15. [DOI] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik M, Sublette E. 1993. Persistent activation of the ζ isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci 90: 8342–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU. 2003. Anisomycin inhibits the late maintenance of long-term depression in rat hippocampal slices in vitro. Neurosci Lett 338: 147–150. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. 1999. Can molecules explain long-term potentiation? Nat Neurosci 2: 597–604. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. 2011. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci 31: 9170–9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AR, Comprido D, Duarte CB. 2010. Regulation of local translation at the synapse by BDNF. Prog Neurobiol 92: 505–516. [DOI] [PubMed] [Google Scholar]
- Serrano P, Yao Y, Sacktor TC. 2005. Persistent phosphorylation by protein kinase M maintains late-phase long-term potentiation. J Neurosci 25: 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, et al. 2008. PKMζ maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol 6: e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabashov D, Shohami E, Yaka R. 2011. Inactivation of PKMζ in the NAc shell abolished cocaine-conditioned reward. J Mol Neurosci 47: 546–553. [DOI] [PubMed] [Google Scholar]
- Shao CY, Sondhi R, van de Nes PS, Sacktor TC. 2012. PKMζ is necessary and sufficient for synaptic clustering of PSD-95. Hippocampus 22: 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Yadin D. 2007. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKMζ. Science 317: 951–953. [DOI] [PubMed] [Google Scholar]
- Shema R, Hazvi S, Sacktor TC, Dudai Y. 2009. Boundary conditions for the maintenance of memory by PKM in neocortex. Learn Mem 16: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ. 2005. Clusters of interacting receptors can stabilize synaptic efficacies. Proc Natl Acad Sci 102: 14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen P, Baxter DA, Byrne JH. 2012. Molecular constraints on synaptic tagging and maintenance of long-term potentiation: a predictive model ed. A. Morrison. PLoS Comput Biol 8: e1002620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert ML. 1999. Insulin activates protein kinases C-ζ and C-λ by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem 274: 25308–25316. [DOI] [PubMed] [Google Scholar]
- Standaert ML, Galloway L, Karnam P, Bandyopadhyay G, Moscat J, Farese RV. 1997. Protein kinase C- as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem 272: 30075–30082. [DOI] [PubMed] [Google Scholar]
- Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. 2001. Insulin and PIP3 activate PKC-ζ by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry 40: 249–255. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. 2006. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: 49–58. [DOI] [PubMed] [Google Scholar]
- Thomson M, Gunawardena J. 2009. Unlimited multistability in multisite phosphorylation systems. Nature 460: 274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P. 2013. Conditional knockout of the PKC/PKMz gene and acute translational blockade by PKMz antisense show new synthesis of PKMz protein is crucial for LTP and long-term memory. Soc Neurosci Abstr 233: G59. [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. 2005. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci 25: 5833–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. 2007. Mitogen-activated protein kinase up-regulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci 27: 5885–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Novák B. 2010. Functional motifs in biochemical reaction networks. Annu Rev Phys Chem 61: 219–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 15: 221–231. [DOI] [PubMed] [Google Scholar]
- Verdugo A, Vinod PK, Tyson JJ, Novak B. 2013. Molecular mechanisms creating bistable switches at cell cycle transitions. Open Biol 3: 120179–120179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt-Eisele A, Krüger C, Duning K, Weber D, Spoelgen R, Pitzer C, Plaas C, Eisenhardt G, Meyer A, Vogt G, et al. 2014. KIBRA (KIdney/BRAin protein) regulates learning and memory and stabilizes protein kinase Mζ. J Neurochem 128: 686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. 2013. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493: 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark P, Westmark C, Wang S, Levenson J, O'Riordan K, Burger C, Malter J. 2010. Pin1 and PKMζ sequentially control dendritic protein synthesis. Sci Signal 3: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR. 2006. Learning induces long-term potentiation in the hippocampus. Science 313: 1093–1097. [DOI] [PubMed] [Google Scholar]
- Yao Y, Shao C, Jothianandan D, Tcherepanov A, Shouval H, Sacktor TC. 2013a. Matching biochemical and functional efficacies confirm ZIP as a potent competitive inhibitor of PKMζ in neurons. Neuropharmacology 64: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsokas P, Jothianandan D, Tcherepanov A, van de Nes P, Sacktor TC. 2013b. Compensation by PKCi/l activation and PKMi/l formation in PKMz knock-out mice. Soc Neurosci Abstr 233: G60. [Google Scholar]
- Zhang Y, Smolen P, Baxter DA, Byrne JH. 2010. The sensitivity of memory consolidation and reconsolidation to inhibitors of protein synthesis and kinases: computational analysis. Learn Mem 17: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. 2013. Epigenetic regulation of memory formation and maintenance. Learn Mem 20: 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.