

Abstract
Memory deficits are common among stroke survivors. Identifying neuroprotective agents that can prevent memory impairment or improve memory recovery is a vital area of research. Glycogen synthase kinase-3β (GSK-3β) is involved in several essential intracellular signaling pathways. Unlike many other kinases, GSK-3β is active only when dephosphorylated and activation promotes inflammation and apoptosis. In contrast, increased phosphorylation leads to reduced GSK-3β (pGSK-3β) activity. GSK-3β inhibition has beneficial effects on memory in other disease models. GSK-3β regulates both the 5′AMP-activated kinase (AMPK) and transforming growth factor-β-activated kinase (TAK1) pathways. In this work, we examined the effect of GSK-3β inhibition, both independently, in conjunction with a TAK inhibitor, and in AMPK-α2 deficient mice, after stroke to investigate mechanistic interactions between these pathways. GSK-3β inhibition was neuroprotective and ameliorated stroke-induced cognitive impairments. This was independent of AMPK signaling as the protective effects of GSK-3β inhibition were seen in AMPK deficient mice. However, GSK-3β inhibition provided no additive protection in mice treated with a TAK inhibitor suggesting that TAK1 is an upstream regulator of GSK-3β. Targeting GSK-3β could be a novel therapeutic strategy for post-stroke cognitive deficits.
Stroke is the primary cause of long-term adult disability and the fourth leading cause of death in the USA (Feigin et al. 2003; Lloyd-Jones et al. 2010; Vaartjes et al. 2013). Ischemic strokes accounts for 80%–85% of all strokes (Go et al. 2014). Despite the global burden of stroke, only one FDA-approved therapy is available to treat ischemic stroke patients, the thrombolytic tissue plasminogen activator (Ziegler et al. 2005). tPA can only be used in a small percentage of patients due to its short therapeutic time window and numerous contraindications (Ziegler et al. 2005). As our population ages the prevalence and incidence of cerebrovascular disease will continue to increase (Lloyd-Jones et al. 2010; Vaartjes et al. 2013), as will the number of individuals with post-stroke cognitive deficits. While hospital costs account for three-fourths of total stroke care costs, the cost of long-term chronic care is a major economic concern. Stroke survivors with physical or cognitive impairments often need community-based care or nursing home placement. No neuroprotective agents have demonstrated benefit in clinical trials, suggesting the growing need to explore novel pathways and targets.
Glycogen synthase kinase-3 (GSK-3) is an evolutionary conserved ubiquitous serine/threonine kinase consisting of two distinct isoforms, GSK-3α and GSK-3β (Liang and Chuang 2007). It is a multifaceted protein that is highly expressed in the mammalian brain and involved in diverse cellular and neurophysiological functions (Chuang et al. 2011). One of the most notable qualities of GSK-3 is the vast number of signaling pathways that converge on it, suggesting that it may be an important biological target (Forde and Dale 2007; Miura and Miki 2009). GSK-3 is constitutively active under normal resting conditions (Peineau et al. 2008). A growing body of evidence indicates that activated GSK-3 is pro-apoptotic (Jendželovský et al. 2012). GSK-3 is inactivated by phosphorylation at Ser9 (McManus et al. 2005; Chuang et al. 2011). Dysregulation of GSK-3-mediated substrate phosphorylation and signaling has been implicated in several pathophysiological conditions including cancer (Luo 2009), Alzheimer's disease (Engel et al. 2006), diabetes (Eldar-Finkelman et al. 1999), and mood disorders (Li and Jope 2010).
GSK-3 acts as a regulator of apoptosis and inflammation, known contributors to stroke-induced cell death (Gao et al. 2008). Loss of GSK-3β, not GSK-3α, suppressed spontaneous neuronal death in extended culture models (Liang and Chuang 2007). Nonselective GSK-3β inhibition with lithium is neuroprotective (Chuang et al. 2011; Wei et al. 2013) and GSK-3β inhibitors are currently being tested in clinical trials for treatment of cognitive deficits and dementia (Hong-Qi et al. 2012). GSK-3β is known to interact with the mitogen-activated protein kinase family (MAPKs) and promotes signaling after stress (Kim et al. 2003). Transforming growth factor-β-activated kinase-1 (TAK1) is a member of the MAPK family that is also known as mitogen-activated protein kinase kinase kinase-7. TAK1 is activated by TGF-β, tumor necrosis factor-α (TNF-α), and other cytokines including interleukin-1 (IL-1) (Takaesu et al. 2001). TAK is also an upstream kinase of 5′ adenosine monophosphate-activated protein kinase (AMPK), a key energy sensing kinase involved in stroke. We have recently found that inhibition of TAK1 is neuroprotective after focal ischemia (White et al. 2012). Our previous work demonstrated that neuroprotective effects of TAK1 inhibition are independent of its activation of AMPK (White et al. 2012).
In the present study, we utilized GSK-3β Inhibitor VIII, a specific and highly potent GSK-3β inhibitor to examine the effects of GSK-3β inhibition on ischemic injury and stroke-induced memory impairment. Furthermore, we investigated interactions between GSK-3β, AMPK, and TAK1 signaling by using combined treatment paradigms and coimmunoprecipitation.
Results
GSK-3β inhibition significantly reduced infarct size
Significantly reduced infarct volumes were seen after ischemic stroke with both early and delayed inhibition of GSK-3β. Immediate treatment with a GSK-3β inhibitor at the onset of stroke led to a significant reduction in cortical (vehicle 51.1 ± 2.8 versus drug 40.1 ± 3.7; P < 0.05), striatal (vehicle 67.8 ± 1.6 versus drug 54.8 ± 3.4; P < 0.05), and total hemisphere (49.4 ± 2.6 versus drug 35.9 ± 2.4; P < 0.05) (n = 10/vehicle group; n = 11/drug group) infarct at 48 h of reperfusion (Fig. 1A,B). Interestingly, similar protective effects were also observed when treatment was delayed, with the first dose administered 2 h after ischemia and a second dose 24 h after ischemic onset, in the cortex (vehicle 54.8 ± 2.8 versus drug 39.2 ± 3.8; P < 0.05), striatum (vehicle 63.8 ± 2.1 versus drug 51.6 ± 3.2; P < 0.05), and in total hemisphere (vehicle 49.7 ± 3.1 versus drug 32.2 ± 1.6; P < 0.05) (Fig. 2A,B) (n = 10/group) even 7 d after injury. Three mice from the vehicle treated and one mouse from the GSK-3β inhibitor treated group died by day 7 of reperfusion. No mortality was observed in either vehicle or in GSK-3β inhibitor treated groups at 48 h after reperfusion.
Figure 1.

TTC analysis of infarct size 48 h after MCAO. The GSK-3β inhibitor was administered at the onset of stroke with a second dose at 24 h after stroke. (A) Representative TTC stained coronal sections showing neuroprotective effects of GSK-3β inhibition compared with vehicle (white = infarcted area; red = viable tissue). (B) Quantification data of infarct analysis Vehicle treated (n = 10); GSK-3β inhibitor VIII (n = 11); (*) P < 0.05. Error bars represent SEM.
Figure 2.

Neuroprotection was seen with delayed treatment with a GSK-3β inhibitor. Infarct size assessment and NORT were performed 7 d after MCAO. (A) Representative TTC stained coronal sections showing neuroprotective effects of delayed GSK-3β inhibitor treatment (initiated 2 h after stroke onset) compared with vehicle group (n = 10/group); (B) Quantification confirms the significant neuroprotective effect with GSK-3β inhibitor VIII compared with vehicle in cortex, striatum, and total hemisphere after stroke; (C) GSK-3β inhibition also significantly improved the percentage time spent with a novel object after stroke ((*) P < 0.05) (n = 10 per group). (D) GSK-3β inhibitor VIII treated mice had significantly improved learning recovery on rotarod compared with vehicle-treated mice (amount of time spent on accelerating rod after stroke). (*) P < 0.05.
GSK-3β inhibition improved neurological deficit scores and post-stroke cognitive performance in the novel object-recognition test (NORT)
Improved neurological deficit scores were seen with GSK-3β inhibition 48 h after stroke with both immediate treatment (Vehicle 3[0.25] n = 10 versus Drug 1[0.25]; n = 11; P < 0.05) and with delayed treatment (Vehicle 2[0] n = 10 versus Drug 1.0[0.5]; n = 10; P < 0.05). At 7 d, delayed treatment (given 2 and 24 h after onset of stroke) significantly reduced stroke-induced cognitive deficits as measured by the novel object-recognition test (NORT); 46.6% ± 3.5% with vehicle versus 64.0% ± 3.1% with GSK-3β inhibitor VIII, P < 0.05, (n = 9/group). No significant differences were seen between drug versus vehicle in sham mice (66.9% ± 6.1% for vehicle versus 75.1% ± 5.8% for drug, P > 0.05, n = 4/vehicle group, n = 4/drug group) which showed the expected preference for a novel versus a familiar object (Fig. 2C; Bevins and Besheer 2006). GSK-3β inhibitor treated mice showed significantly improved time spent on rotarod compared with vehicle-treated group at 6 d after stroke (41 ± 6 s for vehicle versus 67 ± 9 s for GSK-3β inhibitor, P < 0.05, n = 9/group). No differences between drug and vehicle groups were seen in spontaneous locomotor activity in the open field (1332 ± 123 for vehicle versus 1476 ± 198 for drug).
Treatment with a GSK-3β inhibitor significantly increased pGSK-3β levels but did not change pLKB1, β-Catenin, pAMPK, or pTAK1 levels
Western blot analysis was performed on brain lysates 6 h after stroke (Fig. 3). ANOVA for changes in GSK-3β protein levels showed stroke alone significantly increased pGSK-3β protein levels compared with shams in vehicle-treated groups (sham 1.0 ± 0.3 versus stroke 2.2 ± 0.3; P < 0.05). Treatment with the GSK-3β inhibitor further increased pGSK-3β levels after stroke (sham 1.4 ± 0.2 versus stroke 3.2 ± 0.14; P < 0.01), implying significant additional effects of the drug in stroke mice (Fig. 3; n = 4/sham group; n = 6/drug group).). We did not see any significant changes in pLKB1 levels with inhibitor treatment. No significant changes were seen in β-Catenin, pAMPK, or pTAK1 levels with drug treatment at this time point (data not shown).
Figure 3.

Western blot analysis confirmed the specificity and CNS availability of the GSK-3β inhibitor. (A) Representative Western blot images sham (sh) and stroke GSK-3β levels with vehicle and drug treatment (ss) (B) Quantification of GSK-3β levels show that at 6 h after stroke pGSK-3β levels increase with ischemia and this increase is significantly higher after inhibitor treatment. No significant changes in pLKB1 levels were detected. (*) P < 0.05; (n = 4 per sham group and n = 6 per stroke group).
GSK-3β inhibition was neuroprotective and improved memory even in the absence of AMPK
A significant neuroprotective effect of the GSK-3β inhibitor VIII was seen in AMPK-α2 KO mice compared with vehicle-treated AMPK-α2 KO mice (n = 10/group), in the cortex (42.9 ± 3.5 versus 30.0 ± 4.9; P < 0.05), striatum (54.6 ± 5.0 versus 43.1 ± 3.4; P < 0.05), and in the total hemisphere infarcts (36.8 ± 3.4 versus 26.9 ± 3.2; P < 0.05) (Fig. 4A). Significant differences in neurological scores between groups were seen that showed an improved functional outcome in the GSK-3β inhibitor treated group 1.0[0.5], compared with the vehicle group, 2.0[0.5], n = 10/group; P < 0.05. Compared with vehicle (54.6% ± 3.5%), the GSK-3β inhibitor VIII treated cohort (69.0% ± 4.3%) spent significantly more time with novel object; n = 10/group (Fig. 4B). No significant differences were seen in spontaneous locomotor activity.
Figure 4.

GSK-3β inhibition induces neuroprotection and cognitive recovery in AMPK-α2 KO mice. (A) Quantification of infarct volumes reveals a significant neuroprotective effect of GSK-3β inhibition in AMPK-α2 KO mice; (n = 10 per group). (B) Inhibitor treatment also significantly improved the amount of time spent with novel object in AMPK-α2 KO mice. (*) P < 0.05; (n = 10 per group).
No additional neuroprotective or cognitive benefits were seen with combined inhibition of TAK1 and GSK-3β compared with GSK3β treatment alone
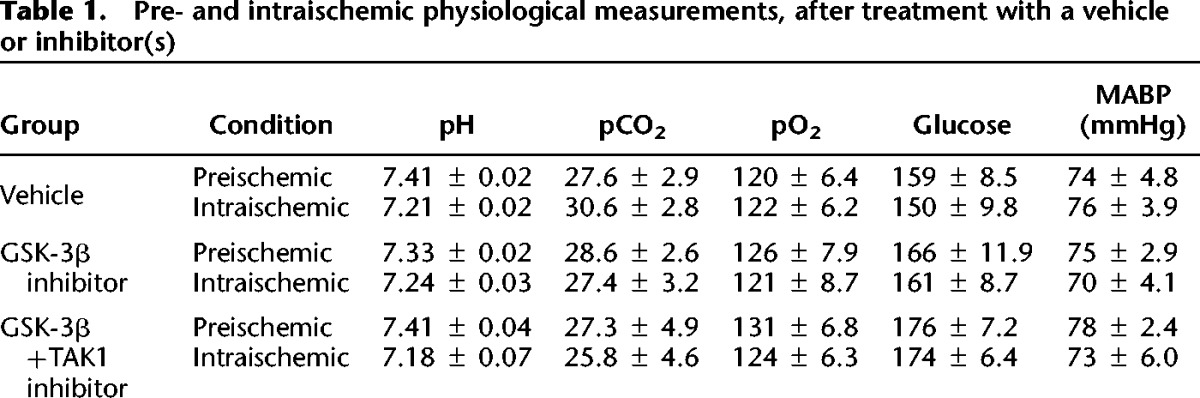
We then investigated whether an additive neuroprotective effect was seen with simultaneous inhibition of both TAK1 and GSK-3β. Compared with GSK-3β inhibition alone, combined GSK-3β + TAK1 inhibition did not offer any additional neuroprotection in the cortex (32.4 ± 4.1 versus 33.6 ± 3.4; P > 0.05), striatum (58.9 ± 3.0 versus 56.0 ± 2.9; P > 0.05), or total hemisphere (33.6 ± 2.8 versus 30.7 ± 3.9; P > 0.05) (Fig. 5A). No significant differences in neurological scores were seen between GSK-3β inhibitor group alone (1.5[0.75] n = 9) compared with (GSK-3β + TAK1) inhibitor treated groups (1.0[0.25]; n = 9; P > 0.05). Furthermore, the GSK-3β + TAK1 treated group showed no additional improvement in memory (65.5% ± 4.0% for GSK-3β inhibitor alone versus 66.2% ± 4.8% for (GSK-3β + TAK1) inhibitors, P > 0.05, (n = 9/group) (Fig. 5B) nor in learning ability on rotarod (64 ± 7 s for GSK-3β inhibitor alone versus 66 ± 6s for (GSK-3β + TAK1) inhibitors, P > 0.05, (n = 9/group). Western blot analysis revealed TAK1 inhibition also inhibited GSK-3β (Supplemental Fig. 1). No significant differences were seen in any physiological parameters in pre- and intraischemic period at acute time point (including pH, pCO2, pO2, glucose, mean arterial pressure) (Table 1) between drug- and vehicle-treated groups.
Figure 5.

GSK-3β inhibition along with TAK1 inhibition has no additional neuroprotective effects compared with GSK-3β inhibition alone. (A) Quantification of infarct volume reveals no significant additional neuroprotective effects of GSK-3β inhibition in combination with TAK1 inhibition; (n = 9 per group). (B) Combined inhibitor treatment did not improve the percentage of time spent with a novel object compared with GSK-3β inhibition alone. (*) P < 0.05; (n = 9 per group). (C) Combined inhibitor treatment did not improve the amount of time spent on rotarod compared with GSK-3β inhibition alone suggesting a lack of additional benefit on post-stroke learning recovery. (*) P < 0.05.
Table 1.
Pre- and intraischemic physiological measurements, after treatment with a vehicle or inhibitor(s)
Coimmunoprecipitation revealed a possible interaction between TAK1 and pGSK-3β
To further investigate if TAK1 interacts with pGSK-3β, brain homogenates from additional cohorts of mice were examined 6 h after sham and stroke surgery to determine if the neuroprotective effect and cognitive improvement seen with GSK-3β and TAK1 inhibition occurs via a direct interaction between TAK1 and pGSK-3β. Coimmunoprecipitation studies with anti-TAK1 and anti-pGSK-3β antibodies in brain homogenates revealed a clear interaction between TAK1 and pGSK-3β in both stroke and sham brain lysates (Fig. 6). IgG controls and whole lysate were run for controls.
Figure 6.

Coimmunoprecipitation followed by Western blot revealed that TAK1 interacts with pGSK-3β. Co-ip of whole-cell lysate obtained from sham (sh) and stroke (ss) hemispheres of TAK1 (total, rabbit) lysate probed with pGSK-3β (mouse) demonstrated a significant interaction of TAK1 and pGSK-3β in both sham and stroke brains. Flow through and IgG controls were used as positive controls.
Discussion
Inhibition of GSK-3β enhances cell survival in several in vitro and in vivo studies (Zeng et al. 2014). pGSK-3β signaling is involved in neuroprotection, and appears to have a role in improved memory in cognitive disorders (Koh et al. 2008; Hong et al. 2012; Llorens-Martin et al. 2013). Most previous studies used nonselective inhibitors given immediately prior to or at stroke onset, which may not translate into benefit in clinical populations where treatment is delayed several hours after ischemia. The results of the present study confirm that pGSK-3β signaling is detrimental in ischemic stroke and also demonstrates several new findings. (1) GSK-3β inhibition either immediately or even with delayed treatment (2 h after stroke), reduced the severity of ischemic lesion; (2) GSK-3β inhibition enhanced post-stroke cognitive recovery suggesting potential for a translatable therapy; (3) the neuroprotective effect is independent of AMPK signaling and finally (4) that TAK1 and pGSK-3β inhibition appear to share a common pathway that leads to improved stroke outcomes.
Accumulating evidence suggests that GSK-3 plays an important role in apoptosis and inflammation. Overexpression of GSK-3 induces cell death and its inhibition is protective (Miura and Miki 2009). As several studies have shown that administration of a GSK-3β inhibitor can protect cultured neurons against excitotoxicity and toxin-induced apoptosis (Liang and Chuang 2007; Chuang et al. 2011), and reduce the presence of pyknotic neurons in a model of Alzheimer's disease (Hu et al. 2009), we questioned if similar benefits could be observed following stroke by using the potent and selective GSK-3β inhibitor VIII (Koh et al. 2008). Earlier studies that utilized GSK-3β inhibitor VIII were performed in in vitro cell culture systems (Nair and Olanow 2008) but in vivo rat models have also demonstrated the efficacy of this agent when administered through the external jugular vein (Koh et al. 2007, 2008). Although a few studies have shown a benefit of nonselective GSK inhibitors in stroke, these were limited by lack of pharmacological specificity of the inhibitor used, and the mechanism was not clearly identified. Most of these studies also only examined inhibition initiated prior to or immediately at the onset of stroke, and assessed only acute endpoints (1–2 d), limiting their translational relevance. Changes in pGSK-3β levels have been previously documented after stroke (Chuang et al. 2011; Wei et al. 2013). Consistent with these earlier studies we found that stroke-induced phosphorylation of GSK-3β, suggesting that GSK-3β activation may be an endogenous neuroprotective response to stroke. Treatment with GSK-3β inhibitor VIII further inhibited GSK-3β as visualized as increased pGSK-3β in drug-treated brain lysates at 6 h after stroke confirming the potency and bioavailability of the inhibitor (Liang and Chuang 2007).
The roles for GSK-3 in neuronal differentiation, maturation, and development are increasingly recognized. For example, overexpression of GSK-3β in the central nervous system of developing mice results in a reduction in the size of the brain and spinal cord (Spittaels et al. 2002), and GSK-3β gene disruption causes embryonic lethality (Hoeflich et al. 2000). One of the most studied mechanisms of GSK-3 regulation is via the canonical Wnt pathway (Valvezan and Klein 2012). Although we observed a significant increase in GSK-3β phosphorylation with drug treatment, we did not find a corresponding decrease in β-Catenin phosphorylation (data not shown). Similar findings were reported in recent study using a traumatic brain injury model (Dash et al. 2011), the reason for this is not clear at present, it is possible that the activity of GSK-3α, which can also phosphorylate β-Catenin, was unaffected by stroke (Soutar et al. 2010; Dash et al. 2011) and maintained β-Catenin phosphorylation levels.
In our previous studies, we found that global AMPK inhibition or genetic deletion of the catalytic α2 isoform confers neuroprotection after stroke, demonstrating that AMPK-α2 is critical in the response to an ischemic challenge (McCullough et al. 2005; Li et al. 2007; Venna et al. 2012a). The mechanisms by which acute AMPK activation increases ischemic damage remains unclear, but increased glucose uptake due to unregulated glucose transporters in the reperfusion phase, enhanced lactate accumulation, and increased autophagy likely contribute to increased neuronal death after ischemic reperfusion (McCullough et al. 2005; Li et al. 2007; Arsikin et al. 2012). A recent study has shown that GSK-3β acts as a critical sensor for anabolic signaling and regulates AMPK; others have found that AMPK activation mediates learning and memory (Suzuki et al. 2013; Kobilo et al. 2014). To address the role of AMPK in the neuroprotective and cognitive benefit of GSK-3β inhibition, we treated AMPK-α2 deficient mice with GSK-3β inhibitor VIII. AMPK-α2 deficient mice have smaller infarcts at baseline compared with WT mice (Li et al. 2007). We hypothesized that the GSK-3β inhibitor would be ineffective in the absence of catalytic AMPK-α2 if signaling was mediated by the same pathway. To our surprise, the GSK-3β inhibitor maintained its neuroprotective effects and increased cognitive recovery in these knockout mice. This suggests that neuroprotective effects of GSK-3β inhibition are mediated through pathways that are independent of AMPK signaling.
Once it was clear that AMPK was not the signaling pathway by which GSK-3 β inhibition exerts its neuroprotective effects in this model, we investigated the role of TAK1. GSK has been recently demonstrated to increase the stabilization of TAK1's interaction with its binding partner, TAB (TAK1-binding protein) (Bang et al. 2013), but no studies have investigated the direct interaction of GSK-3β and TAK1. Acute inhibition of TAK1 is neuroprotective and stroke-induced memory deficits can be ameliorated by TAK1 inhibition (White et al. 2012). Activation of TAK1 enhances caspase activity via downstream JNK signaling (Sanna et al. 2002). TAK1 has also been implicated in pro-inflammatory signaling through induction of the transcription factor AP-1 with the subsequent expression of inflammatory genes such as COX-2 (Sanna et al. 2002; Zhang et al. 2010b). GSK-3β also has a strong regulatory effect on inflammation in neurodegeneration models that have associated impairments in memory. Although GSK3β and TAK1 appear to share similar mechanisms that regulate cell survival or cell death, it is unknown if they interact with each other after injury. Interestingly, no additive neuroprotective effect was seen with combined TAK and GSK inhibition suggesting that these share a common signaling pathway (Supplemental Fig. 2). Although it is possible that there was a “floor” effect, this is unlikely as the residual infarct was still >30% of the ischemic hemisphere even after combination treatment, due to the length of the occlusion. Infarct in this model has the potential to be reduced to as low as 16% (Zhang et al. 2010a). However, treatment with the GSK3β inhibitor had no effect on pTAK1 levels. To directly assess interaction after ischemic injury, we performed co-IP studies. A clear TAK1/pGSK-3β interaction was seen, and this interaction was reduced after stroke. We speculate that this TAK1/pGSK-3β interaction restrains deleterious pro-apoptotic signaling during stroke, and is consistent with our results showing that the inhibition TAK1 or GSK3β is neuroprotective. The downstream effects and targets of this TAK1/pGSK-3β interaction are not yet known but likely include JNK/c-Jun signaling as seen in prior studies (White et al. 2012). While these findings are encouraging, this work has several limitations. This study only used pharmacological methods, as mice with genetic deletion of GSK3β are unavailable due to lethality. In addition the effects seen on cognitive “recovery” may simply reflect the reduction in infarct size, and the acute neuroprotective effects of the inhibitor, rather than enhanced recovery. In addition mice were studied for only 1 wk post stroke. In clinical settings, cognitive deficits may improve even months to years after stroke, unlike in the mouse where recovery is more rapid. Despite these limitations, the results from the current study represent a step forward toward realizing the potential benefits of GSK3β inhibition for post-stroke cognitive deficits.
In conclusion, this study demonstrates that both early and delayed pharmacological inhibition of GSK-3β following stroke leads to neuroprotection and improved post-stroke cognitive recovery. This work also shows the novel finding that TAK1 interacts with pGSK-3β. These molecular pathways appear to share a common signaling pathway. Given that aberrant GSK-3β activity has been implicated in the pathophysiology of stroke, is involved in the pathogenesis of dementia and memory disorders, and that the development of GSK-3β-specific inhibitors is ongoing, GSK-3β inhibitors may become a viable therapeutic intervention for both acute stroke and to treat post-stroke cognitive deficits.
Materials and Methods
Experimental animals
C57Bl/6 male mice were purchased from Charles River laboratories (Wilmington, MA). All mice used for these experiments were 20–25 g, housed five per cage with ad libitum access to food and water. All animal work was approved by the Center for Animal Care at University of Connecticut Health Center and was performed in accordance with National Institutes of Health guidelines. Surgical procedures were performed following the five criteria derived from the stroke therapy academic industry roundtable (STAIR) group guidelines for preclinical evaluation of stroke therapeutics: (1) cerebral blood flow was monitored by using laser Doppler to confirm the MCA occlusion; (2) mice were randomly assigned to treatment groups; (3) investigators were blinded to treatment administration; (4) analysis were performed by researcher blinded to treatments; and (5) core body temperature were monitored and maintained during the surgery period (Philip et al. 2009).
Drugs and treatment groups
The potent GSK-3β Inhibitor, GSK-3β Inhibitor VIII, was purchased from Calbiochem, a highly selective TAK1 inhibitor, 5Z-7-oxozeaenol was purchased from Tocris Bioscience and both inhibitors were diluted in a small quantity (5 mg/130 μL) of DMSO (Sigma) and the desired volume was achieved by adding saline (Hospira) pH balanced to 7.4. Control mice were treated with a corresponding volume of DMSO in saline (vehicle). Dose volumes were 100 μL/10 g body weight. Inhibitors and their doses were carefully selected from the literature and from our previous study using TAK1 inhibitor (Koh et al. 2008; White et al. 2012). All control animals were treated with equal volume of vehicle. For first experiment mice were treated with GSK-3β Inhibitor VIII (4 mg/kg, 10 min before the onset intraperitoneally (i.p.)) and second doses were injected at 24 h from the onset of MCAO. Infarct analysis was performed on brains after 48 h of reperfusion. For the second set of experiments the GSK-3β Inhibitor VIII (4 mg/kg) treatment was delayed and doses were administered at 2 and 24 h after the onset of stroke. To test durability of the effect the infarcts were analyzed at 7 d after stroke. An additional cohort of AMPK-α2 KO mice were used to investigate the efficacy of the GSK-3β inhibitor in a similar paradigm as above, with delayed dosing as in the second set of experiments. A separate cohort of animals was used to test the combined effect of TAK and GSK inhibitors. Mice were either treated with GSK-3β Inhibitor VIII (4 mg/kg) alone or in combination with the TAK inhibitor 5Z-7-oxozeaenol (5 mg/kg) administered i.p. 2 h after the onset of stroke with a second dose at 24 h after stroke. Infarcts analysis and NORT were performed at 7 d of reperfusion.
Middle cerebral artery occlusion model
Cerebral ischemia was induced by 90 min of reversible middle cerebral artery occlusion (MCAO) under Isoflurane anesthesia, as described previously (Venna et al. 2012b; White et al. 2012). In brief, a midline ventral neck incision was made, and unilateral right MCAO was performed by advancing a 6.0 silicone-coated nylon monofilament into the internal carotid artery 6 mm from the internal carotid-pterygopalatine artery bifurcation via an external carotid artery stump. Rectal temperatures were continuously monitored with a Monotherm system (VWR LabShop) and temperature was maintained with a heating pad at ∼37°C during surgery and ischemia. Separate cohorts of mice were used to assess changes in physiological variables such as mean arterial blood pressure and cortical cerebral perfusion data were obtained as previously described (Li et al. 2010). Cerebral blood flow measurements by laser Doppler flowmetry (DRT 4/Moor Instruments Ltd) confirmed ischemic occlusion (reduction to 85% of baseline) during MCAO and restoration of blood flow during reperfusion. Surgical controls are used for Western blot analysis, a sham surgery in which the suture was not advanced into the carotid artery (controls). Additional cohorts (n = 4/group) were used for investigating differences in physiological parameters.
Infarct analysis
After 48 h or 7 d of reperfusion all animals were euthanized, brains for infarct analysis were collected and hardened at −20°C for 5 min, cut into five 2-mm coronal sections and stained with 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) for 8 min at 38°C. Slices were formalin-fixed (4%) and then digitalized and infarct volumes analyzed using Sigma Scan Pro software as previously described (Venna et al. 2012b). The final infarct volumes are presented as percentage volume (percentage of contralateral structures with correction for edema).
Behavioral testing
Neurological scores
Neurological deficit scores (Holmes et al. 2008) were obtained during the intraischemic period and at 24 h post-stroke. Our standard scoring system was as follows: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when held by tail; 2, circling to affected side; 3, unable to bear weight on affected side; and 4, no spontaneous locomotor activity or barrel rolling as described previously (Venna et al. 2012b; White et al. 2012).
Open-field analysis
Mice were acclimatized to the testing room for 1 h prior to the test. Testing was performed during the light phase of the circadian cycle, between 9:00 a.m. and 12:00 p.m. under normal fluorescent room lights. For testing, mice were individually placed in the open-field chamber (15″ × 15″) equipped with 16 infrared beam emitting LEDs on each side for a duration of 20 min. The total number of beam breaks was automatically collected by a computer-operated PAS Open-Field system (San Diego Instruments). The open-field chambers were cleaned after each individual test session using 70% ethanol (White et al. 2012).
Object-recognition test
The novel object-recognition test (NORT) was performed as described earlier (Bevins and Besheer 2006; White et al. 2012). In brief, animals were placed individually in a Plexiglas chamber containing two identical objects and allowed to habituate with the objects for duration of 10 min at 7 d after stroke. Animals should spend at least 38 sec with each object to be selected for test. Mice were returned to their home cages and were reintroduced 1 h later for a 5-min exploration test, at this time one of the objects was replaced with a novel object. Data was recorded by an investigator blinded to treatment conditions and time spent exploring the novel object was expressed as a percentage.
Rotarod
Prior to stroke all animals were trained four trials per day for 3 d on the rotarod apparatus (Med Associates). Latency was assessed by measuring the length of time each mouse remained on the rotating drum as it accelerated from 4 to 40 rpm over a span of 2 min. The latency of each mouse to fall from the rotating drum was recorded for each trial (in seconds), and the average latency was used for further analysis. Animals were then tested on day 2 and day 6 after surgery, with four trials as in the training period, and the average latency was recorded.
Western blotting
Western blots were performed as described previously (Venna et al. 2012b; White et al. 2012). Six hours after the onset of cerebral ischemia or a sham surgery with vehicle and GSK-3β inhibitor treatment, the mice were euthanized; brains were rapidly collected and flash frozen. The brain was homogenized in lysis buffer, a BCA assay was performed to determine protein concentrations and an equal amount of protein was loaded on a 4%–15% gradient sodium dodecyl sulfate–polyacrylamide gel and subsequently transferred to a polyvinylidene difluoride membrane. p-GSK-3β (Ser-9) (1:2500;Santa Cruz Biotechnology), TAK1 (1:500; Cell Signaling Technologies), β-Catenin (1:1000; Cell Signaling Technologies); β-actin (1:5000; Sigma) was used as the loading control. Blots were incubated overnight in primary antibody at 4°C in Tris-buffered saline containing 4% bovine serum albumin in 0.1% Tween20. Secondary antibodies (goat anti-rabbit IgG 1:10,000 and goat antimouse IgG; Chemicon) were diluted and incubated for 45 min at room temperature, ECL (Pico) detection kit (ThermoScientific) was used for signal detection. Densitometry was performed using Adobe Photoshop software.
Coimmunoprecipitation
Coimmunoprecipitation (co-ip) was performed using Protein A-Sepharose beads. Whole-cell lysate (100 µg) from the right hemisphere of sham (Sh) and stroke (ss) mice was incubated for 1 h with 1 μg of the TAK1 antibody at room temperature to form the immune complex. Then, 100 µL of a 50% slurry of Protein A beads in PBS was added to the complex and incubated for 1 h at room temperature as described in Turtzo et al. (2013). Beads were spun down, the unbound fraction removed and then the beads were extensively washed. To elute off the complex, the beads were incubated in sample buffer and boiled. Western blotting was performed (as detailed above) to assess the pGSK-3 interaction.
Statistics
Data were expressed as mean ± SEM except for NDS, which was presented as median (interquartile range). Statistics were performed with analysis of variance (ANOVA) with Tukey's post hoc correction for multiple comparisons, or by Mann–Whitney U test for nonparametric values (Holmes et al. 2008). A probability value P < 0.05 was considered to be statistically significant. Mice were randomly assigned to sham or stroke surgery and drug or vehicle treatments using SPSS program. Investigators performing MCAO, behavioral, and infarct size analysis were blinded to treatment conditions.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (National Institute of Neurological Disorders and Stroke R01 NS050505 and R21 NS088969-01) to L.D.M. and American Heart and Stroke Association fellowship and an honorarium to V.R.V.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.038083.115.
Competing Interest Statement
The authors declare no competing financial interests.
References
- Arsikin K, Kravic-Stevovic T, Jovanovic M, Ristic B, Tovilovic G, Zogovic N, Bumbasirevic V, Trajkovic V, Harhaji-Trajkovic L. 2012. Autophagy-dependent and -independent involvement of AMP-activated protein kinase in 6-hydroxydopamine toxicity to SH-SY5Y neuroblastoma cells. Biochim Biophys Acta 1822: 1826–1836. [DOI] [PubMed] [Google Scholar]
- Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS. 2013. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov 3: 690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. 2006. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1: 1306–1311. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Wang Z, Chiu CT. 2011. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci 4: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Johnson D, Clark J, Orsi SA, Zhang M, Zhao J, Grill RJ, Moore AN, Pati S. 2011. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS One 6: e24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG. 1999. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 48: 1662–1666. [DOI] [PubMed] [Google Scholar]
- Engel T, Hernández F, Avila J, Lucas JJ. 2006. Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26: 5083–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin VL, Lawes CM, Bennett DA, Anderson CS. 2003. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol 2: 43–53. [DOI] [PubMed] [Google Scholar]
- Forde JE, Dale TC. 2007. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 64: 1930–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HK, Yin Z, Zhou N, Feng XY, Gao F, Wang HC. 2008. Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J Cardiovasc Pharmacol 52: 286–292. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al. 2014. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 129: e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. 2000. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 406: 86–90. [DOI] [PubMed] [Google Scholar]
- Holmes T, O’Brien TA, Knight R, Lindeman R, Symonds G, Dolnikov A. 2008. The role of glycogen synthase kinase-3β in normal haematopoiesis, angiogenesis and leukaemia. Curr Med Chem 15: 1493–1499. [DOI] [PubMed] [Google Scholar]
- Hong JG, Kim DH, Lee CH, Park SJ, Kim JM, Cai M, Jang DS, Ryu JH. 2012. GSK-3β activity in the hippocampus is required for memory retrieval. Neurobiol Learn Mem 98: 122–129. [DOI] [PubMed] [Google Scholar]
- Hong-Qi Y, Zhi-Kun S, Sheng-Di C. 2012. Current advances in the treatment of Alzheimer's disease: focused on considerations targeting Aβ and τ. Transl Neurodegener 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. 2009. GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 33: 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendželovský R, Koval J, Mikeš J, Papčová Z, Plšíková J, Fedoročko P. 2012. Inhibition of GSK-3β reverses the pro-apoptotic effect of proadifen (SKF-525A) in HT-29 colon adenocarcinoma cells. Toxicol In Vitro 26: 775–782. [DOI] [PubMed] [Google Scholar]
- Kim JW, Lee JE, Kim MJ, Cho EG, Cho SG, Choi EJ. 2003. Glycogen synthase kinase 3β is a natural activator of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1). J Biol Chem 278: 13995–14001. [DOI] [PubMed] [Google Scholar]
- Kobilo T, Guerrieri D, Zhang Y, Collica SC, Becker KG, van Praag H. 2014. AMPK agonist AICAR improves cognition and motor coordination in young and aged mice. Learn Mem 21: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Kim Y, Kim HY, Hwang S, Lee CH, Kim SH. 2007. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp Neurol 205: 336–346. [DOI] [PubMed] [Google Scholar]
- Koh SH, Yoo AR, Chang DI, Hwang SJ, Kim SH. 2008. Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem Biophys Res Commun 371: 894–899. [DOI] [PubMed] [Google Scholar]
- Li X, Jope RS. 2010. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology 35: 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. 2007. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke 38: 2992–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Benashski SE, Venna VR, McCullough LD. 2010. Effects of metformin in experimental stroke. Stroke 41: 2645–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. 2007. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem 282: 3904–3917. [DOI] [PubMed] [Google Scholar]
- Llorens-Martin M, Fuster-Matanzo A, Teixeira CM, Jurado-Arjona J, Ulloa F, Defelipe J, Rabano A, Hernandez F, Soriano E, Avila J. 2013. GSK-3β overexpression causes reversible alterations on postsynaptic densities and dendritic morphology of hippocampal granule neurons in vivo. Mol Psychiatry 18: 451–460. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, et al. 2010. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121: e46–e215. [DOI] [PubMed] [Google Scholar]
- Luo J. 2009. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett 273: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. 2005. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem 280: 20493–20502. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. 2005. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J 24: 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Miki T. 2009. GSK-3β, a therapeutic target for cardiomyocyte protection. Circ J 73: 1184–1192. [DOI] [PubMed] [Google Scholar]
- Nair VD, Olanow CW. 2008. Differential modulation of Akt/glycogen synthase kinase-3β pathway regulates apoptotic and cytoprotective signaling responses. J Biol Chem 283: 15469–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingridge GL. 2008. The role of GSK-3 in synaptic plasticity. Br J Pharmacol 153 Suppl 1: S428–S437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Benatar M, Fisher M, Savitz SI. 2009. Methodological quality of animal studies of neuroprotective agents currently in phase II/III acute ischemic stroke trials. Stroke 40: 577–581. [DOI] [PubMed] [Google Scholar]
- Sanna MG, da Silva Correia J, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux QL, Ulevitch RJ. 2002. IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol Cell Biol 22: 1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, Snider WD, Gordon-Wks PR, Sutherland C. 2010. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem 115: 974–983. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Terwel D, Vandezande K, Lasrado R, Bruynseels K, Irizarry M, Verhoye M, Van Lint J, et al. 2002. Neonatal neuronal overexpression of glycogen synthase kinase-3 β reduces brain size in transgenic mice. Neuroscience 113: 797–808. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, Inoki K. 2013. Inhibition of AMPK catabolic action by GSK3. Mol Cell 50: 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K. 2001. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol Cell Biol 21: 2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtzo LC, Li J, Persky R, Benashski S, Weston G, Bucala R, Venna VR, McCullough LD. 2013. Deletion of macrophage migration inhibitory factor worsens stroke outcome in female mice. Neurobiol Dis 54: 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaartjes I, O'Flaherty M, Capewell S, Kappelle J, Bots M. 2013. Remarkable decline in ischemic stroke mortality is not matched by changes in incidence. Stroke 44: 591–597. [DOI] [PubMed] [Google Scholar]
- Valvezan AJ, Klein PS. 2012. GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front Mol Neurosci 5: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venna VR, Li J, Benashski SE, Tarabishy S, McCullough LD. 2012a. Preconditioning induces sustained neuroprotection by downregulation of adenosine 5′-monophosphate-activated protein kinase. Neuroscience 201: 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venna VR, Weston G, Benashski SE, Tarabishy S, Liu F, Li J, Conti LH, McCullough LD. 2012b. NF-κB contributes to the detrimental effects of social isolation after experimental stroke. Acta Neuropathol 124: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Yao X, Yang L, Wang S, Guo F, Zhou H, Marsicano G, Wang Q, Xiong L. 2013. Glycogen synthase kinase-3β is involved in electroacupuncture pretreatment via the cannabinoid CB1 receptor in ischemic stroke. Mol Neurobiol 49: 326–336. [DOI] [PubMed] [Google Scholar]
- White BJ, Tarabishy S, Venna VR, Manwani B, Benashski S, McCullough LD, Li J. 2012. Protection from cerebral ischemia by inhibition of TGFβ-activated kinase. Exp Neurol 237: 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Liu D, Qiu Z, Huang Y, Chen B, Wang L, Xu H, Huang N, Liu L, Li W. 2014. GSK3β overexpression indicates poor prognosis and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS One 9: e91231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H, et al. 2010a. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol 184: 4087–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lei T, Chen X, Peng Y, Long H, Zhou L, Huang J, Chen Z, Long Q, Yang Z. 2010b. Resistin up-regulates COX-2 expression via TAK1-IKK-NF-κB signaling pathway. Inflammation 33: 25–33. [DOI] [PubMed] [Google Scholar]
- Ziegler S, Rohrs S, Tickenbrock L, Moroy T, Klein-Hitpass L, Vetter IR, Muller O. 2005. Novel target genes of the Wnt pathway and statistical insights into Wnt target promoter regulation. FEBS J 272: 1600–1615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.